
Abstract
Rapid, selective particle separation and concentration within the bacterial size range (1–3 μm) in clinical or environmental samples promises significant improvements in detection of pathogenic microorganisms in areas including diagnostics and bio-defence. It has been proposed that microfluidic Dean flow-based separation might offer simple, efficient sample clean-up: separation of larger, bioassay contaminants to prepare bioassay targets including spores, viruses and proteins. However, reports are limited to focusing spherical particles with diameters of 5 μm or above. To evaluate Dean flow separation for (1–3 μm) range samples, we employ a 20 μm width and depth, spiral microchannel. We demonstrate focusing, separation and concentration of particles with closely spaced diameters of 2.1 and 3.2 μm, significantly smaller than previously reported as separated in Dean flow devices. The smallest target, represented by 1.0 μm particles, is not focused due to the high pressures associated with focussing particles of this size; however, it is cleaned of 93 % of 3.2 μm and 87 % of 2.1 μm microparticles. Concentration increases approaching 3.5 times, close to the maximum, were obtained for 3.2 μm particles at a flow rate of 10 μl min−1. Increasing concentration degraded separation, commencing at significantly lower concentrations than previously predicted, particularly for particles on the limit of being focused. It was demonstrated that flow separation specificity can be fine-tuned by adjustment of output pressure differentials, improving separation of closely spaced particle sizes. We conclude that Dean flow separation techniques can be effectively applied to sample clean-up within this significant microorganism size range.
Similar content being viewed by others

1 Introduction
The removal of contaminating material from samples, often termed ‘clean-up’, and sample concentration is key elements of sample preparation from environmental, clinical and other sources prior to diverse forms of bioassay and is often achieved by the same process. Integration of these functions within microfluidics has been demonstrated, employing techniques including sedimentation (Huh et al. 2007), centrifugal separation (Riegger et al. 2007) and filtration (Prince et al. 2007). However, implementation of all these approaches requires relatively high precision, fine and complex geometries. Passive microfluidic separation techniques employing inertial flow effects exploiting laminar, microfluidic flow regimes to achieve size-based microparticle separation are being developed. The current state of the art has been well reviewed (Di Carlo 2009; Gossett et al. 2010).
The majority of passive, inertial flow separation techniques can be grouped into one of four categories (Di Carlo 2009): flow in a straight channel, deterministic lateral displacement, pinched flow fractionation and inertial focusing in curved channels (commonly known as Dean flow separation). In general, Di Carlo concludes that inertial focusing in curved microchannels is the most promising technique currently available to implement continuous concurrent separation of differently sized microparticle mixtures.
Dean flow, inertial focusing of spherical microparticles with diameters ranging between 5 and 20 μm has demonstrated the promise of efficient separation as well as increased throughput (Russom et al. 2009; Kuntaegowdanahalli et al. 2009; Xiang et al. 2012; Bhagat et al. 2008a). High throughput separation of 10 μm microparticles has also been performed using the Dean flow effect within ‘U’- and ‘S’-shaped channels (Ardabili et al. 2010). Most recently, the applicability of the technique for separation of non-spherical microparticles has also been examined (Hur 2011). To date, the Dean flow-based inertial focusing technique has not been reported with particle sizes below 5 μm.
For applications involving pathogenic microorganisms, it is desirable to separate and ideally concentrate particles in the range of (1–3 μm) equivalent diameter. There are few reports of particle separation in this range, except those employing inertial migration of particles due to equilibration of drag forces and lift forces acting on particles flowing in straight microchannels (Yamada and Seki 2005; Bhagat et al. 2008b) which successfully separated and concentrated small microparticles (1–3 μm). This technique effectively forms a hydrodynamic cut point whereupon all particles above the cut size are focused into regions near the wall of the straight channel and are then separated at a 4-way junction into side channels. The particles below the cut size remain unfocussed and simply separate in proportion with the geometry of the junction. These techniques do not easily allow individual separation of multiple, differently sized particles. The technique reported by Yamada and Seki (2005) both separated and significantly increased the concentration of 1.0 and 2.1 μm microparticles by employing complex devices consisting of 160 side channels branching from a main flow channel whose cross-section was 5 μm × 10 μm and operating at a flow rate of just 1 μl min−1. Despite its favourable particle handing performance, requirements for small microchannel dimensions (that are relatively close to those of the particles), very low flow rates and a complex arrangement of branching output ports and associated fluidic conditions render it difficult to employ in its current form for many ‘real-world’ applications.
