
Abstract
Zirconia modified SBA-15 becomes a very active catalyst for the selective hydrolysis of cellobiose to glucose after sulfation. Spectroscopic investigations indicate the presence of Brønsted acid sites with similar properties to those present in conventional sulfated zirconia. Indications are found that the sulfate groups attached to zirconia interact with silanol groups of SBA-15. The catalytic activity in cellobiose hydrolysis correlates well with results for temperature-programmed decomposition of i-propylamine for a range of sulfated ZrO2/SBA-15 catalysts. A glucose yield of 60% during cellobiose hydrolysis at a reaction time of 90 min at 160 °C is obtained.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
So far, considerable effort has been dedicated for the economic production of ethanol as it is an excellent alternative transportation fuel and can be blended with gasoline [1, 2]. However, ethanol is currently produced from sugar cane and the starch portion of corn, competing with food production, thus raising ethical concerns [3]. It is also very important to develop technologies for the efficient utilization of cellulose, a major constituent of lignocellulosic agricultural residues, as one of the renewable sources of chemicals [4, 5]. Cellulose is a polymer of glucose monomers connected by β-1,4-glycosidic bonds. It can be hydrolyzed to glucose through enzymatic or acid hydrolysis [6, 7]. The advantage of the enzymatic route is the absence of the degradation products but the reactions proceed rather slowly and separation is cumbersome [8]. Mineral acids can depolymerize cellulose into individual glucose units, accompanied by degradation products such as furfural, hydroxymethylfurfural and tar-like products. These side products are toxic for the microorganisms used for further fermentation of the glucose product. Recovery of the mineral acids is another challenge and in any case mineral acids are not desired because of their corrosive nature [9]. In order to overcome these drawbacks, heterogeneous catalytic systems have been proposed [10–17]. The conversion of cellulose into glucose and the simultaneous hydrogenation of glucose into sugar alcohols, such as sorbitol and mannitol on Ru/C [11], polymer stabilized Ru nanoclusters [12], and Pt and Ru supported by HUSY and γ-Al2O3 [13] has been reported. In order to replace the mineral acids in the acid hydrolysis of cellulose, solid acid catalysts such as sulfonated activated carbon (AC-SO3H) [14], sulfonic acid-mesoporous materials [15, 16], and Brønsted acidic zeolites [13, 14] were used. Higher glucose yields were reported for sulfate modified catalysts than for acidic zeolites. Besides in the hydrolysis of sucrose and starch, sulfonated mesoporous silicas showed higher activities than ion exchange resins and HZSM-5 catalysts [17]. In this sense, sulfated zirconia is expected to show high glucose yields, albeit a significant amount of by-products has been noted [14].
The textural properties of conventional sulfated zirconia are not beneficial with relatively low surfaces areas (100–120 m2/g). Much effort has been spent in order to prepare a high surface area sulfated zirconia. Mesoporous silicas have attracted wide-spread interest due to the high surface area (up to 1000 m2/g), large pore volume, and a hexagonal array of uniform pores. The silanol groups on the surface of these materials are neutral or slightly acidic. Combining these beneficial textural properties of ordered mesoporous silicas with the intrinsic acidity of sulfated zirconia by dispersing the latter in the former would greatly enhance the catalytic activity. SBA-n mesoporous materials [18, 19] have higher stability than other typical mesoporous silicas such as of the M41S family [20] due to their thicker walls, which should be beneficial when the aim is to introduce other components into the framework. The introduction of the zirconia precursor simultaneously with the silicon precursor in the preparation of SBA-15 was shown to be effective for a good distribution of zirconium within the silica framework [21].
