
Abstract
For the preparation of electrically conductive composites, various combinations of cellulose and conducting materials such as polymers, metals, metal oxides and carbon have been reported. The conductivity of these cellulose composites reported to date ranges from 10−6 to 103 S cm−1. Cellulose nanocrystals (CNCs) are excellent building blocks for the production of high added value coatings. The essential process steps for preparing such coatings, i.e. surface modification of CNCs dispersed in water and/or alcohol followed by application of the dispersion to substrate samples using dip coating, are low cost and easily scalable. Here, we present coatings consisting of Ag modified CNCs that form a percolated network upon solvent evaporation. After photonic sintering, the resulting coatings are electrically conductive with an unprecedented high conductivity of 2.9 × 104 S cm−1. Furthermore, we report the first colloidal synthesis that yields CNCs with a high degree of Ag coverage on the surface, which is a prerequisite for obtaining coatings with high electrical conductivity.
Similar content being viewed by others


Introduction
An important class of advanced cellulose materials are electrically conductive composites (Shi et al. 2013). They are prepared by combining conductive materials such as polymers (Qaiser et al. 2011; Carlsson et al. 2012), metal oxides (Jur et al. 2011), metals (Zabetakis et al. 2005) and carbon (Anderson et al. 2010; Weng et al. 2011) with cellulose. The conductivity of the composites strongly depends on the conductive material applied. Using conducting polymers like polypyrrole (Carlsson et al. 2012; Raqaz et al. 2009; Shi et al. 2014), polyaniline (Qaiser et al. 2011; Xu et al. 2016) or PEDOT (Anothumakkool et al. 2015; Tkalya et al. 2013), conductivities ranging from 10−5 to 102 S cm−1 were achieved (Shi et al. 2013). Alternatively, carbon nanotubes (Anderson et al. 2010) or graphene (Weng et al. 2011) were applied yielding composites with a conductivity between 10−5 and 102 S cm−1 (Shi et al. 2013). The highest conductivity reported to date for cellulose composites is 103 S cm−1 (Weng et al. 2011). This was achieved through use of metal oxides like ZnO (Jur et al. 2011) and SnO2 (Aoki et al. 2006), and metals like Ag (Zabetakis et al. 2005; Koga et al. 2014), Au and Cu (Weng et al. 2011).
Recently, our group demonstrated that silicated CNCs are excellent natural building blocks for the production of high added value, antireflective coatings (Buskens et al. 2015, 2016). The essential process steps for preparing these coatings, i.e. surface modification of CNCs dispersed in water and/or alcohol followed by application of the dispersion to glass samples using dip coating, are low cost and easily scalable. Based on this, and our previous work on Ag decoration of glucose-functionalized polystyrene latexes for the formation of Ag nanoshells (Mann et al. 2014, 2016), we hypothesized that it should be possible to decorate CNCs, which are glucose polymers, with a dense Ag shell through controlled reduction of Ag+, and subsequently apply the dispersion to glass substrates. Assuming that, in analogy to the silicated CNCs, the Ag modified crystals form a percolated network upon solvent evaporation, the resulting coatings should be electrically conductive. We anticipated that a high degree of coverage of the CNC surface with Ag was required for obtaining coatings with high electrical conductivity.
Here, we present a novel method for the surface decoration of needle shaped CNCs with Ag, yielding the first CNC-Ag composite particles with a high degree of surface coverage. The resulting coatings on glass, prepared through dip coating, displayed an unprecedented high conductivity when compared to previously reported electrically conductive cellulose composites.
Materials and methods
Materials
CNC slurry (11.5 wt% in water) was commercially obtained from the Process Development Center of the University of Maine and used without further purification. The product specification sheets are available from their website (https://umaine.edu/pdc/facilities-available-for-use/nanocellulose-facility/nanocellulose-spec-sheets-and-safety-data-sheets/). (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl (TEMPO), NaBr, NaOCl (11 wt% in water), NaOH, SnCl2, AgNO3, poly(ethylene glycol) methyl ether thiol (PEG-thiol, Mw 6000 g mol−1), NH3 (28–30 wt% in water), HCl and ethanol (EtOH) were obtained from Sigma-Aldrich and used without further purification.