The objective of this paper is to explore the use of Dean flow separation techniques in this smaller particle size range where they offer the prospect of relaxing to some degree all of the conditions applying to the straight channel technique. Specifically, we investigate the application of Dean flow-based inertial focusing and separation employing polydimethylsiloxane (PDMS) spiral microchannels in two regards. Firstly, in terms of its ability to concentrate and separate to individual fluid output ports, small, closely sized 2.1 and 3.2 μm microspheres. These microspheres are approximately five times smaller in diameter than the most closely ratiometrically spaced microparticle sizes previously reported as separated by Dean flow (10, 15 and 20 μm) (Kuntaegowdanahalli et al. 2009) and significantly more closely ratiometrically spaced by size than the smallest separated particle sizes previously reported (1.9 and 7.32 μm) (Bhagat et al. 2008a). Secondly, we examine its ability to clean-up and direct to an individual output port, 1.0 μm microspheres simulating assay target materials cleaned from 2.1 to 3.2 μm microspheres that represent contamination in the context of the 1.0 μm microspheres output port.
2 Inertial hydrodynamic microfluidic microparticle separation
Within a microchannel, fluid undergoing pressure-driven flow has a parabolic flow velocity profile. This profile results in a shear gradient and thus an inertial lift force (Saffman lift force) which will drive suspended microparticles towards the channel wall. However, in the near-wall region, a microparticle experiences another lift force normal to the wall. This wall-induced lift is a result of the vorticity generated by the microparticle which advects and propagates downstream. However, the adjacent wall results in a distortion of the vorticity’s axisymmetry resulting in a net force that acts to transport the microparticle away from the wall. In straight microchannels, these two forces balance resulting in particles migrating to an equilibrium position displaced from the channel walls, forming a ring of particles in circular cross-section microchannels or four face-centred loci in the case of square cross-section microchannels.
The addition of curvature to a microchannel induces two counter-rotating flow vortices orthogonal to the main flow direction, referred to as ‘Dean flow’ (Dean 1927), which introduce an additional drag force on a microparticle (however, Dean flow is not a microscale-only phenomenon). It is possible to configure flow conditions such that the Dean flow recirculates microparticles suspended within the primary flow until they settle in stable equilibrium positions. These equilibrium positions form where the forces acting away from the channel walls (wall-induced lift and Dean flow drag force) balance the force towards the inner wall (shear-induced, Saffman lift force). It has been shown (Di Carlo et al. 2007; Di Carlo 2009; Guan et al. 2013) that particles can become focussed at one of two locations, either towards the top or bottom of the channel’s cross-section at equal lateral displacements determined by the flow rate, as shown in Fig. 1.

Schematic cross section of a curved microchannel showing the two counter-rotating Dean vortices orthogonal to the main flow direction as a result of channel curvature, where F D = Dean drag force, acting to recirculate non-equilibrated particles (green). The equilibrated particles (red) are subject to F 1 = F D + F WL, where F WL = wall lift force and F 2 = Saffman lift force
The ability to simultaneously focus microparticles of different dimensions at different lateral locations within the flow stream is a unique advantage of inertial focusing in curved microchannels. The variables defining the focusing and position for given particle dimensions are channel dimensions, curvature of the flow channel, microparticle dimensions and flow rate (Di Carlo 2009).
3 Experimental methods
3.1 Design
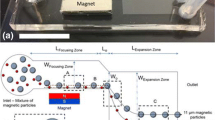
Here, we present a separator design which incorporates a single inlet leading to an Archimedean spiral flow channel which in turn leads to an expanding output channel that splits into four separate sub-channels, each connected to a separate output tube. The spiral consists of a square cross-section channel, 20 μm wide and 20 μm deep, with an initial radius of 2.12 mm and a spacing of 220 μm between spiral loops. The spiral section of the flow channel has a total length of 82 mm along its centre-line. The expanding output channel diverges at an angle of 30º until the channel width is 550 μm wide. Each of the four output channels is 100 μm wide. The four output channel configuration was employed to allow flexibility when separating focused microspheres given the asymmetric bias of particle positions across the flow channel resulting from separation of larger particles commencing at the inner wall. The four outputs, from the inside to the outside of the channel curvature, are referred to as A, B, C and D, respectively, see Fig. 2.