In our previous work, we have found indications for strong Brønsted acidity of sulfate modified ZrO2/SBA-15 by 1H MAS NMR [22]. The origin of the strong surface acidity in sulfated zirconia has not been unequivocally resolved [23, 24]. Several postulates on the acidity, whether it is related to the sulfate group and various binding structures of this group to the zirconia surface, have been proposed [25–31]. Here, we investigated the surface acidity of a range of ZrO2/SBA-15 composites by infrared spectroscopy of adsorbed carbon monoxide. In addition, temperature-programmed decomposition of adsorbed alkylamines was used to probe the density of surface acidic sites. It is known that alkyl ammonium ions form upon protonation of amines by Brønsted sites and decompose into the corresponding alkene and NH3 in a narrow temperature range through a reaction similar to the Hoffman elimination [32]. The hydrolysis reactivity of a range of ZrO2/SBA-15 composites was determined using cellobiose as a model substrate. Cellobiose is a dimer of glucose with β-1,4-glycosidic bonds, providing a useful model compound for the hydrolysis of cellulose.
2 Experimental
2.1 Synthesis of Materials
SBA-15 silica materials were prepared by a well-established procedure [18, 19]. Siliceous SBA-15 was prepared by adding 9 ml of tetraethyl orthosilicate (TEOS) to 150 ml of 1.5 M HCl solution containing 4 g of Pluronic-123 (Aldrich). The mixture was stirred for 24 h at 40 °C and allowed to further react at 100 °C overnight in Teflon bottles. Subsequently, the solid material was obtained by filtration, dried at room temperature overnight and calcined at 500 °C in an air flow for 5 h. Zirconium was introduced in the synthesis solution simultaneously with TEOS in the form of zirconiumoxychloride (ZrOCl2·8H2O, 99.9%, Strem Chemicals). Appropriate amounts of the zirconium precursor were added to obtain final products with 5–25:100 ZrO2:SiO2 ratios. The materials are denoted as Zr(x)SBA-15 with x being the ZrO2 M amounts per 100 mol SiO2. Sulfated catalysts were prepared by sulfation in a 0.25 M H2SO4 solution for 15 min followed by drying overnight at 80 °C. The sulfated catalysts were activated at 350 °C for 3 h in static oven to remove the physisorbed sulfur species. Al-SBA-15 was prepared by prehydrolysing 0.25 g aluminum iso-propoxide (Acros Organics, 98%) in 10 ml of HCl solution. This solution was then added to the synthesis gel of SBA-15 to obtain Al-SBA-15. The Si/Al ratio is 34 in the final catalyst with a surface area of 903 m2/g. Conventional sulfated zirconia was prepared by calcination of the sulfated zirconium hydroxide provided by MEL Chemicals (XZO 1249/01) at 500 °C for 5 h.
2.2 Characterization
Elemental analyses were carried out by ICP-OES (Spectro Ciros CCD ICP optical emission spectrometer with axial plasma viewing). To extract the metals, the catalysts were dissolved in a 1.5 ml solution of HF/HNO3/H2O (1:1:1) acid mixture.
TEM images were taken on a FEI T20 electron microscope operating at 200 kV. The specimens were dispersed in ethanol and placed on holey copper grids.
The surface areas were measured using nitrogen adsorption isotherms at −196 °C on a Micromeritics ASAP 2000 gas sorption and porosimetry system. The samples were prepared for measurement by degassing at 150 °C for 24 h. Surface areas were calculated by the BET (Brunauer-Emmett-Teller) method and the pore volumes were determined by using the BJH (Barrett-Joyner-Halenda) method by using the adsorption cumulative volume of pores between 1.7 and 100 nm.
FT–IR spectra of the samples were recorded in the range of 4000–400 cm−1 by a Bruker IFS 113v instrument. The spectra were acquired at a 2 cm−1 resolution and averaged over 20 scans. The samples were prepared as thin self-supporting wafers of 5–10 mg/cm2 and placed inside a controlled environment infrared transmission cell, capable of heating and cooling, gas dosing and evacuation. Prior to CO adsorption, the catalyst wafer was heated to 450 °C at a rate of 10 °C/min in an oxygen atmosphere. Subsequently, the cell was out-gassed at the final temperature until the residual pressure was below 5 × 10−5 mbar. The sample was then cooled to −190 °C. CO was introduced into the cell via a sample loop (0.4 μmol per dose) connected to a Valco six-port valve.