TEMPO mediated oxidation of CNCs
Similar to the procedure reported by Ifuku et al. (Ifuku et al. 2009), CNC slurry (90.6 g, 11.5 wt% in water) was diluted with demineralized (demi) water (375 ml) and sonicated using a Branson Sonifier for 5 min. The dispersion was added to a solution of TEMPO (25 mg, 0.16 mmol g−1 CNC) and NaBr (250 mg) in demi water (375 ml). NaOCl (40 g of 11 wt% solution in water; 5.0 mmol g−1 CNC) was added to start the oxidation, after which the pH of the resulting dispersion was adjusted to 10.5 through addition of NaOH. The suspension was stirred overnight at room temperature, after which the oxidation was terminated by the addition of ethanol (10 ml). The resulting suspension was dialyzed against demi water for 7 days until neutral pH was achieved in the effluent.
Isolated yield: 7.7 g (74%) of oxidized CNC.
Two step procedure for silver deposition on CNCs
First step: Similar to the procedure reported by our group for Ag deposition on glucose-functionalized polystyrene latexes (Mann et al. 2014, 2016), a dispersion of TEMPO-oxidized CNCs (15.0 g, 0.77 wt% in demi water) was added to a solution of SnCl2 (1.25 g, 5.54 mmol) and HCl (2 ml, 37 wt%) in demi water (100 ml). The mixture was stirred for one hour on a roller bank at room temperature, and subsequently centrifuged for 30 min at 10,000 rpm. Then, the supernatant was removed, and the residue dispersed in demi water (100 ml) using an ultrasonic bath. The resulting dispersion had a solid content of 0.10 wt%. Subsequently, the mixture was added to a solution of silver nitrate (600 mg, 3.53 mmol) and aqueous ammonia (3.75 ml, 28–30 wt% in water) in water (100 ml). The resulting reaction mixture was inverted 10 times, immediately centrifuged at 10,000 rpm for 30 min and dispersed in demi water (50 ml).
Isolated yield: 0.02 g CNC-Ag composite particles (45 wt% Ag, based on thermogravimetric analysis (TGA)).
Second step: A dispersion of CNC-Ag composite particles (16.7 ml, 0.04 wt%, 45 wt% Ag) was added to a freshly prepared Ag deposition bath. The deposition bath was prepared by combining solution 1 and 2 (Table 1) in an ice bath. Solution 2 was prepared by adding glucose and tartaric acid to a mixture of ethanol and demi water, after which the solution was placed in an ultrasonic bath for 30 min. The particle dispersion was added to the deposition bath and stirred overnight on a roller bank. PEG-thiol (Mw 6000, 100 mg, 0.017 mmol) was added to stabilize the metallized CNC particles. The resulting dispersion was stirred for one hour on the roller bank, centrifuged at 10,000 rpm for 30 min, and dispersed in demi water (20 ml) using an ultrasonic tip (2 min, power level 20%).
Isolated yield: 0.13 g CNC-Ag composite particles (94 wt% Ag, based on TGA).
UV–VIS extinction spectrophotometry on CNC-Ag dispersions
For the optical characterization of aqueous CNC-Ag dispersions, we performed UV–VIS extinction spectrophotometry using a Shimadzu 3600 UV–VIS–NIR spectrophotometer. The measurements were performed in quartz cuvettes having an optical path length of 10 mm. Spectra were recorded for the wavelength range from 300 to 700 nm.
Thermogravimetric analysis
TGA was applied to determine the decomposition temperature of CNCs in CNC-Ag composite particles, and the weight ratio CNC to Ag using a Discovery TGA (TA Instruments). Samples were heated to 900 °C under air with a rate of 20 °C min−1.
Coatings on float glass
Coatings comprising CNC-Ag composite particles were applied to float glass slides from an alcoholic dispersion (1.0 wt%) using dip coating (KSV NIMA dipcoater with control software). A withdrawal speed of 500 mm min−1 was applied. The solvent was evaporated under ambient conditions. Up to eight layers were applied on top of each other through sequential dip coating. Photonic sintering was applied to transform the dip coated nanoparticle layers into a conductive coating, using a NovaCentrix PulseForge 1300. The sample was half covered with a metal plate. The other half was exposed to light flashes for photonic sintering using diverse settings (Table 2). One sample, resulting from eight sequential dips, was cured in an oven for 3 h at 450 °C.
