
Abstract
Patients with COVID-19 present a wide spectrum of disease severity, from asymptomatic cases in the majority to serious disease leading to critical care and even death. Clinically, four different scenarios occur within the typical disease timeline: first, an incubation and asymptomatic period; second, a stage with mild symptoms due mainly to the virus itself; third, in up to 20% of the patients, a stage with severe symptoms where a hyperinflammatory response with a cytokine storm driven by host immunity induces acute respiratory distress syndrome; and finally, a post-acute sequelae (PASC) phase, which present symptoms that can range from mild or annoying to actually quite incapacitating. Although the most common manifestation is acute respiratory failure of the lungs, other organs are also frequently involved. The clinical manifestations of the COVID-19 infection support a key role for endothelial dysfunction in the pathobiology of this condition. The virus enters into the organism via its interaction with angiotensin-converting enzyme 2-receptor that is present prominently in the alveoli, but also in endothelial cells, which can be directly infected by the virus. Cytokine release syndrome can also drive endothelial damage independently. Consequently, a distinctive feature of SARS-CoV-2 infection is vascular harm, with severe endothelial injury, widespread thrombosis, microangiopathy, and neo-angiogenesis in response to endothelial damage. Therefore, endothelial dysfunction seems to be the pathophysiological substrate for severe COVID-19 complications. Biomarkers of endothelial injury could constitute strong indicators of disease progression and severity. In addition, the endothelium could represent a very attractive target to both prevent and treat these complications. To establish an adequate therapy, the underlying pathophysiology and corresponding clinical stage should be clearly identified. In this review, the clinical features of COVID-19, the central role of the endothelium in COVID-19 and in other pathologies, and the potential of specific therapies aimed at protecting the endothelium in COVID-19 patients are addressed.
Similar content being viewed by others

Endothelial Damage in COVID-19
Patients affected by COVID-19 display different degrees of disease severity, from asymptomatic and pauci-symptomatic cases to severe disease that can even lead to death [1]. The severity and mortality of the disease is highly related to advanced age and comorbidities [2, 3]. Possibilities of death increase exponentially with age [4]. COVID-19 is characterized by acute respiratory failure caused by several mechanisms. Progression of the disease to an advanced stage, as a consequence of a hyperinflammatory response that causes systemic cytokine release syndrome, is associated with the presence of pulmonary infiltrates and a rapid evolution to respiratory failure. Typically, this clinical situation occurs with a hypercoagulable state and marked elevation of acute-phase reactants in the circulation (including reactive C protein, D-dimer, and ferritin). Although the lung is the main target of the infection, there are extrapulmonary manifestations of the disease. The clinical and laboratory features suggest that endothelial activation and damage play a key role in the pathophysiology of this condition.
The vascular endothelium is considered the largest organ in the body, strategically located between the vessel wall and blood stream. The endothelium is not homogeneous and phenotypic characteristics differ from different vascular beds. Endothelial functions are not limited to the contention of blood, since it actively participates in the control of hemostasis, fibrinolysis, inflammation, oxidative stress, vascular permeability, and vascular structure [5]. The endothelium acts as a paracrine (acting on nearby cells), endocrine (exerting actions on distant cells), and autocrine (regulating its own function) organ. Endothelial cells (ECs) have a profound capability of adapting to their environment and responding to stimuli, and when this capacity is lost, ECs play a critical role in vascular disease [6]. In particular, loss of physiological features leads to endothelial dysfunction (ED), which is characterized by a reduced capacity for vasodilation, enhanced proinflammatory and prothrombotic properties, and abnormal modulation of vascular growth, among other characteristics. ED has been demonstrated in multiple clinical conditions with increased risk of thrombotic complications, such as chronic kidney disease [7] and liver failure [8], diabetes, obesity [9], pulmonary hypertension [10], septic syndromes [11], coronary artery disease, and vascular senescence [12]. In addition, specific therapies such as hematopoietic stem cell transplantation represent a significant toxic insult to the endothelium, which may in turn lead to endothelial-related complications such as veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) [13].
Studies by our group have previously characterized the ED associated with some of these pathological conditions, with alterations in hemostasis, accelerated atherothrombosis, enhanced inflammatory reaction, and an impaired immune response. Despite the causes differ among the mentioned pathological conditions, ED shares common characteristics, such as an increased expression of adhesion receptors at the cell surface, the production of a more thrombogenic extracellular matrix enriched with pro-adhesive proteins, such as von Willebrand factor (VWF) and tissue factor (TF), the activation of intracellular inflammation-related proteins, such as p38 MAPK and the nuclear factor kappa B (NFκB), and the development of oxidative stress [14,15,16]. These features coexist with the activation of monocytes, macrophages, and granulocytes, favoring the inflammatory phenotype.
The evidence generated in COVID-19 points to the endothelial damage as a key element for the progression of the disease to the later complicated stages. Terms like endotheliitis, endotheliopathy, endothelial dysfunction, and endothelial disease have been commonly used in the recent literature in association with COVID-19. Initial clinical and histopathological data [17] prompted hypothetical literature on the role of the endothelial damage in the progression of the disease [18]. Nowadays, there is a plethora of publications reporting different data, and even different results, on a number of biomarkers of endothelial injury.
The host angiotensin-converting enzyme 2, ACE2 [19], constitutes a specific receptor for the SARS-CoV and also for SARS-CoV-2. It is present prominently, other than in the alveolus, in endothelial cells. There is recent evidence demonstrating the mechanisms SARS-CoV-2 uses to infect cells. Co-expression of the protein-transmembrane protease serine 2 (TMPRSS2) seems to be essential for the virus entry to the cell [20]. TMPRSS2 is also present in endothelial cells, with variable expression depending on the vascular beds [21]. In addition, SARS-CoV-2 spike protein interacts with both ACE2 and cellular heparan sulfate through its receptor-binding domain (RBD) [22]. ACE2 contributes to the integrity of the vascular endothelial cells and SARS-CoV-2 infection downregulates its expression. Importantly, ACE2 downregulates the activity of angiotensin-II (Ang-II) at the endothelial cell by facilitating conversion of Ang-II to Ang 1–7. With the loss of ACE2, Ang-II signaling increases and drives activation of inflammatory pathways in the cell, including the p38/MAPK pathway [23]. Furthermore, the elevated systemic cytokine levels in these patients may also have a deleterious effect on the endothelium. Virus entry and exposure to a proinflammatory environment causes intracellular activation of more proinflammatory pathways [23].
The soluble forms of adhesion receptors overexpressed at the cell surface, namely, VCAM-1, the acute phase reactant VWF, the natural anticoagulant thrombomodulin (TM), among other endothelial-derived factors, have been found to be elevated in COVID-19 patients. Some of these markers correlate with critical illness and even death [24, 25]. Endothelial damage is associated with the loss of the anticoagulant properties of the endothelium, which may contribute to the hypercoagulation state in these patients. Elevated levels of the thrombin generated could also enrich the proinflammatory milieu.
Upregulation of plasma heparanase 1 activity has been also reported in COVID-19 [26], which acts by digesting the endothelial cell glycocalyx. This layer covers the luminal face of the endothelium, being essential for the maintenance of its integrity and functions [27]. It consists of a scaffold of proteins and glycosaminoglycan polymers, such as heparan sulfate, chondroitin sulfate, and hyaluronans. The glycocalyx constitutes a platform that facilitates the adhesion of several growth factors, proteases, cytokines and chemokines, coagulation factors, elements of the complement system, membrane receptors, and adhesion proteins [28]. Degradation and shedding of the endothelial glycocalyx occurs in association with many pathological conditions, and septic syndromes are among them [29, 30]. Aggression to the cell surface by glycocalyx degradation facilitates virus infection and leads to a defenseless endothelium. In COVID-19, heparan sulfate levels are found to be increased in plasma in association with the disease severity, being significantly higher in critically ill patients [25]. In addition to the glycocalyx degradation, activation of the complement system potentially heightens the proneness of the endothelium to become dysfunctional in COVID-19.