Temperature programmed decomposition (TPD) of i-propylamine (IPAm) was performed in a packed-bed quartz reactor. The effluent gases were analyzed by a Balzers quadrupole mass spectrometer. 100 mg of catalyst was heated at a rate of 5 °C/min to 450 °C in a He flow of 100 ml/min. The sample was then cooled to 100 °C and exposed to excess gaseous IPAm (99.5%, Aldrich) for 10 min. Physisorbed IPAm was removed at 100 °C by purging in 100 ml/min He for 24 h. TPD of IPAm was carried out by heating the sample to 550 °C at a rate of 5 °C/min in 200 ml/min He. The formation of propene (m/e = 41) and ammonia (m/e = 17), i.e. the decomposition products of IPAm, as well as the amount of desorbed IPAm (m/e = 44, 41, and 17) were followed and compared to well-calibrated standards. The TPD curves were carefully deconvoluted and the peak areas were used to determine the relative contributions.
2.3 Catalytic Activity
Cellobiose hydrolysis was carried out in a lab-scale batch reactor (Autoclave Engineers, 100 ml). Typically, the reactor was charged with a 50 ml solution of 0.5 wt% cellobiose in water to which a predetermined amount of catalyst (0.1 wt% compared to solvent) was added. Kinetic experiments were performed at 160 °C under a nitrogen pressure of 20 bar. Nitrogen overpressure was employed to ensure that the reaction medium (water) remained as liquid during the reaction. The start of the reaction was taken as the moment when the reactor contents reached the desired reaction temperature. Although the reactors were heated as fast as possible, heating took about 10 min, during which period some reaction already took place. When the loss of cellobiose was traced during heating, it was found that the loss was not more than 10% for each catalytic system under study amounting significantly lower than the initial rates. Samples were withdrawn every 30 min for 1.5 h. Analysis was performed offline using a Waters HPLC system equipped with a H+-column (Polymer Lab.) and RI detector. The column temperature was kept constant at 60 °C. The mobile phase was 10 mM sulfuric acid solution which was fed at a flow rate of 0.6 ml/min. The glucose and the cellobiose concentrations were determined from calibration curves prepared by using external standards.
The sulfur elution of the catalysts were determined at the reaction conditions as well. In a typical test, 10 mg catalyst was added to 5 ml of water for 0–60 min at 160 °C. The catalyst was recovered by centrifugal separation under vacuum and the elemental analysis was performed by using a Perkin-Elmer Series II 2400 CHNS analyzer.
3 Results and Discussion
3.1 Physicochemical Properties
The intended compositions, Zr and S contents determined from the elemental analysis, SO4/Zr ratio and BET surface areas of the catalysts are collected in Table 1. Pure SBA-15 has a surface area of nearly 800 m2/g, which is consistent with the highly ordered mesostructure seen from TEM image (Fig. 1). Upon introduction of zirconia, the BET surface areas decreased to values between 500 and 600 m2/g, indicating that the introduction of zirconia in SBA-15 has resulted in a less ordered structure than siliceous SBA-15. The zirconium contents in the calcined materials were lower than the amounts in the synthesis gel, implying that not all Zr was built into SBA-15 and remained dissolved in the synthesis solution. Initially, we used 1 M H2SO4 to sulfate the Zr(x)SBA-15 materials, but this had a strong adverse effect on the textural properties (surface areas below 100 m2/g). A sulfuric acid concentration of 0.25 M was sufficient to provide good sulfation without causing too much loss of surface area. The surface area of the sulfated Zr(x)SBA-15 materials was above 200 m2/g and still higher than that of conventional sulfated zirconia. The sulfur content of the calcined sulfated samples was about 3.3 wt% and did not change much with the Zr content. The SO4/Zr ratio decreases with Zr content. While this ratio is in excess of 2 at low Zr content, it decreases strongly above a Zr content of 5 wt%. This may point to the gradual agglomeration of Zr ions into larger domains, of which only the outer surface can be sulfated. The XRD patterns did not show clear reflections in the low angle region for the zirconium-containing samples, indicating that the long range order of mesopores was absent. No reflections were observed in the high-angle region, implying that either the zirconium atoms were well dispersed or that the zirconia domains remain very small. Elemental analysis of the catalysts at various time under hydrothermal conditions at 160 °C (Table 2) indicated that a substantial part of the sulfur species are eluted under reaction conditions in the commercial sulfated zirconia catalyst as well as the SZr(x)SBA-15 catalysts.