Left is a photograph of an assembled PMMA–PDMS inertial separator. Right is a schematic of the spiral channel separator design showing the positions of the inlet and outputs A, B, C and D. (i) represents the region interrogated by fluorescence microscopy in Fig. 3, and (ii) represents the region interrogated by fluorescence microscopy in Fig. 7
First-order design rules for inertial focusing in rectangular channels were compiled by Di Carlo (2009), who discusses the optimal parameters for required channel length, volumetric flow rate and, in the case of curving channels, the limiting criteria for the channel radius. The microchannel dimensions reported in this paper satisfy the conditions for the required radius of curvature for all microsphere sizes tested. In terms of channel length requirements, our design satisfies them for both the 2.1 and 3.2 μm microspheres. However, for 1.0 μm microspheres, for a flow rate range of 1–100 μl min−1, the channel length requirement could range from 1.5 to 1.5 cm. Therefore, for devices which operate in low flow rate regimes, the channel length requirement cannot be met for the smallest microspheres. The predicted minimum requirements for volumetric flow rate are 6, 17 and 92 μl min−1 for 3.2, 2.1 and 1.0 μm microspheres, respectively. For the microchannels employed in this study, the limiting concentrations for which Di Carlo predicts successful focusing to a single stream of particles are 2.5 × 109, 1.2 × 109 and 7.6 × 108 ml−1 for 1.0, 2.1 and 3.2 μm diameter microspheres, respectively. The highest concentrations employed in this study (see Sect. 3.3) are at least an order of magnitude below Di Carlo’s design-rule conditions necessary to avoid crowding effects.
3.2 Fabrication
Separator microstructures were fabricated using rapid prototyping processes previously reported (Johnston et al. 2005). SU-8 25 (MicroChem Corp., USA) moulds were fabricated on silicon wafers. PDMS structures were then cast from Sylgard 184 elastomer (Dow Corning, USA) mixed in the standard ratio of 10 parts A to 1 part B. The PDMS was then cured at 100 °C. Cast Poly(methyl methacrylate) (PMMA) sheet was used to close the fluid channels and provide fluid connectivity. The PMMA (Clarex, Nitto Jushi Kogyo Co., Ltd., Japan) was drilled under computer numerical control (CNC) to implement accurately located through vias with integral end stop locations for inserting locating tubing; 300-μm bore holes were drilled for connecting to the PDMS microstructure and then partially counter bored at 800 μm from the outer PMMA face to locate connecting tubing.
The PDMS and PMMA components were then bonded by modification of the PMMA substrate using a silane. Several techniques have been reported (Vlachopoulou et al. 2009; Kim et al. 2010; Sunkara et al. 2011); however, we employed our closely related, in-house protocol, as follows. Clean, dry PMMA was exposed to UV-Ozone using a PSD-UVT system (Novoscan Technologies Inc., USA) for 5 min. The PMMA was then silanised using aminopropyltriethoxysilane (APTES, 100 μl in a gas tight 100 ml container) vapour for 1.5 h at 60 °C at atmospheric pressure. The PMMA was immediately rinsed with isopropanol and dried with filtered nitrogen gas. Clean, dry PDMS was then exposed to UV-Ozone for 3 min with the PSD-UVT system. The two treated components were carefully aligned and brought together. The composite device was then baked at 60 °C for 12 h to create a strong irreversible covalent bond between the two materials.
The fluid connections were then made by fitting 0.01″ i.d., 1/32″ o.d. PEEK tubing (IDEX® Health and Science, USA) into the counter-drilled 800-μm holes and bonding with general purpose epoxy adhesive. The assembled device is shown in Fig. 2.
3.3 Sample preparation
Fluorescent polystyrene microspheres were used to evaluate separation. Stock solutions of green 1.0 μm, blue 2.1 μm and red 3.2 μm diameter microspheres were supplied at concentrations of 1.9 × 1010, 2.1 × 109 and 6.4 × 108 ml−1, respectively (G0100, R0200, B0300, Thermo Scientific, USA). Experimental solutions (herein referred to as 1.00, 0.10 and 0.01 %) containing equal quantities of all three microsphere sizes by volume were obtained by diluting stock solutions in 0.1 μm filtered de-ionised water by 100×, 1,000× and 10,000×, respectively.