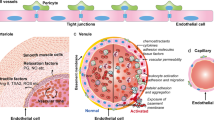
The endothelial injury caused by COVID-19 is therefore in the crossroad of the hypercoagulative state, an impaired fibrinolysis, the activation of the complement system, and the degradation of the glycocalyx layer, and all of which are processes linked in the pathogenesis of COVID-19 complications (Fig. 1). As endothelial injury seems to be a key element for the complications of SARS-CoV2 infection, strategies aimed at protecting the endothelium may be effective to treat them.

Endothelial damage in the crossroad of infection, inflammation, and immunity in COVID-19. SARS-CoV-2 infection causes an endotheliopathy associated with a state of hyperinflammation and dysregulated immunity, in which different factors are involved. Soluble factors secreted by cells and tissues to the circulation, such as cytokines, adhesion receptors, coagulation proteins, elements of the activated complement system, and products released from the degradation of the endothelial glycocalyx. Cellular response involves macrophages and leukocytes, and to a lesser extent platelets and potentially circulating microvesicles secreted from injured cells. Signaling mediators may play a role, with activation of transcription factors, engagement of inflammasome, and TLR4 overexpression, promoting further activation of proinflammatory mediators. TLR, Toll-like receptor; HS, heparan sulfate
Evidence of Neo-angiogenesis
In patients suffering from severe COVID-19, infection by SARS-CoV-2 produces many additional specific effects not observed in influenza A infections. Among these effects, it is important to highlight the severe endothelial injury reported with the disruption of the endothelial cell membranes; a widespread vascular thrombosis with microangiopathy and occlusion of alveolar capillaries, and significant new vessel growth, mainly through a mechanism of intussusceptive and sprouting angiogenesis, was never observed in influenza cases [18].
The main evidence of the angiogenesis occurring in association with COVID-19 comes from a small number of cases [31], and a large study evaluating circulating markers of angiogenesis [32]. Histologic samples show elongated capillaries in the lungs with sudden changes in caliber and the presence of intussusceptive pillars within the capillaries. In the lungs from patients with COVID-19, the density of intussusceptive angiogenesis was significantly higher than the observed in lungs from patients with influenza or in uninfected healthy lungs. The density of features of conventional sprouting angiogenesis (much less frequent) was also higher in the COVID-19 group than in the influenza group. Although tissue hypoxia was a common feature in the lungs from both SARS-CoV-2 and influenza A (H1N1) patients, probably the greater degree of endotheliitis and thrombosis in the lungs from patients with COVID-19 may contribute to the relative frequency of sprouting and intussusceptive angiogenesis observed in these patients. Additionally, the rates of the pulmonary angiogenic features were plotted as a function of the length of the hospital stay. Interestingly, the degree of intussusceptive angiogenesis was found to increase significantly with longer duration of hospitalization. The relationship between these findings and the clinical course of COVID-19 requires further research.
Circulating vascular markers associated with disease severity and mortality have been recently analyzed in a series of patients admitted to Yale-New Haven Hospital with confirmed diagnosis of COVID-19 [32]. This study demonstrates a significant increase in markers of angiogenesis and endotheliopathy in patients hospitalized with COVID-19. The main findings of this study were as follows: multiple circulating proangiogenic factors, including VEGF-A, PDGF-AA, and PDGF-AB/BB, significantly elevated in non-intensive care unit (ICU) COVID-19 patients compared to healthy donors, which could contribute to the recently described vascular remodeling processes in COVID-19; significantly increased levels of angiopoietin-2, follistatin, and PAI-1 in patients at the ICU, reflecting ongoing endotheliopathy and supporting emerging data implicating endothelial involvement in critical illness; and Kaplan–Meier survival analyses indicating that markers of endotheliopathy segregated significantly with in-hospital mortality (angiopoietin-2, follistatin, PAI-1), suggesting that endotheliopathy may be a predictor of mortality in COVID-19.
Whether neo-angiogenesis occurring in association with COVID-19 is a reparative process for the endotheliopathy developed with the disease or may be a way to spread infection remains to be elucidated.
Thrombophilic Endotheliopathy in COVID-19
Morbidity and mortality in patients admitted for COVID-19 is not only directly associated with pneumonia and acute respiratory distress syndrome (ARDS), but also related to coagulopathy, clinically evidenced by a higher incidence of both venous and arterial thromboembolism, especially in critically ill patients [33]. Elevations or alterations of some laboratory markers related to both hemostasis and/or fibrinolysis have been variably found associated with these conditions. Normal or elevated fibrinogen levels, near normal platelet counts, moderate prolongations in prothrombin time (PT) and activated partial thromboplastin time (aPTT), and the lack of a manifest bleeding phenotype (a thrombophilic state predominates) suggest that COVID-19-associated coagulopathy could be an individualized entity [34, 35]. Several studies have also showed the use of D-dimer as a stratification tool between mild and severe forms of COVID-19 and also as mortality predictive marker.
Klok et al. [36] described the characteristics of 184 COVID-19 patients admitted to ICU and 31% of them had thrombotic complications despite being treated with prophylactic doses of low molecular weight heparins (LMWH). The most frequent thrombotic complication observed was pulmonary thromboembolism (81%), whereas the least prevalent was arterial thrombosis presented as ischemic stroke in all cases. Also, an increased incidence of thrombotic complications has been demonstrated in non-ICU patients (12%) despite thromboprophylaxis, being the most frequent complication the asymptomatic deep vein thrombosis [37]. Although COVID-19-associated coagulopathy analytically shares some characteristics with classical sepsis-induced coagulopathy and disseminated intravascular coagulation (DIC), several reports highlighted significant differences between them [38, 39].
In addition to coronavirus infection, admission into hospital emergency units or ICU has been widely accepted as a risk factor for the development of coagulopathy [40, 41]. However, presence of laboratory indicators even in patients with moderate disease, as well as the potential organic dysfunction derived from the coagulopathy itself, suggests that specific pathophysiological mechanisms must be considered in COVID-19 patients.
Several mechanisms could be implicated in the activation of the coagulation cascade following SARS-CoV-2 infection. The innate immune system is triggered unspecifically by the recognition of pathogen-associated molecular patterns (PAMPs), through Toll-like receptors (TLRs) [42], promoting the initiation of a storm of proinflammatory cytokines, mainly interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF). Consequently, cytokine-related endothelial damage and enrollment of neutrophil and monocyte cells could be the main responsible of the tissue-factor pathway activation [43, 44]. Moreover, recruited neutrophils release DNA strands, assembled by histones, altogether with fibrin threads forming a complex extracellular net which acts by intercepting leukocytes and activated platelets. These neutrophil extracellular traps (NET) stabilize oxidant and cytotoxic molecules in the extracellular space and participate in the activation of thrombin generation. In fact, cell-free DNA and myeloperoxidase-DNA complexes were found to be higher in COVID-19 patients than in healthy donors and showed correlation with their respiratory severity [45].
Activation of endothelial cell lineages can induce the generation and expression of TF [46] and destruction of activated endothelial cells could result in the expression of procoagulant membrane phospholipids. Furthermore, impaired antithrombin III production, disrupted activated protein C-TM interaction, and a fostered platelet pro-aggregant state, due to increased VWF release [47], would generate the conditions for a local procoagulant tendency. Interestingly, unlike in septic patients, ADAMTS-13 has been reported to be strictly normal in COVID-19 patients [25], despite the increased release of VWF from the injured endothelial cells. Furthermore, platelet activation through the different pathways described would participate potentiating thrombin generation.