Temperature monitoring during photonic sintering
To monitor the increase in coating temperature during photonic sintering, a sample with an electrical circuit containing a molybdenum line with known dimensions and temperature coefficient of resistance was covered with a thick layer of CNC-Ag particles (eight sequential dips). The line resistance was measured during photonic sintering, and directly correlated to the sintering temperature.
Zeta potential
The zeta potential of the CNC dispersions was measured using a Malvern nano Zetasizer. All measurements were performed in translucent disposable zeta cuvettes (DTS1060C) at 25 °C with an equilibration time of 2 min. Each measurement was repeated three times with a delay of 10 s in order to confirm the quality of the obtained measuring data.
X-ray diffraction
Samples are measured in Bragg–Brentano geometry with a Bruker D8 Advance diffractometer, equipped with a motorized slit with opening angle of 0.30°, primary and secondary soller slit of 2.5° and Lynxeye detector with opening angle of 2.94°. Data collection is carried out at room temperature using Cu Kα radiation (λ = 0.15406 nm) in the 2θ region between 10° and 40°, step size 0.02 degrees 2θ. The sample is deposited on a zero background holder (Si single crystal 〈510〉 wafer) and was rotated at 15 rpm during the measurement. Generator settings are 40 kV and 40 mA. Phase identification is carried out with the Bruker program “EVA4.2.”
Fourier transform-infrared spectroscopy
Infrared spectroscopy was used to characterize the CNCs before and after TEMPO oxidation. Infrared spectra were recorded on a FT-IR Perkin Elmer spectrophotometer.
Electron microscopy
The dimensions and composition of CNCs and CNC-Ag composite particles were determined by transmission electron microscopy (TEM) using a JEOL ARM200F operated at 80 kV. Dried dispersions of CNC typically display a high degree of aggregation as a result of drying during sample preparation. Therefore, cryogenic dispersion samples were prepared on copper-supported lacey carbon by rapid vitrification in liquid ethane.
High resolution scanning electron microscopy (SEM) images were recorded with a FEI Nova NanoSem 650 with the following settings: an acceleration voltage of 4 kV, spot size 3, chamber vacuum of 60 Pa and a gaseous analytical detector, or with an acceleration voltage of 5 kV, spot size 3, chamber vacuum of circa 1 mPa and a through lens detector.
Titration to determine the degree of oxidation of CNCs
The carboxyl content of the CNCs after TEMPO oxidation was determined by suspending of freeze dried oxidized CNCs (50–55 mg) into 0.01 M hydrochloric acid solution (15 ml). The suspension was treated with an ultrasonic tip for 2 min, after which the sample was titrated with 0.01 M NaOH. The carboxyl groups content, or degree of oxidation (DO), was calculated using following equation:
where V 1 and V 2 are the volumes of NaOH (in l), c is the concentration NaOH (in mol/l) and w is the weight of the freeze dried sample (Habibi et al. 2006).
UV–VIS–NIR extinction spectrophotometry
Transmission of the dip coated glass samples was measured using a Shimadzu 3600 UV–VIS–NIR spectrophotometer. Spectra were recorded for the wavelength range from 300 to 1200 nm.
Sheet resistance
Four point probe measurements for determination of sheet resistance are not reliable on highly porous coatings on glass. The probes penetrate the coating and destroy the three-dimensional network of nanoparticles, resulting in irreproducible values. Therefore, resistances were measured between small dots of conductive silver glue, applied 8 mm apart from each other, on top of the CNC-Ag coatings. The sheet resistance R S can be calculated as described by Wolf and Tauber (1986).
with W as distance between the dots of silver glue (i.e. 8 mm), R T as total measured resistance and R C as contact resistance. The conductivity is calculated from the sheet resistance R S of the coating and its layer thickness t
Layer thickness
Layer thickness measurements were performed using a Dektak XT profilometer from Bruker, with 1 mg stylus force. The coating thickness was determined on five different positions for each coated sample. Average values are presented in this report.