3.4 Characterisation
We have employed both florescence imaging and flow cytometry. Imaging allowed us to monitor variation in hydrodynamic behaviour such as output flow division and allowed real-time operation of the experimental system. However, it could not accurately quantify microsphere concentrations within the device due to variations in both the fluorescence intensity of the three fluorophores labelling the microspheres, the microsphere size and the spectral response of the imaging apparatus employed. Furthermore, scattering and absorption of photons are probable in zones of high particle concentration leading to a nonlinear intensity particle–count relationship. In contrast, flow cytometry allows quantitative, but non-real-time characterisation of separation behaviour.
3.5 Imaging
Fluorescence imaging was used to monitor microsphere separation. An EXi Aqua CCD camera (QImaging, USA) was mounted on an Axiovert 40 CFL microscope (Zeiss, USA). A Zeiss filter set number 25 (with excitation wavelengths of 400, 495 and 570 nm and emission wavelengths of 460, 530 and 625 nm) allowed simultaneous imaging of green, red and blue microspheres. Images were captured using QCapture Pro 6 (QImaging, USA). Exposure times were adjusted for each sample concentration to obtain clear, time-averaged fluorescence images within the separator structure resulting in exposure times of 150 ms, 1 s and 2 s for the 1.00, 0.10 and 0.01 % samples, respectively. In order to improve the visual representation of the average flow behaviour of the microspheres, ImageJ (National Institutes of Health, public domain) software was employed to create Z-stacked composite images by overlaying 25 consecutively captured images.
3.6 Operation and sample collection
Particle suspensions were pumped through the device using a PHD 2000 syringe pump (Harvard Apparatus, USA). During initial testing, it was found that the pressure conditions at the flow separation point were low enough that modest pressure differentials could be employed to accurately manipulate the locations of the flowing particles despite the high pressure conditions required at the inlet of the device. In order to accurately set relatively small pressure differentials between the outputs, the samples were collected into separate Safe-Lock tubes (Eppendorf, Germany) each of which was mounted on a retort stand. The individual tubes were then positioned with appropriate vertical fluid meniscus displacements (with 10 cm of height differential approximating to a pressure differential of 980 Pa), so as to allow necessary hydrostatic pressure differentials to be established between them.
3.7 Flow cytometric characterisation
To investigate the effectiveness of the inertial focusing and subsequent separation, the collected samples were analysed using a Partec PAS flow cytometer (Partec, Germany). The flow cytometer underwent full flushing procedures between runs so as to minimise carry-over, a contributing component of background counts. Control microsphere solutions containing monodisperse microspheres were used to pre-define gated regions (data filters) for size-specific particle counting within samples containing differently sized microspheres (Kuntaegowdanahalli et al. 2009). Background counts, processed via the gated regions, were then conducted for the 0.1 μm filtered DI water. Worst case counts of below 1 % of sample microsphere concentration were obtained for the 1 μm microsphere gated region, the larger microsphere gated regions displayed even lower counts. The samples were then analysed, and the gates applied to the data sets in order to obtain accurate count data for each of the outputs. Collected samples were analysed on the day of collection to minimise counting inaccuracies due to possible agglomeration or bacterial growth.
4 Results
4.1 Operating limitations of the device
Design calculations indicated that operating flow rates in the region of 92 μl min−1 would be required focus the smallest 1.0 um microspheres. These flow rates might result in pressures exceeding the capacity of PDMS–PMMA devices. Accordingly, initial experiments were conducted to establish the maximum flow rate capability of the device. This was found to be 10 μl min−1, at which point devices visibly deformed and, with further pressure, catastrophically failed due to bursting in the fluid introduction port region where the largest area of un-bonded PDMS fluid structure is exposed to full driving pressure and hence the greatest deformational forces are generated. Whilst small refinements to dimensions in the through via region were made, the minimum, 300 μm, diameter of the CNC-drilled PMMA through via and its associated alignment tolerances ultimately set a lower limit to this exposed area.
Accordingly, given the significant gap between predicted ‘ideal’ flow rates for 1 μm microsphere focussing and the maximum experimental flow rate, the possibility of focussing 1 μm microspheres was not pursued further. Therefore, subsequent experimentation concentrated upon the primary objective of cleaning-up 1 μm ‘target’ material from larger ‘contaminating’ microspheres and the separation of the larger microspheres from each other.