Dysregulation of the fibrinolytic system could be an additional cause for COVID-19 coagulopathy. Plasmin production by viruses and other pathogens for their infective purposes and immune evasion of host immunity has been reported [48, 49]. The initial plasmin production in early stages of the infection, in which antifibrinolytic treatment would have a role [50, 51], in parallel to the thrombin generation, may be exceeded at some point and, as a consequence, fibrin deposits are perpetuated. Wright et al. described the fibrinolytic shutdown in COVID-19 patients tested by thrombo-elastography [52]. The lack of lysis of the clot after 30 min, even with D-dimer levels above 2600 ng/mL, predicted venous thromboembolic events [47]. Nougier et al. demonstrated increased plasma PAI-1 levels in COVID-19 patients and the coexistence of both hypercoagulability (despite heparin prophylaxis) and hypofibrinolysis, evidenced also by thrombo-elastography [53]. However, a recent publication described high levels of PAI-1 and tissue plasminogen activator (tPA) in COVID-19 patients, both correlating with worsening respiratory status, specially tPA, whose extreme high levels were correlated with higher mortality [54]. Moreover, the coexistence of two parallel fibrinolytic systems has been proposed: the systemic and the local in bronchoalveolar parenchyma, whose enhancement or shutdown can be independent [55]. Therefore, the balance between fibrinolysis and hypofibrinolysis mechanisms seems complex in the COVID-19 setting.
A possible link between the COVID-19 coagulopathy and presence of acquired antiphospholipid antibodies has been also reported. Zhang et al. firstly described the presence of autoantibodies IgA and IgG anti-cardiolipin and anti-β2-glycoprotein-I in three COVID-19 patients with arterial thrombotic events [56].
A higher prevalence of autoantibodies IgA anti-β2-glycoprotein-I was confirmed in a larger cohort of COVID-19 patients with the additional finding of significantly lower levels of β2-glycoprotein-I in COVID-19 patients than in healthy controls, with an inverse correlation with ventilation failure [57]. More recently, several studies have informed an elevated incidence of lupus anticoagulant presence in COVID-19 patients, by coagulometric tests [47, 58, 59]. The majority of these findings did not show any straightforward correlation with the presence of antiphospholipid antibodies (measured by immunoassay) or with thromboembolic events [60]. The transient development of lupus anticoagulant or antiphospholipid antibodies concomitant to a viral infection is a well-known fact, resulting from a collateral and unspecific activation of the immune system [61, 62]. In addition, Wang et al. described in their preliminary data the presence of diverse autoantibodies in patients with COVID-19, which could range from those against immune-related proteins, to immune-cell surface proteins and to tissue-associated antigens, such as endothelial adhesion molecules [63]. Nevertheless, the true role of these antibodies and their impact on the prothrombotic profile of these patients remains to be elucidated.
The convincing evidence for a thrombophilic state developing in COVID-19 patients fully supports the use of anticoagulant treatment to reduce the overall morbidity and mortality of COVID-19. It remains to be defined the group of patients that can benefit more from anticoagulation. Anticoagulant treatment will be discussed below.
Endothelial Damage and Complement System in COVID-19
The complement system is a part of the innate immune surveillance system and plays a critical role in the host response to bacterial and viral infection. However, activation of the complement cascade can also promote acute and chronic inflammation and intravascular coagulation together with endothelial cell injury, leading to a thrombotic microangiopathy (TMA) with multiorgan affection [64]. Atypical hemolytic uremic syndrome (aHUS) is the gold standard disease in which complement dysregulation could be inherited or acquired [65]. There is growing evidence suggesting that other pathological conditions, such as pre-eclampsia, HELLP syndrome, and catastrophic antiphospholipid syndrome, could be included in the spectrum of complementopathies [66]. The different pathways of the complement system are highly regulated to ensure that the propagation of complement activation is appropriately restricted, avoiding the potential injury to vascular endothelium and other bystander host tissues [67]. Of note, certain elements of the complement cascade facilitate coagulation and interfere with anticoagulation [66, 68]. Therefore, crosstalk between the complement and coagulation dysregulated cascades, both converging at the endothelial level, may create a prothrombotic environment associated with adverse outcomes in COVID-19 [69].
Preclinical and clinical studies strongly suggest an overactivation of complement cascade in SARS-CoV-2 infection. Although the pathogenesis of COVID-19 is likely multifactorial, it definitely includes endothelial damage in parallel to complement activation. Not only are C5b9 and C5a plasma levels elevated in moderate and severe COVID-19 disease compared with healthy controls [70], but C5b9 levels also correlate with those of vWF and are associated with disease severity [71]. Autopsy findings from 5 COVID-19 non-survivors revealed multi microthrombi and fibrin deposition within alveolar capillaries, among other organs, suggesting the complement implication in this TMA development. The analysis of lung tissues from these patients with severe COVID-19 revealed significant deposits of complement components such as C5b9, C4d, and the mannose-binding lectin (MBL)–associated serine protease (MASP)2, consistent with systemic activation of the alternative and lectin-based complement pathways [72]. Furthermore, results from the proteomic and metabolomic analysis of the serum from COVID-19 patients also suggest a complement involvement in the pathogenesis of this infection [73]. Interestingly, a preclinical work has demonstrated that SARS-CoV-2 spike proteins bind directly to heparan sulfate to activate the alternative complement pathway on cell surfaces [74]. Finally, the published clinical observations of severe COVID-19 (elevated D-dimer, lactate dehydrogenase, bilirubin, and cardiac and renal injury, among other TMA signs) are also consistent with excessive complement activation.
Despite the evidence of complement involvement in endothelial damage and sustained inflammatory response in COVID-19 patients, the complement fundamental role is the early control of virus replication. It constitutes an essential step before the activation of a more effective adaptive immune response [75]. Then, in order to elucidate whether complement system mainly plays a protective or pathogenic role in coronavirus infection, two studies investigated complement activation in Middle East Respiratory Syndrome (MERS) and SARS-CoV infection in murine models. In those studies, MERS-CoV infection–induced lung damage and C5a and C5b9 complement activation products were detected in sera and lung tissues, respectively. Blocking C5a production, by targeting its receptor, decreased tissue damage, cytokine response, and viral replication [76]. Another study using mice deficient in C3, the central component of the complement system, found that, compared with control mice, mice lacking C3 exhibited significantly less weight loss and less respiratory dysfunction despite equivalent viral loads in the lung [77], identifying the complement system as an important host mediator of SARS-CoV-induced disease.
Therapeutic Strategies to Protect the Endothelium
As previously commented, the SARS-CoV-2 enters the host via the respiratory tract and alveolar epithelial cells, infecting and damaging also vascular endothelial cells and alveolar macrophages, all of them expressing ACE2 cells [78, 79]. As with other CoV, SARS-CoV-2 has mechanisms of evasion of the primary immune response, mainly suppressing interferon expression and action [80, 81], which seems to contribute to productive viral replication and severe pathogenesis.
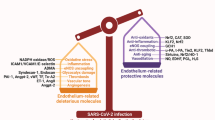
As a consequence of the interaction of infected cells and immune cells (mainly macrophages and T cells), a high concentration of inflammatory cytokines (IL-6, IL-8, IL-1β, GM-CSF, TNF-α, among others) is released at the end of the first week [82]. This has been called “cytokine storm” and is also described as cytokine release syndrome (CRS) which is a phenomenon not exclusive of SARS-CoV-2 infection but it can also be seen in a variety of other settings [83]. This contributes to endothelial dysfunction and promotes pulmonary vascular leakage and ARDS in the most severe patients [78], which is the main cause of death. Consequently, therapeutic strategies to improve outcomes in COVID-19 patients may include molecules addressing different targets. The battery of compounds includes the following: antivirals, molecules with effects against the inflammatory pathways, anticoagulants, inhibitors of complement system activation, or compounds reducing endothelial damage (Fig. 2). Which one (or what combination), when to administer, and whom are questions currently under investigation.