Electron micrographs of a SBA-15 and b SZr(15)SBA-15
3.2 Infrared Spectroscopy of Adsorbed Carbon Monoxide
A FT-IR spectrum in the CO stretching region of SBA-15 after carbon monoxide adsorption at liquid nitrogen temperature is shown in Fig. 2a. The spectra exhibit two distinct bands at 2138 and 2158 cm−1. The latter is attributed to CO adsorbed on weakly acidic silanol groups. The band at 2138 cm−1 is due to a weakly perturbed condensed carbon monoxide phase on the surface of SBA-15 [33, 34]. The weak feature at 2110 cm−1 derives from carbon monoxide coordinating with its oxygen atom to the silanol group. The FT-IR spectra in the CO stretching region of Zr(15)SBA-15 catalyst are given in Fig. 2b. In the presence of zirconia, an additional band at 2185 cm−1 was also observed. This band was assigned to Lewis acid sites formed by coordinatively unsaturated Zr4+ sites [32]. Increasing CO coverage saturated these sites earlier than the silanol groups, implying that the former sites bind CO stronger than the latter in accordance with the larger shift of the carbon monoxide stretching frequency.

FT–IR spectra of adsorbed carbon monoxide at liquid nitrogen temperature for a SBA-15, b Zr(15)SBA-15 and c SZr(15)SBA-15 upon increasing CO coverage (doses of 0.4 μmol CO)
Infrared spectra of CO adsorption on SZr(15)SBA-15 are shown in Fig. 2c. Similar to the non-sulfated catalyst, a band at 2138 cm−1 was observed due to the weakly physisorbed CO. The band around 2160 cm−1 was attributed to CO adsorbed on silanol groups. It was located at a slightly higher wavenumber than that in SBA-15 and Zr(15)SBA-15 (νCO = 2158 cm−1). The band at 2186 cm−1 due to CO coordinating to low coordinated Zr4+ sites disappeared on sulfated samples. This was attributed to complete coverage of the Lewis acid sites by sulfate groups. Scheme 1 gives a schematic representation of the low coordinated Zr4+ sites and silanol groups on the surface of the Zr-SBA-15 catalysts and the suggested formation of strong acid groups in the presence of sulfate groups for SZr-SBA-15 catalysts.

The infrared spectra of CO adsorption on conventional sulfated zirconia are shown in Fig. 3. The band at 2170 cm−1 was attributed to the CO adsorbed on terminal hydroxyl groups on the zirconia surface. The high frequency band was red-shifting with increasing CO coverage in the region between 2192 and 2205 cm−1. This band was assigned to the C–O stretching mode of carbon monoxide interacting with Lewis acid sites, i.e. the Zr4+ sites, of varying acidity.