4.2 Separation
The dependence of inertial focusing of microspheres on flow rate was investigated for each of the three samples containing 1.00, 0.10 and 0.01 % concentrations of all three sizes of polymer microspheres. As volumetric flow rate was increased from 1 μl min−1 (Re = 0.83) to 10 μl min−1 (Re = 8.3), microsphere focusing was maintained, as shown in Fig. 3. However, at volumetric flow rates above 10 μl min−1, the previously focused streams began to defocus prior to device failure. Such effects are discussed by Di Carlo et al. (2009) as occurring above the ‘order of magnitude’ of the ‘ideal’ flow rate calculated by their methodology. However, the maximum flow rate of 10 μl min−1 we employ only mildly exceeds the ideal flow rate of 6 μl min−1 for the largest 3.2 μm microparticles. Accordingly, the flow rate employed is well within an ‘order of magnitude’ of the ideal and unlikely to be the primary cause of defocusing. Instead, we believe that pressure-induced expansion of the spiral PDMS flow channel (as distinct from the flow entry region) begins to manifest at the pressure associated with the 10 μl min−1 flow rate resulting in distortion and increased channel cross-section and hence unbalancing of the forces acting on the microspheres. If all other conditions remain the same, then this results in a reduction in both resultant velocity and Dean force.

Z-stacked fluorescence images showing time averaged distribution of the focusing of 1.0 μm (green), 2.1 μm (blue) and 3.2 μm (red) microspheres with increasing flow rate
At low flow rates (1 μl min−1), viscous drag effects dominate and Dean flow recirculation is not strong enough to focus the microspheres towards the inside wall of the curving microchannel. At medium flow rates (5 μl min−1), the Dean flow recirculation increased such that the force balancing of the 2.1 and 3.2 μm microspheres resulted in a migration of the majority of the 2.1 and 3.2 μm microspheres towards the inside wall. As the flow rates were further increased to 10 μl min−1, the force balance improved further and the 2.1 and 3.2 μm microspheres were more distinctly moved towards the inside wall, with a lower percentage of the larger microspheres being observed in the region towards the outside of the channel wall: thus providing a flow regime which would most efficiently separate and concentrate the 2.1 and 3.2 μm microspheres. At the higher flow rates, it was visually apparent that the flow patterns were better defined with fewer of the larger microspheres observed in the unfocussed outer regions leading to output channels C and D. As predicted, the Dean forces acting on the 1.0 μm microspheres were insufficient to provide a force balance, and thus, the 1.0 μm microspheres remain evenly distributed across the channel.
Output samples were collected using an inlet volumetric flow rate of 10 μl min−1 in order to quantify microsphere separation of the 1.00, 0.10 and 0.01 % sample concentrations. The output fractions were then analysed by flow cytometry (see Fig. 4).

Representative image of flow cytometry data and gated regions for the 0.01 % concentration sample containing 1.0 μm (green), 2.1 μm (blue) and 3.2 μm (red) microspheres for outputs A, B, C and D
The data for particle counting across the four outputs of the spiral separator for each of the sample concentrations are presented in Fig. 5. For 3.2 μm microspheres, the data indicate that a high level of separation, between 94 and 96 %, could be achieved across all sample concentrations, resulting in a concentration increase in approximately 375 % in the output A sample. The data for the 2.1 μm microspheres are in agreement with the visual observation that the 2.1 μm microspheres did not form a narrowly focused band. Approximately, 80 % of the 2.1 μm microspheres were separated into outputs A and B, yielding sample concentration increases in up to approximately 185 %. As the sample concentration increased, the most densely packed region of the 2.1 μm microspheres shifted further away from the inside wall of the microchannel.

Particle count data displaying concentrations of 1.0 μm (green), 2.1 μm (blue) and 3.2 μm (red) microspheres at sample outputs A, B, C and D after flowing through the spiral separator structure at a volumetric flow rate of 10 μl min−1 with initial concentrations of 1.00, 0.10 and 0.01 %
The data for the 1.0 μm microspheres show a relatively uniform distribution across all four outputs with minimal inertial focusing. This is consistent with the observations of the fluorescence images which show that wall lift effects move the 1.0 μm microspheres away from the channel walls but that no further force balance was achieved. In terms of small target clean-up, the data from Fig. 5 for the C = 0.10 % case show that device output D yields 1 μm microspheres at unchanged concentration from the input sample; however, 87 % of 2.1 μm microspheres and 93 % of 3.2 μm microspheres, with respect to the input concentration, have been removed (cleaned-up) from them.