Therapeutic strategies to improve outcomes in COVID-19 patients may include molecules addressing different targets involved in the pathogenesis of the disease. The battery of therapeutic compounds include the following: antivirals (remdesivir, convalescent plasma), molecules with effects against the inflammatory pathways (corticosteroids, tocilizumab, baricitinib), anticoagulants (heparins, DOAC), complement inhibitors (eculizumab, anti-complement C3 antibodies), or compounds reducing endothelial damage (defibrotide, Ang-II pathway antagonists, ACE inhibitors)
Antivirals
Antivirals, if administered early enough in the illness course, could avoid or decrease the hyperinflammatory response. The only antiviral currently commercialized that has been shown to be effective against the SARS-CoV-2 is remdesivir. Although it does not appear to reduce mortality among hospitalized patients overall, it is associated with a reduced time to recovery in severe COVID-19 patients and it could decrease mortality in those who only require low-flow supplemental among this group [84, 85]. Other drugs with antiviral properties in vitro, such as lopinavir-ritonavir or hydroxychloroquine, have been used but they have not been demonstrated to be consistently effective and so are not currently recommended outside of clinical trials [86, 87]. Convalescent plasma from recovered COVID-19 patients has also been used so leveraging any effect of neutralizing antibodies, but results to date are both contradictory and without clear clinical benefit, maybe dependent on their antibody-containing titers [88,89,90]. Additional trials with monoclonal antibodies, convalescent sera, and additional antivirals are ongoing [91, 92].
Therapies with antiviral properties could reduce endothelial dysfunction limiting the entry of the virus into the endothelial cells and avoiding its further replication. Of note, while effective antiviral agents are desirable for treatment in the acute phase, it is worth considering that the chronic use of some antiviral drugs has been recognized to induce endothelial dysfunction [93, 94].
Anti-inflammatory and Immunomodulatory Compounds
Therapeutics targeting the inflammatory cascade, or one of its components, have been assessed. They are classified as anti-inflammatory and/or immunomodulatory drugs and include corticosteroids, cytokine pathway inhibitors, and Janus kinase (JAK)-2 inhibitors. The hyperinflammatory state is associated with certain inflammatory biomarkers, such as those related to macrophage activation (C-reactive protein (CRP), ferritin, sCD25/sCD163, and triglycerides), lymphocyte activation (with lymphopenia), and endothelial dysfunction with increases in D-dimer and fibrinogen. Many of these parameters have been associated with worse outcomes and may help to guide when to initiate treatment to inhibit this hyperinflammatory state [38]. However, if they are initiated too soon, they may impair the immune response against the virus or even promote its replication [95]. Further to this, if the treatment is initiated when ARDS is already well established, it could be too late. Glucocorticoids, specifically dexamethasone, are the only anti-inflammatory compounds that have shown a reduction in mortality when administering to a specific population with COVID-19. The use of 6 mg/day of dexamethasone during 10 days in the RECOVERY trial reduced 28-day mortality compared with standard of care alone in those patients receiving some ventilatory support or oxygen, mainly after day 7 of symptoms onset [96,97,98]. This treatment had no effect or it was deleterious among those patients without an oxygen requirement.
Previous evidence supports the fact that glucocorticoids are able to modulate endothelial function, acting differently depending on whether they are used in healthy conditions, where they have been shown to promote endothelial dysfunction in in vitro or animal studies [99, 100]; or if they are administered in situations where inflammation is present, like septic shock, atherosclerosis, or inflammatory diseases [101, 102]. Positive effects over endothelial function in this scenario have been attributed to a reduction in endothelial expression of different cytokines and biomarkers (such as IL-6, IL-8, VEGF, endothelin-1, NFκB, and COX-2) [103].
Among cytokine action inhibitors, the preferred target has been IL-6, mainly with the use of the monoclonal antibody tocilizumab, a potent IL-6 receptor blocker. After very positive results in observational studies, some randomized trials had shown no clear benefit in mortality. However, recently, three different trials have suggested benefits in clinical outcomes among different subgroups of patients, including critically ill patients [104,105,106,107,108]. In vitro data indicate that endothelial IL-6 trans-signaling is associated with an inflammation circuit for robust IL-6, IL-8, and MCP-1 production and promoted PAI-1 production and that IL-6 signaling blockade by tocilizumab can blunt this endothelial cell activation [109]. Previously, IL-6 blockade with tocilizumab had been demonstrated to improve endothelial function in a cardiovascular high-risk population [110]. Interestingly, median elevation of IL-6 in this scenario is generally lower than seen in other settings of hyperinflammation, such as the CRS associated with patients with lymphoid neoplasms treated with chimeric antigen receptor (CAR)-T cells, which have shown a better response to tocilizumab [111]. This observation suggests that other pathways are involved in COVID-19 pathobiology.
Other cytokine inhibitors have shown positive results in observational studies and are under evaluation in as part of randomized clinical trials: anakinra and canakinumab against IL-1 [112, 113], mavrilimumab against GM-CSF [114], and emapalumab against IFN-γ [115] are examples. Furthermore, JAK-2 inhibitors, as baricitinib or ruxolitinib, are also promising drugs with already proven benefits and ongoing prospective studies underway [116,117,118]. Specifically, in a limited series of severe COVID-19 patients, administration of baricitinib was related to the attenuation of circulating biomarkers associated with cytokine storm and vascular endothelial damage [119]. Also, treatment with Bruton tyrosine kinase (BTK) inhibitors, such as ibrutinib, may provide protection against lung injury and even improve pulmonary function in hypoxic patients with COVID-19 [120]. Clinical studies are now ongoing.
Anticoagulant and Antiplatelet Therapies
Treatments addressing coagulation pathways are useful to treat the coagulopathy developing in severe cases of COVID-19. The most studied molecules in this regard are thromboprophylaxis and treatment of the thrombotic events with heparin. Tang et al. have described the protective role of prophylactic doses of LMWH in COVID-19 patients with a sepsis-induced coagulopathy score ≥ 4 or D-dimer levels above six-fold the upper limit of normal by decreasing their mortality rate [121]. In fact, the International Society of Thrombosis and Haemostasis (ISTH) interim guidelines [122] advise prophylaxis with LMWH in all COVID-19 hospitalized patients if there is no contraindication and, in subsequent updates, several guidelines have suggested the intensification of anticoagulation based on severity of the disease and thrombotic risk [123, 124]. The rationale for the use of LMWH, in addition to its widely described anticoagulant effect, is based on both impaired ACE2-mediated internalization of the virus in the presence of heparin [22] and on its proven anti-inflammatory effects [125]. Moreover, heparin may have a beneficial effect protecting the endothelium in the setting of SARS-CoV-2 infection since patients treated with therapeutic doses of anticoagulation for other reasons before their admission for COVID-19 had lower levels of circulating endothelial cells, analyzed as a marker of endothelial damage in an observational study [126]. Early studies suggested that not only heparins but also direct oral anticoagulants (DOACs) could be beneficial for severe COVID-19 patients [127] though this indication has been challenged in later studies on critically ill COVID-19 patients [128].
DOACs can be an option of anticoagulation in ambulatory COVID-19 patients, despite the need for awareness about interactions with other drugs that could be used for the COVID-19 treatment (as antivirals in the beginning of the pandemic) as well as the importance of monitoring and dose adjustment in the setting of renal failure [129]. It also remains to be defined the group of patients that can benefit most from anticoagulation [130]. Of note, some DOACs may exert anti-inflammatory action at lower doses protecting from endothelial dysfunction while still exhibiting some anticoagulant action [131]. Although antiplatelet therapy has been under consideration, its beneficial role in COVID-19 hypercoagulability has not been proved, while their interference with some antiviral drugs used during the pandemic has prompted physicians to replace the antiplatelet therapy [132].
Likewise, other treatments have been proposed. Wang et al. reported respiratory improvement in three ICU COVID-19 patients after the administration of tissue plasminogen activator (tPA) [133], and clinical assays are ongoing to evaluate the role of fibrinolysis in patients with COVID-19-induced ARDS [134]. As explained above, although there is a systemic hypofibrinolysis, as evidenced by plasma biomarkers, it can be a local pulmonary hyper-fibrinolysis. Administration of plasminogen has been demonstrated to be beneficial in patients with COVID-19 pneumonia, and, for this treatment to be effective, presence of u-PA is necessary to convert the administered plasminogen into plasmin [135]. In a recent meta-analysis, recombinant human soluble TM (rhsTM) also proved to reduce 28-day mortality in non-COVID-19 septic patients with sepsis-associated coagulopathy [136]. Also, treatment with antithrombin and activated C protein is under consideration since nebulized administration has been successfully used in lung injury and sepsis models, respectively [137, 138]. However, further studies are needed in the specific COVID-19 context.
