
Abstract
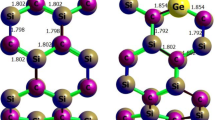
In the present investigation, the feasibility of detecting the chlorofluoromethane (CFM) gas molecule onto the outer surface of pristine single layer boron nitride nanosheet (BNNS), as well as its aluminum (Al)– and gallium (Ga)–doped structures, was carefully evaluated. For achieving this goal, a density functional theory level of study using the Perdew, Burke, and Ernzerhof exchange–correlation (PBEPBE) functional together with a 6-311G(d) basis set has been used. Subsequently, the B3LYP, CAM-B3LYP, wB97XD, and M062X functionals with a 6-311G(d) basis set were also employed to consider the single-point energies. Natural bond orbital (NBO) and quantum theory of atoms in molecules (QTAIM) were implemented by using the B3LYP-D3/6-311G(d) method, and the results were compatible with the electronic properties. In this regard, the total density of states (TDOSs), the Wiberg bond index (WBI), natural charge, natural electron configuration, donor–acceptor natural bond orbital interactions, and the second-order perturbation energies are performed to explore the nature of the intermolecular interactions. All of the energy calculations and population analyses denote that by adsorbing of the gas molecule onto the surface of the considered nanostructures, the intermolecular interactions are of the type of strong chemical adsorption. Among the doped nanosheets, Ga-doped nanosheet has very high adsorption energy compared with other elements (i.e., Ga-doped > Al-doped > pristine). Generally, it was revealed that the sensitivity of the adsorption will be increased when the gas molecule interacts with decorated nanosheets and decrease the HOMO-LUMO band gap; therefore, the change of electronic properties can be used to design suitable nanosensors to detect CFM gas.

Graphical abstract







Similar content being viewed by others

References
Abbasi M, Nemati-Kande E, Mohammadi MD (2018) Doping of the first row transition metals onto B12N12 nanocage: a DFT study. Comput Theoretical Chem 1132:1–11
Ahmadi R (2012) Computational study of chemical properties of captopril drug and the connected form to fullerene (C60) as a medicine nano carrier. J Phys Theor Chem 9:185–190
Ahmadi R, Salmaniha M (2014) Investigation of chemical properties in fullerene derivatives of fluoxetine drug: a DFT study. Int J New Chem 1:151–159
Esrafili MD, Mousavian P, Rad FA (2018) Adsorption of formamide over pristine and Al-doped boron nitride nanosheets: a dispersion-corrected DFT study. J Mol Graph Model 82:101–107
Mohammadi MD, Hamzehloo M (2018) The adsorption of bromomethane onto the exterior surface of aluminum nitride, boron nitride, carbon, and silicon carbide nanotubes: a PBC-DFT, NBO, and QTAIM study. Comput Theoretical Chem 1144:26–37
Nemati-Kande E, Abbasi M, Doust Mohammadi M (2018) DFT, QTAIM and NBO investigation of the interaction of rare gases with pristine and decorated boron nitride nanotube. ChemistrySelect 3:9833–9840
Nemati-Kande E, Abbasi M, Mohammadi MD (2020) DFT studies on the interactions of pristine, Al and Ga-doped boron nitride nanosheets with CH3X (X= F, Cl and Br). J Mol Struct 1199:126962
Nemati-Kande E, Abbasi M, Mohammadi MD (2019) Feasibility of pristine and decorated AlN and SiC nanotubes in sensing of noble gases: a DFT study. ChemistrySelect 4:2453–2462
Nemati-Kande E, Karimian R, Goodarzi V, Ghazizadeh E (2020) Feasibility of pristine, Al-doped and Ga-doped boron nitride nanotubes for detecting SF4 gas: a DFT, NBO and QTAIM Investigation. Appl Surf Sci:145490
Li LH, Xing T, Chen Y, Jones R (2014) Boron nitride nanosheets for metal protection. Adv Mater Interfaces 1:1300132
Fu L, Chen G, Jiang N, Yu J, Lin C-T, Yu A (2016) In situ growth of metal nanoparticles on boron nitride nanosheets as highly efficient catalysts. J Mater Chem A 4:19107–19115
Luo W, Wang Y, Hitz E, Lin Y, Yang B, Hu L (2017) Solution processed boron nitride nanosheets: synthesis, assemblies and emerging applications. Adv Funct Mater 27:1701450
Raza MA, Nadeem A, Ilyas MT (2019) Corrosion study of boron nitride nanosheets deposited on copper metal by electrophoretic deposition. TMS 2019 148th Annual Meeting & Exhibition Supplemental Proceedings. Springer, pp 681–685
Omidirad R, Azizi K (2019) DFT study of charge-controlled mechanism of water molecule dissociation on vacancy defected boron nitride nanosheets. J Mol Graph Model 93:107448
Esrafili MD, Saeidi N (2018) Carbon-doped boron nitride nanosheet as a promising catalyst for N2O reduction by CO or SO2 molecule: a comparative DFT study. Appl Surf Sci 444:584–589