4.3 Concentration effects
Increasing microsphere concentration increased the width of the resulting band of focused microspheres. The flow cytometry data of Fig. 5 objectively confirm concentration-dependent changes in distribution. The 3.2 μm microspheres remained well focused with a slight increase in the width of the channel occupied as the concentration was increased; however, the effect was small in the context of the 100× increase in microsphere concentration. The 2.1 μm microspheres behaved similarly but at the highest concentration this increase degraded the flow focusing, resulting in a considerably wider occupancy region for the 2.1 μm microspheres compared to that at the lower concentrations.
Figure 6 illustrates the concentration dependence of focusing by means of intensity profiles of the microsphere streams for two concentration cases shown within Fig. 3: specifically 0.10 and 1.00 % microsphere concentrations at 10 μl min−1. It can be observed that the clarity of focus between the two flow streams markedly worsens as concentration is increased and that the widening of the tail of the distribution indicated by flow cytometry is apparent. However, whilst illustrative of concentration-dependent defocusing, Fig. 6 is not suitable for more quantitative interpretation for reasons discussed in Sect. 3.4. Furthermore, total intensity was measured without optical colour filtering or post-processing of the composite image.

Intensity profiles of 0.10 and 1.00 % microsphere concentrations at 10 μl min−1 from two corresponding sub-images of Fig. 3. The illustrative inset image indicates the profile area. For each cross-sectional point, the mean of the associated row of pixel intensities forms the intensity value. The red stream comprises 3.2 μm microspheres, and the blue comprises 2.1 μm microspheres
4.4 Optimising separation
When seeking to perform precise separations of particles with small differences in hydrodynamic drag, it becomes difficult to deterministically design separator structures that are intrinsically correct-by-design: rather particle populations are likely to be misaligned with desired outputs. Accordingly, the possibility of improving separation and/or concentration of the collected microspheres by manipulating the output flow conditions was investigated. The pressure at output A was increased slowly so as to visually optimise alignment of the interface region between the inertially focused 3.2 and 2.1 μm microspheres, see Fig. 7. When optimised, the pressure differential between output A and the other outputs was 2.0 kPa. Decreasing the pressure at output A by 2.0 kPa with respect to the other outputs resulted in a repositioning of the focused 3.2 and 2.1 μm microspheres such that their joint separation from the remaining sample was optimised. Decreasing or increasing the flow velocities within output A had a broadening or narrowing effect on the band of focused microspheres.

Z-stacked fluorescence image showing particle separation in output channels A, B, C and D with equal pressure at A (centre), minus 2.0 kPa pressure at A (left) and plus 2.0 kPa pressure at A (right)
The particle count data shown in Fig. 8 for the samples collected with a ±2.0 kPa pressure differential between output A and the remaining outputs B, C and D agree with the fluorescence images. Particle counts for the sample collected when the pressure at output A was increased show that the separation between the 2.1 and 3.2 μm microspheres was improved yielding concentrations of 350 % of 3.2 μm microspheres in output A and 250 % of 2.1 μm microspheres in output B. When the pressure at output A was decreased, the concentration of 2.1 μm microspheres separated into output A could be increased slightly to 200 % at the cost of a reduced degree of inertial focusing reducing their separation and concentration solely into output A, but rather spreading them across A and B.

Particle count data displaying concentrations of 1.0 μm (green), 2.1 μm (blue) and 3.2 μm (red) microspheres at sample outputs A, B, C and D with a ±2.0 kPa pressure differential between output A and the other outputs at a volumetric flow rate of 10 μl min−1 with initial concentrations of 1.00 %
As previously discussed, the geometry of the separator structure employed limits the maximum possible increase in concentration to 400 %. In practice for the well-focused 3.2 μm microspheres, this is very nearly achieved. The data show that by altering the pressure conditions applied to the individual outlets, it was possible to modify the output flow conditions such that separation and concentration of the less well focused 2.1 μm microspheres was also improved.
5 Discussion
We have presented the results from an experimental investigation of Dean flow-based inertial focusing and separation over a narrow range of sizes: specifically, we have separated 2.1 and 3.2 μm microspheres into separate output ports whilst concurrently cleaning-up 1.0 μm microspheres from 2.1 and 3.2 μm ‘contaminating’ microspheres with flow to a separate output port. Microsphere sizes were necessarily chosen to simulate real-world environmental samples. Each output represents the analyte feed to a separate bioassay in a system realisation.