FT-IR spectra of adsorbed carbon monoxide at liquid nitrogen temperature for commercial sulfated zirconia upon increasing CO coverage (doses of 0.4 μmol CO)
The infrared spectra in the silanol stretching region of SBA-15, Zr(15)SBA-15 and SZr(15)SBA-15 catalysts are shown in Fig. 4a–c, respectively. The spectrum of SBA-15 (Fig. 4a) exhibits a sharp feature at 3747 cm−1 which is ascribed to the isolated, non-hydrogen bonded silanol groups on the surface. The broad tail extending towards 3400 cm−1 is due to the hydrogen bonded silanol groups. CO adsorption on isolated silanol groups weakens the hydroxyl bond and leads to a perturbed band around 3653 cm−1 with a frequency shift of 94 cm−1. The silanol stretching region of the Zr(15)SBA-15 catalyst (Fig. 4b) is similar to pure SBA-15 with the exception of a very weak feature around 3794 cm−1 attributed to isolated terminal hydroxyl groups bonded to Zr. The shift of the hydroxyl groups upon CO adsorption is very similar to SBA-15. This indicates the silanol groups are still only weakly acidic in the zirconia-substituted SBA-15.

FT–IR spectra of adsorbed carbon monoxide at liquid nitrogen temperature in the hydroxyl stretching region for a SBA-15, b Zr(15)SBA-15 and c SZr(15)SBA-15 upon increasing CO coverage (doses of 0.4 μmol CO). The red line represents the spectra before the addition of CO. The arrow indicates the increasing CO coverage
The corresponding hydroxyl region for SZr(15)SBA-15 is shown in Fig. 4c. The intensity of the band at 3747 cm−1 is less pronounced than the intensity of the corresponding bands on SBA-15 or Zr/SBA-15 samples. The IR spectra as a function of CO coverage clearly evidence the presence of two types of silanol groups. At low CO coverage, part of the hydroxyl band at 3745 cm−1 is shifted to 3601 cm−1, corresponding to a shift of 144 cm−1. With increasing CO coverage, the band of perturbed OH broadens and ∆υOH at high CO coverage is 106 cm−1. The evolution of the spectra evidence the presence of stronger acidic sites giving rise to ∆υOH = 144 cm−1 next to weakly acidic sites (∆υOH = 94 cm−1). An IR frequency shift of 144 cm−1 upon CO adsorption on SZr(15)SBA-15 is very close to the 139 cm−1 shift reported earlier for a conventional sulfated zirconia catalyst [35].
Previous FT-IR investigations provide evidence that sulfation of zirconia enhances the strength of the bridging zirconia hydroxyl groups, that is Zr-(OH)-Zr, and eliminates the terminal ones (i.e., ZrOH) [27]. In addition, it creates a new type of Brønsted acid site, presumably protons forming multicentered or single bonds with sulfate ions. These protons are hydrogen bonded to the surface [28]. Similarly, when zirconia included SBA-15 was modified by sulfation, the Lewis acid sites were covered by sulfate groups and the terminal hydroxyls of zirconia were eliminated (Fig. 5). As a result of the interaction of these sulfate ions with the silanol groups, strong Brønsted acidity is developed.

FT–IR spectra of adsorbed carbon monoxide at liquid nitrogen temperature in ZrOH stretching region for a non sulfated Zr(25)SBA-15 and b sulfated Zr(25)SBA-15 upon increasing CO coverage (doses of 0.4 μmol CO)
The presence of strong Brønsted acidity was indicated by the sulfate and the silanol stretching regions. The red shift (∆υOH = 144 cm−1) observed in the silanol stretching region for SZr-SBA-15 catalysts upon CO adsorption is similar to that for conventional sulfated zirconia reported in literature (∆υOH = 139 cm−1) [35]. This suggests that our catalysts have similar acidity as conventional sulfated zirconia. The ∆υOH of 144 cm−1 is higher than the shift for weakly acidic silanol, yet lower than that of strongly acidic zeolites (stabilized faujasite zeolite, ∆υOH ≈ 300 cm−1) [36, 37]. The IR spectra of the S=O stretching region for SZr(15)SBA-15 are presented in Fig. 6. CO adsorption brings about a positive frequency shift of about 26 cm−1. This shift indicates an increase of the S=O bond strength and is attributed to the interaction of CO with the strongly acidic proton, as weakening of the SO–H bond upon interaction of the proton with CO causes an increase of the S=O bond strength. This observation provides an indirect indication for the presence of protonic sites connected to the sulfate groups. Indeed, direct observation of the strong Brønsted acid protons in sulfated zirconia catalysts is not straightforward, likely because of the strong hydrogen bonding to adjacent surface groups [22].