Esrafili MD (2018) NO reduction by CO molecule over Si-doped boron nitride nanosheet: a dispersion-corrected DFT study. Chem Phys Lett 695:131–137
Moladoust R, Esrafili MD, Hosseinian A, Alkorta I, Vessally E (2019) Adsorption sensitivity of pristine and Al-or Si-doped boron nitride nanoflake to COCl2: a DFT study. Mol Phys 117:626–634
Esrafili MD (2018) Epoxidation of ethylene over carbon and silicon-doped boron nitride sheets: a comparative DFT study. Solid State Commun 284:35–39
Esrafili MD, Asadollahi S, Heydari S (2019) A DFT study on NO reduction to N2O using Al-and P-doped hexagonal boron nitride nanosheets. J Mol Graph Model 89:41–49
Dorn RW, Ryan MJ, Kim T-H, Goh TW, Venkatesh A, Heintz PM, Zhou L, Huang W, Rossini AJ (2020) Identifying the molecular edge termination of exfoliated hexagonal boron nitride Nanosheets with solid-state NMR spectroscopy and plane-wave DFT calculations. Chem Mater 32:3109–3121
Ai Z, Chang B, Xu C, Huang B, Wu Y, Hao X, Shao Y (2019) Interface engineering in the BNNS@ Ti 3 C 2 intercalation structure for enhanced electrocatalytic hydrogen evolution. New J Chem 43:8613–8619
Azamat J, Khataee A, Joo SW (2016) Separation of copper and mercury as heavy metals from aqueous solution using functionalized boron nitride nanosheets: a theoretical study. J Mol Struct 1108:144–149
Esrafili MD, Saeidi N (2017) A DFT study on the healing of N-vacancy defects in boron nitride nanosheets and nanotubes by a methylene molecule. Int J Quantum Chem 117:e25450
Esrafili MD, Saeidi N, Nematollahi P (2016) The healing of B-or N-vacancy defective BNNTs by using CO molecule: a DFT study. New J Chem 40:8024–8031
Li H, Chen Z, Fang X, Tie D (2015) Absorption of NH3 on pristine and defected boron nitride nanosheets: a first principle study. Superlattice Microst 88:371–376
Lin S, Huang J, Gao X (2015) A Cu (111) supported h-BN nanosheet: a potential low-cost and high-performance catalyst for CO oxidation. Phys Chem Chem Phys 17:22097–22105
Binbrek OS, Torrie BH, Swainson IP (2002) Neutron powder-profile study of chlorofluoromethane. Acta Crystallogr Sect C: Cryst Struct Commun 58:o672–o674
U.N.E.P.O. Secretariat, Handbook for the Montreal protocol on substances that deplete the ozone layer, UNEP/Earthprint2006
Favero LB, Maris A, Melandri S, Ottaviani P, Caminati W (2019) Non covalent interactions stabilizing the chiral dimer of CH 2 ClF: a rotational study. Phys Chem Chem Phys 21:3695–3700
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865
Hehre WJ, Ditchfield R, Pople JA (1972) Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys 56:2257–2261
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Petersson G, Nakatsuji H (2016) Gaussian 16. Gaussian, Inc., Wallingford CT (2016)
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592
Lu T, Chen F (2012) Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J Mol Graph Model 38:314–323
Lu T, Chen Q (2020) A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure. Theor Chem Accounts 139:25
J.B. Foresman, A. Frisch, Exploring chemistry with electronic structure methods: a guide to using Gaussian, (1996)
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem 27:1787–1799
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132:154104
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32:1456–1465
Parr RG, Donnelly RA, Levy M, Palke WE (1978) Electronegativity: the density functional viewpoint. J Chem Phys 68:3801–3807
von Szentpály L (1998) Valence states in molecules. 3. Transferable vibrational force constants from homonuclear data. J Phys Chem A 102:10912–10915
Janak J (1978) Proof that∂ e∂ n i= ε in density-functional theory. Phys Rev B 18:7165
Parr RG (1980) Density functional theory of atoms and molecules. Horizons of Quantum Chemistry, Springer, Berlin
Chattaraj P, Perennial RD, Parr RG (1991) HSAB principle. J Am Chem Soc 113:1855–1856
Parr RG (1922-1924) L.v. Szentpaly, S. Liu, Electrophilicity index. J Am Chem Soc 121(1999)
Noorizadeh S, Maihami H (2006) A theoretical study on the regioselectivity of Diels–Alder reactions using electrophilicity index. J Mol Struct THEOCHEM 763:133–144
Weinhold F (2012) Discovering chemistry with natural bond orbitals. John Wiley & Sons, New York
Weinhold F, Landis CR (2005) Valency and bonding: a natural bond orbital donor-acceptor perspective. Cambridge University Press, Cambridge
Carpenter J, Weinhold F (1988) Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J Mol Struct THEOCHEM 169:41–62
Foster AJ, Weinhold F (1980) Natural hybrid orbitals. J Am Chem Soc 102:7211–7218
Glendening E, Badenhoop J, Reed A, Carpenter J, Bohmann J, Morales C, Landis C, Weinhold F (2013) NBO 6.0. Theoretical Chemistry Institute, University of Wisconsin, Madison