Flow cytometry results with 2.1 and 3.2 μm microspheres display a significant reduction from previously reported inertially focusable particle size for Dean flow-based separation. This was achieved using a PDMS–PMMA composite device with spiral microchannels of a width and depth of 20 μm fabricated in the PDMS.
In agreement with the design rules of Di Carlo (2009), the device was unable to focus the 1 μm microspheres due to the extremely high pressures associated with necessary focusing conditions resulting in device deformation and ultimate failure at lower pressures. However, under acceptable operating conditions, the device displayed good ‘clean-up’ performance removing 87 % of 2.1 μm microspheres and 93 % of 3.2 μm microspheres, with respect to the input concentration, from the output port assigned to cleaned 1.0 μm microspheres.
The device presented has an outlet section consisting of a diverging manifold leading to four symmetrical outlet channels. Accordingly, with equal outlet pressure and flow conditions, a theoretical maximum of four times initial concentration was possible. We have shown that at a volumetric flow rate of 10 μl min−1, equivalent to a throughput of 2 million particles per minute, it was possible to achieve concentration of 375 % for 3.2 μm microspheres. Depending on the application requirements, these results indicate that concentration increases for the larger microsphere sizes (2.1 and 3.2 μm) can be achieved.
It has been postulated (Di Carlo 2009) that as the concentration of particles increases, inter-particle interaction reduces the effectiveness of inertial focusing into single streams. We examined some of the effects of sample concentration on focusing. The effect of concentration over the range investigated (two orders of magnitude) varied with microsphere size. The data support the view that the 3.2 μm microspheres are comfortably within the focusing regime and as such were not affected by the increasing concentration. The 2.1 μm microspheres were on the limit of meeting focusing criteria and as a result were sensitive to increasing the concentration which resulted in reduced focusing due to increasing inter-microsphere interactions. The 1.0 μm microspheres remained unfocussed as anticipated. However, referring to Fig. 5, we observe that the concentrations of 1 μm microspheres in outputs A and B appear to be influenced by the concentration and location of the focussed, larger, 3 μm microspheres. This was not investigated further, but we hypothesise that it may be due to physical particle–particle interactions in the region of concentration. These results suggest that in the case of devices seeking to focus the smallest materials and necessarily working at the limits of feasible inertial focusing conditions, inter-particle interactions can become complex and potentially detrimental at concentrations well below the guideline values predicted by Di Carlo (2009).
The separation characteristics of the device have been evaluated experimentally, and the results have been assessed in terms of the design rules of Di Carlo (2009). We have shown that increasing the flow constraints of the physical separation with precise modification of the output pressure conditions can enable a significant increase in the deliverable concentration and specificity of separation of specific microparticles to device fluid ports for subsequent use, such as bioassays. In this case, it was found that a minor 2.0 kPa pressure differential across the four outlet channels optimised the flow separation to output ports between the focused particle paths.
We have demonstrated that simple Dean flow-based PMMA–PDMS microstructures have the potential be used to concentrate and separate small, closely sized 2.1 and 3.2 μm microparticles and to ‘clean up’ smaller 1.0 μm microparticles.
We conclude that concentration and separation of pathogenic microorganisms with equivalent diameters of 2.0 μm and above in conjunction with clean-up of smaller target material is possible using Dean flow separation devices. However, further optimisation would be possible employing refined channel dimensions in conjunction with increased channel lengths. Whilst this would be at the expense of reduced throughputs, they would remain in a deployable range. Such optimisation would also allow the recovery of a greater proportion of cleaned-up small material (evidenced here by 1 μm microspheres) with a yet lower level of larger particulate contamination. We believe that optimisation of the rapid prototyping process reported would facilitate this work; however, alternative, hard polymer, rapid prototyping and bonding technologies would be better suited to further developments.