FT–IR spectra of adsorbed carbon monoxide at liquid nitrogen temperature in the region of S=O stretch for SZr(15)SBA-15 upon increasing CO coverage (doses of 0.4 μmol CO)
3.3 Temperature-Programmed Decomposition of IPAm
The evolution of propene during temperature programmed IPAm decomposition for the various catalysts is shown in Fig. 7. No IPAm decomposition is observed for siliceous SBA-15. For the zirconia-containing catalysts, decomposition of adsorbed IPAm takes place between 310 and 360 °C with a maximum around 340 °C. In contrast, the decomposition in the sulfated catalysts occurred in the interval 290–340 °C with a maximum around 312 °C. In Table 3, the amounts of decomposed IPAm as well as the decomposition temperature are given for these catalysts. Introduction of zirconia in the silica results in the creation of sites capable of chemisorbing and decomposing IPAm. Upon sulfation, the decomposition temperature for Zr(15)SBA-15 decreases considerably. The difference is due to the increased acidity of the catalysts. This leads to activation of the chemisorbed IPAm complex at lower temperature. Table 3 also gives the results for two reference samples HZSM-5 and Al-SBA-15. The former contains exclusively aluminum in the zeolite framework and the amount of IPAm decomposed corresponds to the aluminum density. The latter contains mainly Lewis acidic Al sites on the surface of SBA-15. IPAm chemisorbed to these Lewis acidic sites decomposes at a higher temperature and the result is very similar to that of the non-sulfated Zr(x)/SBA-15 samples. It is then straightforward to conclude that IPAm in the non-sulfated catalyst adsorbs to Lewis acid Zr4+ sites, whereas this adsorption state should be absent after sulfation. Instead, the Brønsted acidic sites adsorb IPAm more strongly through protonation of the alkylamine, which results in a lower decomposition temperature. For the series of sulfated Zr(x)/SBA-15, the amount of decomposed IPAm first increases up to a value of 0.5 mmol/g for SZr(15)SBA-15 and then decreases with increasing Zr content. The increase of the acidity at lower Zr content indicates the increase of the number of Zr ions in or at the SBA-15 surface and points to their dispersed nature. At higher Zr content, zirconia domains are formed, which have a lower number of Zr ions at their surface. This is in line with the lower SO4/Zr ratios. In principle, the decrease of the acidity with increasing Zr content may thus be due to the formation of segregated domains of zirconia. An additional reason may lie in the importance of the interaction of sulfate groups attached to Zr ions with silanol groups in acidity generation as indicated by the IR results. Obviously, agglomeration of Zr at the surface of SBA-15 surface results in a decrease of the interaction of the grafted sulfate with the silanol groups and may thus decrease overall acidity. For SZr(20)SBA-15 and the samples with higher Zr loading, we find that there are two peaks in propene production, one main peak around 310 °C and the other smaller contribution around 340 °C. The contribution of the latter peak increases somewhat with the zirconium loading. As its location is similar to the peak observed for the non-sulfated sample, we infer that these samples contain a small amount of Lewis acidic Zr4+ sites.