Reed AE, Weinhold F (1983) Natural bond orbital analysis of near-Hartree–Fock water dimer. J Chem Phys 78:4066–4073
Reed AE, Weinhold F (1985) Natural localized molecular orbitals. J Chem Phys 83:1736–1740
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746
Glendening ED, Landis CR, Weinhold F (2012) Natural bond orbital methods. Interdiscip Rev Comput Mol Sci 2:1–42
Glendening ED, Landis CR, Weinhold F (2019) NBO 7.0: new vistas in localized and delocalized chemical bonding theory. J Comput Chem 40:2234–2241
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Weinhold F (2002) Natural bond orbital methods, encyclopedia of computational chemistry, 3. Hoboken, New Jersey
Weinhold F (2012) Natural bond orbital analysis: a critical overview of relationships to alternative bonding perspectives. J Comput Chem 33:2363–2379
Weinhold F, Landis C, Glendening E (2016) What is NBO analysis and how is it useful? Int Rev Phys Chem 35:399–440
Mulliken RS (1955) Electronic population analysis on LCAO–MO molecular wave functions. I. J Chem Phys 23:1833–1840
Mayer I (1983) Charge, bond order and valence in the AB initio SCF theory. Chem Phys Lett 97:270–274
Mayer I (2012) Improved definition of bond orders for correlated wave functions. Chem Phys Lett 544:83–86
Giambiagi M, de Giambiagi MS, Mundim KC (1990) Definition of a multicenter bond index. Struct Chem 1:423–427
Matito E (2016) An electronic aromaticity index for large rings. Phys Chem Chem Phys 18:11839–11846
Wiberg KB (1968) Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24:1083–1096
Lu T, Chen F (2013) Bond order analysis based on the Laplacian of electron density in fuzzy overlap space. J Phys Chem A 117:3100–3108
Mayer I, Salvador P (2004) Overlap populations, bond orders and valences for ‘fuzzy’atoms. Chem Phys Lett 383:368–375
Sizova OV, Skripnikov LV, Sokolov AY (2008) Symmetry decomposition of quantum chemical bond orders. J Mol Struct THEOCHEM 870:1–9
Bader R, Nguyen-Dang TT, Tal Y (1981) A topological theory of molecular structure. Rep Prog Phys 44:893
Bader RF (1985) Atoms in molecules. Acc Chem Res 18:9–15
Bader RF (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928
Bader RF, Matta CF (2013) Atoms in molecules as non-overlapping, bounded, space-filling open quantum systems. Found Chem 15:253–276
Biegler-könig FW, Bader RF, Tang TH (1982) Calculation of the average properties of atoms in molecules. II. J Comput Chem 3:317–328
Cortés-Guzmán F, Bader RF (2005) Complementarity of QTAIM and MO theory in the study of bonding in donor–acceptor complexes. Coord Chem Rev 249:633–662
Howard S, Krygowski T (1997) Benzenoid hydrocarbon aromaticity in terms of charge density descriptors. Can J Chem 75:1174–1181
Noorizadeh S, Shakerzadeh E (2010) Shannon entropy as a new measure of aromaticity, Shannon aromaticity. Phys Chem Chem Phys 12:4742–4749
Balanarayan P, Gadre SR (2003) Topography of molecular scalar fields. I. Algorithm and Poincaré–Hopf relation. J Chem Phys 119:5037–5043
Roy D, Balanarayan P, Gadre SR (2008) An appraisal of Poincaré–Hopf relation and application to topography of molecular electrostatic potentials. J Chem Phys 129:174103
Matta CF (2006) Hydrogen–hydrogen bonding: the non-electrostatic limit of closed-shell interaction between two hydro, hydrogen bonding—new insights. Springer, Berlin
Bohórquez HJ, Boyd RJ, Matta CF (2011) Molecular model with quantum mechanical bonding information. J Phys Chem A 115:12991–12997
Grabowski SJ (2012) QTAIM characteristics of halogen bond and related interactions. J Phys Chem A 116:1838–1845
Tal Y, Bader R (1978) Studies of the energy density functional approach. I. Kinetic energy. Int J Quantum Chem 14:153–168
Keith T, Bader R, Aray Y (1996) Structural homeomorphism between the electron density and the virial field. Int J Quantum Chem 57:183–198
Contreras-García J, Johnson ER, Keinan S, Chaudret R, Piquemal J-P, Beratan DN, Yang W (2011) NCIPLOT: a program for plotting noncovalent interaction regions. J Chem Theory Comput 7:625–632
Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W (2010) Revealing noncovalent interactions. J Am Chem Soc 132:6498–6506
Acknowledgments
The authors would like to thank the Solid-State Theory Group at the Physics Department at the Universita‘degli Studi di Milano-Italy for providing computational facilities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Doust Mohammadi, M., Abdullah, H.Y. The adsorption of chlorofluoromethane on pristine, and Al- and Ga-doped boron nitride nanosheets: a DFT, NBO, and QTAIM study. J Mol Model 26, 287 (2020). https://doi.org/10.1007/s00894-020-04556-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04556-5