References
Ardabili S, Gantelius J, Kowalewski J, Brismar H, Russom A (2010) Dean flow-coupled inertial focussing for ultra-highthroughput particle filtration. In: 14th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Groningen, The Netherlands, 3–7. pp 1586–1588
Bhagat AAS, Kuntaegowdanahalli SS, Papautsky I (2008a) Continuous particle separation in spiral microchannels using dean flows and differential migration. Lab Chip 8(11):1906–1914
Bhagat AAS, Kuntaegowdanahalli SS, Papautsky I (2008b) Inertial microfluidics for continuous particle filtration and extraction. Microfluid Nanofluid. doi:10.1007/s10404-008-0377-2
Dean WR (1927) XVI. Note on the motion of fluid in a curved pipe. Philosophical Magazine Series 7 4(20):208–223. doi:10.1080/14786440708564324
Di Carlo D (2009) Inertial microfluidics. Lab Chip 9(21):3038–3046
Di Carlo D, Irimia D, Tompkins RG, Toner M (2007) Continuous inertial focusing, ordering, and separation of particles in microchannels. Proc Natl Acad Sci 104(48):18892–18897. doi:10.1073/pnas.0704958104
Gossett D, Weaver W, Mach A, Hur S, Tse H, Lee W, Amini H, Di Carlo D (2010) Label-free cell separation and sorting in microfluidic systems. Anal Bioanal Chem 397(8):3249–3267. doi:10.1007/s00216-010-3721-9
Guan G, Wu L, Bhagat AA, Li Z, Chen PCY, Chao S, Ong CJ, Han J (2013) Spiral microchannel with rectangular and trapezoidal cross-sections for size based particle separation. Sci Rep 3. doi:http://www.nature.com/srep/2013/130318/srep01475/abs/srep01475.html#supplementary-information
Huh D, Bahng JH, Ling Y, Wei H-H, Kripfgans OD, Fowlkes JB, Grotberg JB, Takayama S (2007) Gravity-driven microfluidic particle sorting device with hydrodynamic separation amplification. Anal Chem 79(4):1369–1376. doi:10.1021/ac061542n
Hur SC (2011) Inertial focusing of non-spherical microparticles. Appl Phys Lett 99(4):044101
Johnston ID, Tracey MC, Davis JB, Tan CKL (2005) Microfluidic solid phase suspension transport with an elastomer-based, single piezo-actuator, micro throttle pump. Lab Chip 5(3):318–325
Kim K, Park S, Yang S (2010) The optimization of PDMS-PMMA bonding process using silane primer. BioChip J 4(2):148–154. doi:10.1007/s13206-010-4210-0
Kuntaegowdanahalli SS, Bhagat AAS, Kumar G, Papautsky I (2009) Inertial microfluidics for continuous particle separation in spiral microchannels. Lab Chip 9(20):2973–2980
Prince M, Ma X, Docker P, Ward M, Prewett P (2007) The development of a novel Bio-MEMS filtration chip for the separation of specific cells in fluid suspension. P I Mech Eng H 221(2):113–128. doi:10.1243/09544119jeim190
Riegger L, Grumann M, Steigert J, Lutz S, Steinert C, Mueller C, Viertel J, Prucker O, Rühe J, Zengerle R, Ducrée J (2007) Single-step centrifugal hematocrit determination on a 10-$ processing device. Biomed Microdevices 9(6):795–799. doi:10.1007/s10544-007-9091-1
Russom A, Gupta AK, Nagrath S, Di Carlo D, Edd JF, Toner M (2009) Differential inertial focusing of particles in curved low-aspect-ratio microchannels. New J Phys 11:075025. doi:10.1088/1367-2630/11/7/075025
Sunkara V, Park D-K, Hwang H, Chantiwas R, Soper SA, Cho Y-K (2011) Simple room temperature bonding of thermoplastics and poly(dimethylsiloxane). Lab Chip 11(5):962–965
Vlachopoulou M-E, Tserepi A, Pavli P, Argitis P, Sanopoulou M, Misiakos K (2009) A low temperature surface modification assisted method for bonding plastic substrates. J Micromech Microeng 19(1):015007. doi:10.1088/0960-1317/19/1/015007
Xiang N, Chen K, Sun D, Wang S, Yi H, Ni Z (2012) Quantitative characterization of the focusing process and dynamic behavior of differently sized microparticles in a spiral microchannel. Microfluid Nanofluid:1–11. doi:10.1007/s10404-012-1025-4
Yamada M, Seki M (2005) Hydrodynamic filtration for on-chip particle concentration and classification utilizing microfluidics. Lab Chip 5(11):1233–1239
Acknowledgments
We acknowledge the support of the UK Ministry of Defence’s Centre for Defence Enterprise under Grant CDE19611.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Johnston, I.D., McDonnell, M.B., Tan, C.K.L. et al. Dean flow focusing and separation of small microspheres within a narrow size range. Microfluid Nanofluid 17, 509–518 (2014). https://doi.org/10.1007/s10404-013-1322-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-013-1322-6