Complement Inhibitors
Although complement activation plays a protective role in front of infection by favoring viral clearance, complement inhibition treatment is worthy of consideration in order to avoid inflammatory over-response and endothelial injury in COVID-19 patients. Actually, blockage of C5 may be therapeutically beneficial without affecting the protective immune response. In fact, some anecdotal results point out to complement blockage as a potential efficient strategy to treat patients with COVID-19-associated ARDS. Treatment with eculizumab (Soliris; Alexion, Boston, MA), a monoclonal antibody that binds with high affinity to the complement protein C5, has been proved to be efficient in 4 cases with COVID-19-associated ARDS, showing a marked clinical improvement within the first 48 h after the first administration [139]. Moreover, a patient with severe ARDS due to COVID-19 pneumonia was successfully treated with the compassionate use of the complement C3 inhibitor AMY-101 (Amyndas Pharmaceuticals, Glyfada, Greece) [140], and upstream complement inhibition at the C3 or C5 level has proved to lead to a rapid decline in the concentration of serum inflammatory markers in patients with COVID-19 [141]. Whether blockade of terminal complement effectors at different levels of the cascade leads to similar outcomes remains under study [142]. It is worth mentioning that a recent study suggested inefficacy of IFX-1, an anti-human C5a monoclonal antibody, in patients with severe COVID-19 in an exploratory, randomized phase 2 trial. However, as acknowledged by the authors, the trial was not powered to show statistically significant differences in clinical primary endpoints (percentage change in PaO2/FiO2 in the supine position between baseline and day 5), eventually jeopardizing any conclusion, even on secondary endpoints (mortality at 28 days and treatment-emergent and serious adverse events) [143]. To date (April 2021, www.clinicaltrials.gov), there are 18 active clinical trials testing complement inhibition across a spectrum of COVID-19 affected patients.
Endothelial Protection
Finally, therapeutic strategies for COVID-19 and its severe manifestations could include agents that decrease endothelial damage and ED, including defibrotide (DF), angiotensin-converting enzyme inhibitors, angiotensin-II receptor blockers, and novel p38/MAPK inhibitors.
DF is a biologically derived, complex mixture of polydeoxyribonucleotides with pleiotropic properties, including anti-thrombotic, pro-fibrinolytic, and anti-inflammatory effects [144]. DF is currently approved for the treatment of VOD/SOS in adults and children [145, 146], with proven safety and efficacy, and favorable tolerability. As an endothelial protective agent, DF is believed to limit the endothelial damage that underlies VOD/SOS, wherein endothelial cells undergo apoptosis and extrusion into hepatic sinusoids [147]. DF interacts directly with endothelial cells at the cell membrane, and becomes internalized by macropinocytosis [148]. DF subsequently affects multiple signaling cascades within endothelial cells, including inhibition of both the p38/MAPK pathway [148] and the PI3K/Akt pathway [149], with the decreased expression of numerous endothelial cell adhesion molecules, including but not limited to P-selectin, E-selectin, VCAM-1, and ICAM-1 [150]. DF also reduces serum cytokines and proinflammatory molecules including IL-6, IL-12, TNF-α, IFN-γ, VEGF, thromboxane A2, leukotriene B4, and ROS, furthering DF’s anti-inflammatory properties relevant in the treatment of hyperinflammatory syndromes and so targeting CRS [149, 150]. On the other hand, DF increases tissue plasminogen activator and TM expression, while simultaneously decreasing VWF and PAI-1, thereby reducing thrombosis and enhancing fibrinolysis [148, 149]. DF also markedly inhibits heparanase expression and activity [151], promoting restoration of endothelial integrity and function. It competes with heparan sulfate to diminish heparanase-mediated degradation, preserving heparan sulfate on the subendothelial basement membrane [152], and, as a consequence, it may interfere with direct interactions between SARS-CoV-2 and heparan sulfate that facilitate viral adherence and entry. This effect may also both diminish viral adherence and complement activation.
In patients with post-transplant thrombotic microangiopathy, treatment with DF significantly reduced complement activation and associated vascular damage [153]. DF may similarly regulate complement hyperactivation observed in patients with severe COVID-19. In fact, two patients with pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 infection received DF, leading to symptomatic resolution and normalization of complement levels [154].
For these reasons, and for its demonstrated long-term safety even in high-risk pediatric populations [155], DF stands as a promising candidate for the treatment of COVID-19, based on the complex pathophysiology of COVID-19 and DF’s pleiotropic mechanisms of action [156].
Several studies of DF in COVID-19 are in process and actively enrolling patients internationally. These include the Spanish DEFACOVID phase 2b randomized, double-blind, placebo-controlled, clinical trial (clinicaltrials.gov: NCT04348383) and single-arm, phase 2 studies in Italy, Ireland, Germany, and the UK, with over 150 patients enrolled and treated to date across these various protocols. Clinical trials in the USA are underway at the University of Michigan in Ann Arbor, MI, USA (clinicaltrials.gov: NCT04530604) and Brigham and Women’s Hospital in Boston, MA, USA (clinicaltrials.gov: NCT04652115). Patients receiving DF are being comprehensively assessed for inflammatory markers as well as other biomarkers associated with the endotheliitis of COVID-19; these data will hopefully confirm the effects of DF on multiple targets fundamental to the pathophysiology of COVID-19 and demonstrate associated clinical benefit.
In addition to DF, other pharmacologic agents have potential as treatments targeting ED through effects on the Ang-II and p38/MAPK signaling axis. As antagonists of the Ang-II pathway, ACE inhibitors and ARBs stand to inhibit the signaling cascade driven by high levels of Ang-II, which leads to p38/MAPK activation in endothelial cells. ACE inhibitors and ARBs are currently being studied for their capacity to downregulate this axis and mitigate the contribution of Ang-II to inflammatory processes in COVID-19. Direct inhibitors of p38/MAPK may also be considered for this reason, and some of these have already been developed and studied in several contexts [157]. These strategies might achieve both immediate and long-term protective effects for endothelia in patients with COVID-19, and so further improve patient outcomes.
Concluding Remarks
-
The endothelium is a strategic component of the pathophysiology of COVID-19 complications.
-
Endothelial injury in COVID-19 is at the crossroad of the hypercoagulative state, the impaired fibrinolysis, the activation of the complement system, and the degradation of the glycocalyx layer.
-
A precise correlation between the patient clinical information and the pathological findings is basic to select an effective treatment for COVID-19.
-
Antiviral therapies will be most effective when viral activity is the main pathogenic factor. Directed anti-inflammatory, anticoagulant, anti-complement, and endothelial protective agents are indicated when the vascular endothelium is affected. Use of preventive strategies could avert development of endothelial-related complications.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med. 2020;382:1199–207.
Wynants L, Van Calster B, Collins GS et al. Prediction models for diagnosis and prognosis of covid-19: systematic review and critical appraisal. BMJ. 2020;369.
Pijls BG, Jolani S, Atherley A et al. Demographic risk factors for COVID-19 infection, severity, ICU admission and death: a meta-analysis of 59 studies. BMJ Open. 2021;11.
Risk for COVID-19 Infection, hospitalization, and death by age group | CDC. Available from: https://www.cdc.gov/coronavirus/2019-ncov/covid-data/investigations-discovery/hospitalization-death-by-age.html
Aird WC. Endothelium in health and disease. Pharmacol Reports Pharmacol Rep. 2008;60:139–43.
Cines DB, Pollak ES, Buck CA, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–61.