Production of propene from i-propylamine decomposition during TPD for the various catalysts. Actual Zr loadings determined by elemental analysis for zirconium-containing silicas are indicated left
3.4 Catalytic Activity
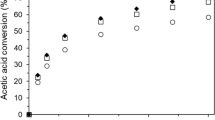
The conversion of cellobiose and the yield to glucose at a reaction temperature at 160 °C are given in Fig. 8. Comparing the SBA-15 based catalysts; the sulfated catalysts are more active in cellobiose conversion. The cellobiose conversion is about 80% after 90 min for SZr(15)SBA-15 and SZr(25)SBA-15. In comparison, Zr(15)SBA-15 displays a much lower activity. The activity of an Al-SBA-15 reference catalyst is also very low. The lower activities of Zr(15)SBA-15 imply that the hydrolysis of cellobiose is more favorable on Brønsted acid sites than on Lewis acid sites. The activity of the conventional sulfated zirconia material is very close to the sulfated Zr(x)SBA-15 materials. On the other hand, the glucose yields of the sulfated Zr(x)SBA-15 are much higher than conventional sulfated zirconia. Catalysts with Brønsted acid sites showed high selectivity for glucose in previous studies [16, 38]. Hence, the higher yields of glucose on sulfated Zr(x)SBA-15 in comparison to conventional sulfated zirconia is most likely due to the small amount of Lewis acid sites, known to catalyze the dehydration of glucose. The hydrolysis reactions are generally followed by glucose degradation reactions, including condensation, fragmentation and resinification reactions producing polyacids, sugar derivatives and polymeric residues [14]. Therefore, we limit ourselves here to follow cellobiose conversion and glucose yield.

Cellobiose conversion (left) and glucose yield (right) as a function of reaction time for a large set of catalysts (T = 160 °C; p N2 = 30 bar; autoclave; 0.5 wt% cellobiose and 0.1 wt% catalyst in water)
The sulfur content of all the catalysts decreased under reaction conditions. A comparison of the two composite catalysts SZr(15)SBA-15 and SZr(25)SBA-15 shows that the sulfur contents during catalytic operation are very similar (Table 2). The SZr(15)SBA-15 catalyst is more active and this is undoubtedly related to the higher amount of Brønsted acid sites. Our IPAm decomposition results show that the SZr(15)SBA-15 catalyst has the highest Brønsted acidity content. The incorporation of sulfated zirconia into SBA-15 structure does not substantially improve hydrothermal stability as sulfur species leach during reaction.
4 Conclusions
Strong Brønsted acidity of sulfated zirconia-SBA-15 catalysts was demonstrated by FTIR spectroscopy of surface hydroxyls upon CO adsorption and TPD of IPAm decomposition. The strong acidity of these materials results from the sulfation of zirconia patches of varying dispersion included in or at the surface of SBA-15. The acidity before sulfation stems from Lewis acidic Zr4+ sites. The higher intrinsic Brønsted acidity of a sulfated zirconia modified SBA-15 is close to that of conventional sulfated zirconia. IR spectroscopy suggests that the Brønsted acidity derives from interactions between sulfate groups attached to zirconia and silanol groups. An optimum in the acidity with respect to Zr content is observed. Sulfated zirconia dispersed throughout SBA-15 is an active catalyst for the scission of the 1,4-β glycosidic bonds in cellobiose, which is a model reaction for the critical step in the depolymerization of cellulose into valuable chemicals. The present study suggests that the isolated Brønsted acid sites catalyze cellobiose hydrolysis resulting in high glucose yields.