Results and discussion
CNC-Ag composite particles
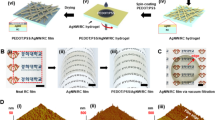
The aim of our study was to prepare CNC-Ag composite particles with a high degree of Ag coverage on the surface of the CNCs, and to use these Ag-decorated CNCs for the preparation of electrically conductive coatings. Very recently, our group reported a two-step procedure for the deposition of Ag on glucose-functionalized polystyrene particles, yielding polystyrene-Ag core–shell particles with a high degree of Ag coverage on the latex surface (>90%) (Mann et al. 2016). We selected this metallization procedure for Ag deposition on CNCs (Fig. 1). At the start of the reported procedure for metallization of the latex particles, Sn2+ ions were coordinated to the surface of glucose-functionalized polystyrene particles to serve as nucleation points for the reductive deposition of Ag from Ag+ (Mann et al. 2014, 2016). This pretreatment step is commonly used to deposit Ag on macroscopic surfaces, and has first been applied as pretreatment in a multistep process for deposition of Ag on spherical colloids by Liz-Marzán and coworkers (Kobayashi et al. 2001).

Schematic representation of TEMPO oxidation of CNCs, and subsequent two-step metallization procedure for Ag deposition on oxidized CNCs
They demonstrated that Sn2+ ions coordinated to the surface of spherical silica particles and reduced Ag+ to Ag, while Sn2+ oxidized to Sn4+. Coordination of Sn2+ ions to the polystyrene particle surface was demonstrated by the strong increase in zeta potential (Mann et al. 2014, 2016). However, after pretreatment of CNCs with SnCl2 and subsequent removal of excess salt through centrifugation and washing, we noticed that the zeta potential remained unchanged at −42.1 mV. Ergo, the Sn2+ ions did not coordinate to the CNC surface.
Because it is well known that Sn2+ coordinates to carboxylate groups, we decided to use surface oxidized CNCs as templates for reductive Ag deposition. Following the procedure developed by Ifuku et al. (Ifuku et al. 2009), we subjected the CNCs to TEMPO mediated oxidation (Saito and Isogai 2004). The use of this technique has attracted a number of investigations, which demonstrated that only the hydroxymethyl groups of polysaccharides were oxidized, whereas the secondary hydroxyls remained unaffected (Ifuku et al. 2009). After TEMPO oxidation and subsequent purification of the CNCs, we analyzed the surface-oxidized CNCs using cryo-TEM (Fig. 2a), XRD (Fig. 2b) and FT-IR spectrometry (Fig. 2c), and compared their properties to those of the non-oxidized crystals. As demonstrated by the cryo-TEM (Fig. 2a) and XRD analyses (Fig. 2b) of untreated and oxidized CNCs, the oxidation did not significantly affect the CNC size and crystallinity, respectively. The average length and width of both the untreated and oxidized needle-shaped CNCs is 150–200 and 5 nm, respectively. Both materials show the X-ray fingerprint which is typical for crystalline cellulose (Buskens et al. 2015). In the FT-IR spectrum, the presence of carboxylic acid groups on the surface of oxidized CNCs is confirmed by the occurrence of the absorption band at 1730 cm−1 representing the C=O stretching vibration (Fig. 2c, Online Resource 1a) (Li et al. 2015).

Characterization of TEMPO oxidized CNCs in comparison to untreated CNCs; a cryo-TEM images, b X-ray diffractograms (y axis are arbitrary units), and c FT-IR spectra
Furthermore, we noticed an additional absorption band at 1601 cm−1 for the TEMPO oxidized CNCs, representing the COO– antisymmetric stretching in carboxylic acid salts. From the FT-IR spectrum of TEMPO oxidized CNCs, we calculated a degree of oxidation (DO) of 0.110 (Habibi et al. 2006). Furthermore, we noticed from the FT-IR spectra that neither the total crystallinity index (\( TCI = 1.00 \) and \( TCI_{ox} = 0.99 \)) nor the lateral order index (\( LOI = 1.00 \) and \( LOI_{ox} = 1.01 \)) changed upon TEMPO oxidation (Li et al. 2015). The hydrogen bond intensity increased upon TEMPO oxidation from \( HBI = 0.06 \) to \( HBI_{ox} = 0.11 \) (Li et al. 2015), which can be expected upon formation of carboxylic acid groups.