Diaz-Ricart M, Torramade-Moix S, Pascual G, et al. Endothelial damage, inflammation and immunity in chronic kidney disease. Toxins (Basel). 2020;12:361.
Berzigotti A, Erice E, Gilabert R, et al. Cardiovascular risk factors and systemic endothelial function in patients with cirrhosis. Am J Gastroenterol. 2013;108:75–82.
Hanzu FA, Palomo M, Kalko SG, et al. Translational evidence of endothelial damage in obese individuals: inflammatory and prothrombotic responses. J Thromb Haemost J Thromb Haemost. 2011;9:1236–45.
Tuder RM, Cool CD, Yeager M, Taraseviciene-Stewart L, Bull TM, Voelkel NF. The pathobiology of pulmonary hypertension: endothelium. Clin Chest Med. 2001;22:405–18.
Ince C, Mayeux PR, Nguyen T, et al. The Endothelium in Sepsis. Shock. 2016;45:259–70.
Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. 2018;123:825–48.
Carreras E, Diaz-Ricart M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplant. 2011;46:1495–502.
Palomo M, Diaz-Ricart M, Carbo C, et al. The release of soluble factors contributing to endothelial activation and damage after hematopoietic stem cell transplantation is not limited to the allogeneic setting and involves several pathogenic mechanisms. Biol Blood Marrow Transplant. 2009;15:537–46.
Caballo C, Palomo M, Cases A, et al. NFκB in the development of endothelial activation and damage in uremia: an in vitro approach. PLoS One. 2012;7:e43374.
Palomo M, Vera M, Martin S, et al. Up-regulation of HDACs, a harbinger of uraemic endothelial dysfunction, is prevented by defibrotide. J Cell Mol Med. 2020;24:1713–23.
Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8.
Teuwen L-A, Geldhof V, Pasut A, Carmeliet P. {COVID}-19: the vasculature unleashed. Nat Rev Immunol. 2020;20:389–91.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46:586–90.
Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-280.e8.
Aimes RT, Zijlstra A, Hooper JD, et al. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb Haemost. 2003;89:561–72.
Clausen TM, Sandoval DR, Spliid CB, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183:1043-1057.e15.
Grimes JM, Grimes KV. p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J Mol Cell Cardiol. 2020;144:63–5.
Goshua G, Pine AB, Meizlish ML, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7:e575–82.
Moreno-Castaño AB, Fernandez S, Palomo M, et al. Circulating biomarkers of COVID-19-triggered endotheliopathy: from conjecture to certainty. Blood. 2020;136:31–2.
Buijsers B, Yanginlar C, de Nooijer A et al. Increased plasma heparanase activity in COVID-19 patients. Front Immunol. 2020;11.
Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–67.
Ori A, Wilkinson MC, Fernig DG. A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J Biol Chem. 2011;286:19892–904.
Chelazzi C, Villa G, Mancinelli P, De GA, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care. 2015;19:26.
Song JW, Zullo J, Lipphardt M, et al. Endothelial glycocalyx-the battleground for complications of sepsis and kidney injury. Nephrol Dial Transplant. 2018;33:203–11.
Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med. 2020;383:120–8.
Pine AB, Meizlish ML, Goshua G, et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm Circ. 2020;10:1–4.
Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–7.
Fogarty H, Townsend L, Ni Cheallaigh C, et al. COVID19 coagulopathy in Caucasian patients. Br J Haematol. 2020;189:1044–9.
Ranucci M, Ballotta A, Di Dedda U, et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. 2020;18:1747–51.
Klok FA, Kruip MJHA, van der Meer NJM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb Res. 2020;191:148–50.
Porfidia A, Santoliquido A, Cammá G, PorcedduPola ER, et al. Incidence of deep vein thrombosis among non-{ICU} patients hospitalized for {COVID}-19 despite pharmacological thromboprophylaxis. J Thromb Haemost. 2020;18:3110–1.
Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62.
Lippi G, Favaloro EJ. D-dimer is associated with severity of coronavirus disease 2019: a pooled analysis. Thromb Haemost. 2020;120:876–8.
Levi M, Opal SM. Coagulation abnormalities in critically ill patients. Crit Care Crit Care 2016. p. 463–71.
Giannis D, Ziogas IA, Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J Clin Virol. 2020;127:104362.
Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–73.
Levi M, Van Der Poll T, Ten Cate H, Van Deventer SJH. The cytokine-mediated imbalance between coagulant and anticoagulant mechanisms in sepsis and endotoxaemia. Eur J Clin Invest. 1997;27:3–9.
Iba T, Levy JH, Levi M, Thachil J. Coagulopathy in COVID-19. J Thromb Haemost. 2020;18:2103–9.
Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:e138999.
Bautista-Vargas M, Bonilla-Abadía F, Cañas CA. Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J Thromb Thrombolysis. 2020;50:479–83.
Helms J, Tacquard C, Severac F, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–98.
Izaguirre G. The proteolytic regulation of virus cell entry by furin and other proprotein convertases. Viruses. 2019;11:837.
Berri F, Rimmelzwaan GF, Hanss M, et al. Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog. 2013;9:e1003229.
Medcalf RL, Keragala CB, Myles PS. Fibrinolysis and COVID-19: a plasmin paradox. J Thromb Haemost. 2020;18:2118–22.
Jacob G, Aharon A, Brenner B. COVID-19-associated hyper-fibrinolysis: mechanism and implementations. Front Physiol. 2020;11.
Wright FL, Vogler TO, Moore EE, et al. Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 infection. J Am Coll Surg. 2020;231:193-203.e1.
Nougier C, Benoit R, Simon M, et al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J Thromb Haemost. 2020;18:2215–9.
Zuo Y, Warnock M, Harbaugh A et al. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci Rep. 2021;11.
Keragala CB, Medcalf RL, Myles PS. Fibrinolysis and COVID-19: a tale of two sites? J Thromb Haemost. 2020;18:2430–2.
Zhang Y, Xiao M, Zhang S, et al. Coagulopathy and antiphospholipid antibodies in patients with COVID-19. N Engl J Med. 2020;382:e38.
Serrano M, Espinosa G, Lalueza A, et al. Beta-2-glycoprotein-I deficiency could precipitate an antiphospholipid syndrome-like prothrombotic situation in patients with coronavirus disease 2019. ACR Open Rheumatol. 2021;3:267–76.
Bowles L, Platton S, Yartey N, et al. Lupus anticoagulant and abnormal coagulation tests in patients with COVID-19. N Engl J Med. 2020;383:288–90.
Harzallah I, Debliquis A, Drénou B. Lupus anticoagulant is frequent in patients with Covid-19. J Thromb Haemost. 2020;18:2064–5.
Devreese KMJ, Linskens EA, Benoit D, Peperstraete H. Antiphospholipid antibodies in patients with COVID-19: a relevant observation? J Thromb Haemost. 2020;18:2191–201.
Abdel-Wahab N, Talathi S, Lopez-Olivo MA, Suarez-Almazor ME. Risk of developing antiphospholipid antibodies following viral infection: a systematic review and meta-analysis. Lupus. 2017;27:572–83.
Uthman IW, Gharavi AE. Viral infections and antiphospholipid antibodies. Semin Arthritis Rheum. 2002;31:256–63.
Wang EY, Mao T, Klein J et al. Diverse functional autoantibodies in patients with COVID-19. medRxiv Prepr Serv Heal Sci. medRxiv; 2020.
Blasco M, Guillén E, Quintana LF et al. Thrombotic microangiopathies assessment: mind the complement. Clin Kidney J. 2020;1–12.
Noris M, Remuzzi G. Atypical hemolytic–uremic syndrome. N Engl J Med. 2009;361:1676–87.
Palomo M, Blasco M, Molina P, et al. Complement activation and thrombotic microangiopathies. Clin J Am Soc Nephrol. 2019;14:1719–32.
Kerr H, Richards A. Complement-mediated injury and protection of endothelium: lessons from atypical haemolytic uraemic syndrome. Immunobiology. 2012;217:195–203.