References
Farrell AE, Plevin RJ, Turner BT, Jones AD, O’Hare M, Kammen DM (2006) Science 311:506
Lynd LR, Cushman JH, Nichols RJ, Wyman CE (1991) Science 251:1318
Service RF (2007) Science 315:1488
Huber GW, Iborra S, Corma A (2006) Chem Rev 106:4044
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T (2006) Science 311:484
Zhang YHP, Lynd LR (2004) Biotechnol Bioeng 88:797
Mok WSL, Antal MJ, Varhegyi G (1992) Ind Eng Chem Res 31:94
Saha BC, Bothast RJ (1999) Appl Biochem Biotechnol 76:65
Wyman CE (1994) Bioresour Technol 50:3
Takagaki A, Tagusagawa C, Hayashi S, Harad M, Domen K (2010) Energ Environ Sci 3:82
Luo C, Wang SA, Liu HC (2007) Angew Chem Int Ed 46:7636
Yan N, Zhao C, Luo C, Dyson PJ, Liu HC, Kou Y (2006) J Am Chem Soc 128:8714
Fukuoka A, Dhepe PL (2006) Angew Chem Int Ed 45:5161
Onda A, Ochi T, Yanagisawa K (2008) Green Chem 10:1033
Nakajima K, Okamura M, Kondo JN, Domen K, Tatsumi T, Hayashi S, Hara M (2009) Chem Mater 21:186
Bootsma JA, Shanks BH (2007) Appl Catal A Gen 327:44
Dhepe PL, Ohashi M, Inagaki S, Ichikawa M, Fukuoka A (2005) Catal Lett 102:163
Zhao DY, Huo QS, Feng JL, Chmelka BF, Stucky GD (1998) J Am Chem Soc 120:6024
Zhao DY, Feng JL, Huo QS, Melosh N, Fredrickson GH, Chmelka BF, Stucky GD (1998) Science 279:548
Beck JS, Vartuli JC, Roth WJ, Leonowicz ME, Kresge CT, Schmitt KD, Chu CTW, Olson DH, Sheppard EW, McCullen SB, Higgins JB, Schlenker JL (1992) J Am Chem Soc 114:10834
Du YC, Sun YY, Di Y, Zhao L, Liu S, Xiao FS (2006) J Porous Mater 13:163
Degirmenci V, Erdem OF, Yilmaz A, Michel D, Uner D (2007) Catal Lett 115:79
Song XM, Sayari A (1996) Cat Rev Sci Eng 38:329
Kanougi T, Atoguchi T, Yao S (2002) J Mol Catal A Chem 177:289
Bensitel M, Saur O, Lavalley JC, Morrow BA (1988) Mater Chem Phys 19:147
Clearfield A, Serrette GPD, Khazisyed AH (1994) Catal Today 20:295
Riemer T, Spielbauer D, Hunger M, Mekhemer GAH, Knozinger H (1994) J Chem Soc Chem Commun 10:1181
Kustov LM, Kazansky VB, Figueras F, Tichit D (1994) J Catal 150:143
Adeeva V, Dehaan JW, Janchen J, Lei GD, Schunemann V, Vandeven LJM, Sachtler WMH, van Santen RA (1995) J Catal 151:364
Li XB, Nagaoka K, Lercher JA (2004) J Catal 227:130
Olindo R, Li XB, Lercher JA (2006) Chem Ing Tech 78:1053
Palkhiwala AG, Gorte RJ (1999) Catal Lett 57:19
Beebe TP, Gelin P, Yates JT (1984) Surf Sci 148:526
Hadjiivanov K, Reddy BM, Knozinger H (1999) Appl Catal A Gen 188:355
Manoilova OV, Olindo R, Arean CO, Lercher JA (2007) Catal Commun 8:865
Crepeau G, Montouillout V, Vimont A, Mariey L, Cseri T, Mauge F (2006) J Phys Chem B 110:15172
Morterra C, Cerrato G, Bolis V, DiCiero S, Signoretto M (1997) J Chem Soc Faraday Trans 93:1179
Shimizu K, Furukawa H, Kobayashi N, Itaya Y, Satsuma A (2009) Green Chem 11:1627
Acknowledgments
VD is grateful for the doctoral scholarship and exchange grants from the Scientific and Research Council of Turkey (TUBITAK) through the BDP program. National Science Foundation supported this work through grant CTS-0455965. This work has been partially supported by Dutch Technology Foundation (STW) under a VIDI grant. We thank the MEL Chemicals for providing the sulfated zirconium hydroxide. We would like to acknowledge Jie Fan and Galen Stucky (UC, Santa Barbara) for transmission electron microscopy measurements.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Degirmenci, V., Uner, D., Cinlar, B. et al. Sulfated Zirconia Modified SBA-15 Catalysts for Cellobiose Hydrolysis. Catal Lett 141, 33–42 (2011). https://doi.org/10.1007/s10562-010-0466-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-010-0466-1