For a more accurate determination of the DO, we performed a conductometric acid–base titration using the method reported by Vignon and coworkers (Fig. 3) (Da Silva Perez et al. 2003; Habibi et al. 2006).

Titration curve of the conductometric acid–base titration of TEMPO oxidized CNCs (amount of CNCs \( w = 0.0495 {\text{g}} \); concentration NaOH \( c = 0.0110\;{\text{mol}}\;{\text{l}}^{ - 1} \))
Based on these results, we obtained a carboxyl group content of 0.60 mmol g−1, which corresponds to a DO of 0.105. This is in good agreement with the DO obtained through FT-IR analysis.
The stability of the dispersions of unmodified and oxidized CNCs at neutral pH was determined through measurement of the zeta potential. For the unmodified and oxidized CNCs, we determined a zeta potential of −42.1 and −43.5 mV, respectively, which indicates that both dispersions are stable at neutral pH.
To validate the surface coordination of Sn2+ ions, the dispersion of oxidized CNCs was mixed with an aqueous solution of SnCl2. After one hour at room temperature, excess SnCl2 was removed through centrifugation and washing, and the resulting CNCs were dispersed in demi water. After treatment, we measured a significant increase in zeta potential from −43.5 to +2.14 mV, demonstrating successful coordination of Sn2+ ions to the carboxylate groups of the oxidized CNCs. This was confirmed by TEM-EDX analysis of the TEMPO oxidized CNCs after treatment with SnCl2 (Fig. 4).

TEM of the TEMPO oxidized CNCs after treatment with SnCl2 and TEM-EDX analysis for Sn (red). (Color figure online)
In the first metallization step, the Sn2+ modified CNCs were subjected to an aqueous solution of AgNO3 and NH3 at room temperature. An immediate colour change due to the formation of Ag nanoparticles on CNCs, with their distinct localized plasmon resonance at about 420 nm, indicated that Ag was formed rather quickly (Fig. 5a). The reaction mixture was inverted 10 times at room temperature, after which the Ag-modified CNCs were isolated through centrifugation. The resulting supernatant was transparent in the visible, indicating that no free Ag particles were formed, and that the metallization procedure is highly selective for CNC surface decoration. The Ag-modified CNCs were then washed and dispersed in demi water. The Ag content was determined using TGA (Fig. 5b). The UV–Vis spectrum displays a rather broad extinction peak with its maximum at a wavelength of 420 nm, which represents the localized plasmon resonance of the Ag nanoparticles deposited on CNCs (Fig. 5a). In the TGA, the Ag-decorated CNCs display approximately 5% mass loss due to dehydration, followed by a distinct temperature profile for degradation with degradation temperatures of 190 and 350 °C (Fig. 5b). The Ag residue obtained after full decomposition of cellulose at a temperature above 550 °C represents 45 wt% of the original CNC-Ag composite particles.

a UV–VIS spectrum of Ag-modified CNCs dispersed in demi water and b TGA of Ag-modified CNCs; both after the first metallization step
Morphology and architecture of the CNC-Ag composite particles, obtained after the first metallization step, were analyzed using high resolution cryo-TEM (Fig. 6). The cryo-TEM image displays a bundle of maximum 5 CNCs inhomogeneously decorated with individual spherical Ag nanoparticles of a size between 5 and 10 nm (pointed out by the red arrow in Fig. 6). This matches the UV–VIS extinction peak at 420 nm.