Markiewski MM, Nilsson B, Nilsson Ekdahl K, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–92.
Java A, Apicelli AJ, Liszewski MK, et al. The complement system in COVID-19: friend and foe? JCI insight. 2020;5:e140711.
Cugno M, Meroni PL, Gualtierotti R, et al. Complement activation in patients with COVID-19: a novel therapeutic target. J Allergy Clin Immunol. 2020;146:215–7.
Cugno M, Meroni PL, Gualtierotti R, et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J Autoimmun. 2021;116:102560.
Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13.
Shen B, Yi X, Sun Y, et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182:59-72.e15.
Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D Inhibition. Blood. 2020;136:2080–9.
Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6:257.
Jiang Y, Zhao G, Song N, et al. Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg Microbes Infect. 2018;7:77.
Gralinski LE, Sheahan TP, Morrison TE, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio. 2018;9:e01753-e1818.
Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 2020. p. 1100–15.
Ziegler CGK, Allon SJ, Nyquist SK, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181:1016-1035.e19.
Zhou P, YangLou X, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3.
Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036-1045.e9.
Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science (80-. ). 2020. p. 473–4.
Shimabukuro-Vornhagen A, Gödel P, Subklewe M et al. Cytokine release syndrome. J Immunother Cancer. 2018.
Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of COVID-19 — final report. N Engl J Med. 2020;383:1813–26.
Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–78.
Cao B, Wang Y, Wen D, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe COVID-19. N Engl J Med. 2020;382:1787–99.
Horby P, Mafham M, Linsell L, et al. Effect of hydroxychloroquine in hospitalized patients with COVID-19. N Engl J Med. 2020;383:2030–40.
Li L, Zhang W, Hu Y, et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA - J Am Med Assoc. 2020;324:460–70.
Agarwal A, Mukherjee A, Kumar G, Chatterjee P, Bhatnagar T, Malhotra P. Convalescent plasma in the management of moderate COVID-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ. 2020; 371.
Libster R, Pérez Marc G, Wappner D et al. Early high-titer plasma therapy to prevent severe COVID-19 in older adults. N Engl J Med. 2021;384.
Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. N Engl J Med. 2021;384:238–51.
Lundgren JD, Grund B, Barkauskas CE et al. A neutralizing monoclonal antibody for hospitalized patients with COVID-19. N Engl J Med. 2020;NEJMoa2033130.
Skowyra A, Zdziechowicz I, Mikuła T, Wiercińska-Drapało A. Endothelial dysfunction - an important factor in the progression of atherosclerosis in HIV-infected persons. HIV AIDS Rev. 2012. p. 57–60.
Jiang B, Hebert VY, Zavecz JH, Dugas TR. Antiretrovirals induce direct endothelial dysfunction in vivo. J Acquir Immune Defic Syndr. 2006;42:391–5.
Lee N, Allen Chan KC, Hui DS, et al. Effects of early corticosteroid treatment on plasma SARS-associated Coronavirus RNA concentrations in adult patients. J Clin Virol. 2004;31:304–9.
Rosas IO, Bräu N, Waters M et al. Tocilizumab in hospitalized patients with severe COVID-19 pneumonia. N Engl J Med. 2021; Available from: http://www.ncbi.nlm.nih.gov/pubmed/33631066
Salama C, Han J, Yau L, et al. Tocilizumab in patients hospitalized with COVID-19 pneumonia. N Engl J Med. 2021;384:20–30.
Horby P, Lim WS, Emberson JR, et al. Dexamethasone in hospitalized patients with COVID-19. N Engl J Med. 2021;384:693–704.
Schäfer SC, Wallerath T, Closs EI et al. Dexamethasone suppresses eNOS and CAT-1 and induces oxidative stress in mouse resistance arterioles. Am J Physiol Heart Circ Physiol; 2005; 288.
Iuchi T, Akaike M, Mitsui T, et al. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ Res. 2003;92:81–7.
Goodwin JE, Feng Y, Velazquez H, Zhou H, Sessa WC. Loss of the endothelial glucocorticoid receptor prevents the therapeutic protection afforded by Dexamethasone after LPS. PLoS ONE. 2014;9:e108126.
Verhoeven F, Totoson P, Maguin-Gaté K, et al. Glucocorticoids improve endothelial function in rheumatoid arthritis: a study in rats with adjuvant-induced arthritis. Clin Exp Immunol. 2017;188:208–18.
Verhoeven F, Prati C, Maguin-Gaté K, Wendling D, Demougeot C. Glucocorticoids and endothelial function in inflammatory diseases: focus on rheumatoid arthritis. Arthritis Res. 2016. p. 258.
Stone JH, Frigault MJ, Serling-Boyd NJ, et al. Efficacy of tocilizumab in patients hospitalized with COVID-19. N Engl J Med. 2020;383:2333–44.
Hermine O, Mariette X, Tharaux PL, Resche-Rigon M, Porcher R, Ravaud P. Effect of tocilizumab vs usual care in adults hospitalized with COVID-19 and moderate or severe pneumonia: a randomized clinical trial. JAMA Intern Med. 2021;181:32–40.
Salvarani C, Dolci G, Massari M, et al. Effect of tocilizumab vs standard care on clinical worsening in patients hospitalized with COVID-19 pneumonia: a randomized clinical trial. JAMA Intern Med. 2021;181:24–31.
Horby PW, Pessoa-Amorim G, Peto L et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): preliminary results of a randomised, controlled, open-label, platform trial. medRxiv. 2021; 2021.02.11.21249258.
Gordon AC, Mouncey PR, Al-beidh F et al. Interleukin-6 receptor antagonists in critically ill patients with COVID-19. medRxiv. N Engl J Med; 2021;NEJMoa2100433.
Kang S, Tanaka T, Inoue H, et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A. 2020;117:22351–6.
Bacchiega BC, Bacchiega AB, Usnayo MJG, Bedirian R, Singh G, Pinheiro G da RC. Interleukin 6 inhibition and coronary artery disease in a high-risk population: a prospective community-based clinical study. J Am Heart Assoc. 2017;6.
Leisman DE, Ronner L, Pinotti R et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020. p. 1233–44.
Cavalli G, De Luca G, Campochiaro C, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2:e325–31.
Ucciferri C, Auricchio A, Di Nicola M et al. Canakinumab in a subgroup of patients with COVID-19. Lancet Rheumatol. 2020. p. e457-ee458.
De Luca G, Cavalli G, Campochiaro C, et al. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: a single-centre, prospective cohort study. Lancet Rheumatol. 2020;2:e465–73.
Efficacy and safety of emapalumab and anakinra in reducing hyperinflammation and respiratory distress in patients with COVID-19 infection. ClinicalTrials.gov.. Available from: https://clinicaltrials.gov/ct2/show/NCT04324021
Cantini F, Niccoli L, Nannini C et al. Beneficial impact of baricitinib in COVID-19 moderate pneumonia; multicentre study. J. Infect. 2020. p. 647–79.
Cao Y, Wei J, Zou L, et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): a multicenter, single-blind, randomized controlled trial. J Allergy Clin Immunol. 2020;146:137-146.e3.
Kalil AC, Patterson TF, Mehta AK et al. Baricitinib plus remdesivir for hospitalized adults with COVID-19. N Engl J Med. 2020;384.
Sims JT, Krishnan V, Chang CY, et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J Allergy Clin Immunol. 2021;147:107–11.
Treon SP, Castillo JJ, Skarbnik AP et al. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19 infected patients. Blood. 2020. p. 1912–5.
Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18:1094–9.
Thachil J, Tang N, Gando S, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020;18:1023–6.
Thachil J, Juffermans NP, Ranucci M, et al. ISTH DIC subcommittee communication on anticoagulation in COVID-19. J Thromb Haemost. 2020;18:2138–44.
Vivas D, Roldán V, Esteve-Pastor MAMA et al. Recommendations on antithrombotic treatment during the {COVID}-19 pandemic. Position statement of the Working Group on Cardiovascular Thrombosis of the Spanish Society of Cardiology. Rev Española Cardiol English Ed. 2020;73:749–57.
Poterucha TJ, Libby P, Goldhaber SZ. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb Haemost. 2017;117:437–44.
Khider L, Gendron N, Goudot G, et al. Curative anticoagulation prevents endothelial lesion in COVID-19 patients. J Thromb Haemost. 2020;18:2391–9.
Nadkarni GN, Lala A, Bagiella E, et al. Anticoagulation, bleeding, mortality, and pathology in hospitalized patients with COVID-19. J Am Coll Cardiol. 2020;76:1815–26.
NIH ACTIV Trial of blood thinners pauses enrollment of critically ill COVID-19 patients | National Institutes of Health (NIH). Available from: https://www.nih.gov/news-events/news-releases/nih-activ-trial-blood-thinners-pauses-enrollment-critically-ill-covid-19-patients
Thachil J, Tang N, Gando S, et al. DOACs and “newer” hemophilia therapies in COVID-19: Reply. J Thromb Haemost. 2020;18:1795–6.
Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033–40.
Torramade-Moix S, Palomo M, Vera M et al. Apixaban downregulates endothelial inflammatory and prothrombotic phenotype in an in vitro model of endothelial dysfunction in uremia. Cardiovasc Drugs Ther. 2020; 1–12.
Watson RA, Johnson DM, Dharia RN, Merli GJ, Doherty JU. Anti-coagulant and anti-platelet therapy in the COVID-19 patient: a best practices quality initiative across a large health system. Hosp Pract. 1995;2020(48):169–79.
Wang J, Hajizadeh N, Moore EE, et al. Tissue plasminogen activator ({tPA}) treatment for {COVID}-19 associated acute respiratory distress syndrome ({ARDS}): a case series. J Thromb Haemost. 2020;18:1752–5.
Fibrinolytic therapy to treat ARDS in the setting of COVID-19 infection. Available from: https://clinicaltrials.gov/ct2/show/NCT04357730
Wu Y, Wang T, Guo C et al. Plasminogen improves lung lesions and hypoxemia in patients with COVID-19. SSRN Electron J. 2020.
Valeriani E, Squizzato A, Gallo A, et al. Efficacy and safety of recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy: a systematic review and meta-analysis. J Thromb Haemost. 2020;18:1618–25.
Uchiba M, Okajima K, Murakami K. Effects of various doses of antithrombin III on endotoxin-induced endothelial cell injury and coagulation abnormalities in rats. Thromb Res. 1998;89:233–41.
Cornet AD, Hofstra JJ, Vlaar AP, et al. Activated protein C attenuates pulmonary coagulopathy in patients with acute respiratory distress syndrome. J Thromb Haemost. 2013;11:894–901.
Diurno F, Numis FG, Porta G et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 2020. p. 4040–7.
Mastaglio S, Ruggeri A, Risitano AM, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215:108450.
de Latour RP, Bergeron A, Lengline E et al. Complement C5 inhibition in patients with COVID-19 - a promising target?. Haematologica. 2020. p. 2847–50.
Mastellos DC, Pires da Silva BGP, Fonseca BAL et al. Complement C3 vs C5 inhibition in severe COVID-19: early clinical findings reveal differential biological efficacy. Clin Immunol. 2020;220.
Vlaar APJ, de Bruin S, Busch M, et al. Anti-C5a antibody IFX-1 (vilobelimab) treatment versus best supportive care for patients with severe COVID-19 (PANAMO): an exploratory, open-label, phase 2 randomised controlled trial. Lancet Rheumatol. 2020;2:e764–73.
Richardson PG, Carreras E, Iacobelli M, Nejadnik B. The use of defibrotide in blood and marrow transplantation. Blood Adv. 2018;2:1495–509.
Richardson PG, Grupp SA, Pagliuca A, Krishnan A, Ho VT, Corbacioglu S. Defibrotide for the treatment of hepatic veno-occlusive disease/sinusoidal obstruction syndrome with multiorgan failure. Int J Hematol Oncol. 2017;6:75–93.
Richardson PG, Riches ML, Kernan NA, et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood. 2016;127:1656–65.
Richardson PG, Soiffer RJ, Antin JH, et al. Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol Blood Marrow Transplant. 2010;16:1005–17.
Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood. 2016;127:1719–27.
Martinez-Sanchez J, Hamelmann H, Palomo M et al. Acute graft-vs.-host disease-associated endothelial activation in vitro is prevented by defibrotide. Front Immunol. 2019;10:2339.
García-Bernal D, Palomo M, Martínez CM, et al. Defibrotide inhibits donor leucocyte-endothelial interactions and protects against acute graft-versus-host disease. J Cell Mol Med. 2020;24:8031–44.
Mitsiades CS, Rouleau C, Echart C, et al. Preclinical studies in support of defibrotide for the treatment of multiple myeloma and other neoplasias. Clin Cancer Res. 2009;15:1210–21.
Koganti R, Suryawanshi R, Shukla D. Heparanase, cell signaling, and viral infections. Cell Mol Life Sci. 2020;77:5059–77.
Calbi V, Fumagalli F, Consiglieri G, et al. Use of Defibrotide to help prevent post-transplant endothelial injury in a genetically predisposed infant with metachromatic leukodystrophy undergoing hematopoietic stem cell gene therapy. Bone Marrow Transplant. 2018;53:913–7.
Lang P, Eichholz T, Bakchoul T, et al. Defibrotide for the treatment of pediatric inflammatory multisystem syndrome temporally associated with severe acute respiratory syndrome coronavirus 2 infection in 2 pediatric patients. J Pediatric Infect Dis Soc. 2020;9:622–5.
Corbacioglu S, Cesaro S, Faraci M, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379:1301–9.
Richardson E, Carlo-Stella C, Jara R, et al. Response to Maccio et al Multifactorial pathogenesis of COVID-19-related coagulopathy: can defibrotide have a role in the early phases of coagulation disorders? J Thromb Haemost. 2020;18(3111):3.
Bühler S, Laufer SA. p38 MAPK inhibitors: a patent review (2012–2013). Expert Opin Ther Pat. 2014;24:535–54.
Funding
We would like to acknowledge support from Fundació Clínic, Barcelona (HCB/2020/0401), and grants from Jazz Pharmaceuticals Plc (IST-16–10355), German José Carreras Leukaemia Foundation (03R/2019), Instituto de Salud Carlos III from Spanish Government (PI19/00888), Fundació La Marató de TV3 (202026–10), Bristol Myers-Squibb (ERISTA 15), and Generalitat de Catalunya (2017-SGR671 and CERCA Program).
Author information
Authors and Affiliations
Contributions
PC reviewed the bibliography and wrote the treatment section, with the assistance of SF, AT, and JMN. MP reviewed and wrote the complement system section. ABMC reviewed and wrote the coagulation section. STM was responsible for the references and the design of Fig. 1. GP and JMS reviewed the COVID-19 pathophysiology and designed Fig. 2. ER and PGR contributed to the review and writing of the treatment section. EC, JJB, and GE contributed to the manuscript with their critical review and comments, and final editing. MDR is responsible for the design, writing, and final editing of the review.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Research Involving Human Participants and/or Animals
Not applicable.
Conflict of Interest
PC and SF have collaborated with Jansen, Gilead, Kite, MSD, Alexion, and Pfizer, outside of the submitted work. ABMC is an advisory board member for Siemens, outside of the submitted work. MDR and EC have been granted by and received honoraria from Jazz Pharmaceuticals. PGR has served on advisory committees for Jazz Pharmaceuticals. The rest of the coauthors have no actual or potential conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Castro, P., Palomo, M., Moreno-Castaño, A.B. et al. Is the Endothelium the Missing Link in the Pathophysiology and Treatment of COVID-19 Complications?. Cardiovasc Drugs Ther 36, 547–560 (2022). https://doi.org/10.1007/s10557-021-07207-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-021-07207-w