Cryo-TEM image of CNC-Ag composite particles obtained after the first metallization step
In analogy to the procedure reported by Buskens and co-workers for Ag deposition on glucose-modified polystyrene latex particles (Mann et al. 2016), we applied a second step for further deposition and growth of Ag nanoparticles. In this step, the CNC-Ag composite particles were subjected to a Ag deposition bath at 0 °C. This bath was prepared through combination of two individually prepared solutions: an aqueous solution of Ag[NH3] +2 complex and a solution of glucose and tartaric acid in a mixture of ethanol and demi water. After finishing the Ag deposition, PEG-SH was added to ensure colloidal stability of the CNC-Ag composite particles. The particles were purified through centrifugation and washing, and dispersed in demi water. Similarly to the first process step, the supernatant was transparent indicating that Ag was selectively deposited on the CNC surface. TEM analysis of the resulting CNC-Ag composite particles revealed an unprecedented high level of surface coverage with Ag particles of a size up to about 50 nm (Fig. 7).

TEM analysis of CNC-Ag composites formed after the second process step
This unprecedented high level of surface coverage was achieved both through the formation of new Ag particles on the CNC surface and through the growth of previously existing Ag nanoparticles. It is well known that metal nanoparticles such as Ag and Pd deposited on the surface of a dielectric particle, act as a catalyst for further Ag deposition and thereby ensure that reduction of further Ag+ ions only takes place on the nanoparticle surface (Kobayashi et al. 2001). TGA analysis showed that the amount of Ag in the composite particles increased during the second process step from 45 to 94 wt% (Online Resource 1b). UV–VIS spectrophotometry yielded an extinction spectrum (Online Resource 1c) almost identical to the one for the CNC-Ag composites after the first process step (Fig. 5a).
Conductive coatings
For the preparation of conductive coatings comprising the CNC-Ag composite particles obtained after the second process step, we prepared a 1.0 wt% dispersion in ethanol. The particles were applied to float glass slides through dip coating using the particle dispersion as coating liquid. We used a withdrawal speed of 500 mm min−1, and applied multiple layers of particles on top of each other through sequential dipping and drying. After evaporation of all volatiles, we photonically sintered the thin film using pulsed light from a flash lamp. This is a common technique for sintering of metal inks, and is frequently used in printed electronics (Schroder 2011). Sintered samples resulting from 2, 4, 6 and 8 sequential dips were analyzed using SEM (Fig. 8). As depicted in the SEM images, the degree of surface coverage and the coating layer thickness t increase with increasing number of dips. For the sample resulting from 8 sequential dips, we performed SEM analysis before photonic sintering (Fig. 9a). From comparison between the sintered (Fig. 8d) and non-sintered coating, we conclude that the morphology of the structures did not significantly change during photonic sintering. Furthermore, we performed a reference experiment through application of an oven cure (Fig. 9b).

Coated samples resulting from a 2, b 4, c 6 and d 8 sequential dips after photonic sintering

Coating from 8 sequential dips a before photonic sintering, and b after 3 h oven cure at 450 °C
By applying a coating (8 sequential dips) on top of an electrical circuit containing a molybdenum line with known dimensions and temperature coefficient of resistance, we determined that the maximum temperature reached during photonic sintering was approximately 450 °C (Online Resource 1d). Therefore, we performed the oven cure at that temperature for a duration of 3 h.
After removal of the coating from one side of the substrate, we determined the UV–Vis transmission using spectrophotometry (Fig. 10). The average transmission in the visible (425–675 nm) T vis decreased with increasing layer thickness from 92.7% (uncoated float glass) to 18.0% (4 dips), 12.7% (6 dips) and 5.9% (8 dips) (Fig. 10, Table 3, Online Resource 1e). The morphology change of the oven cured samples is reflected in the transmission curve yielding an average visible transmission of 28.6%. Using a Dektak profilometer, we determined the average layer thickness t of the coatings obtained after 4, 6, and 8 dips (Table 3). As expected, the average layer thickness increased with increasing number of sequential dips from 70.5 nm (4 dips) to 92.6 nm (6 dips) and 99.4 nm (8 dips). For flat and dense coating layers of the same material applied on top of each other in sequential dips, the coating thickness should linearly increase with the number of dips, provided that the same liquid coating formulation and withdrawal speed are used. Apparently, this is not the case for the porous coatings consisting of CNC-Ag nanoparticles. With increasing number of dips, the average coating thickness achieved per dip decreases (17.6 nm for 4 dips, 15.4 nm for 6 dips, 12.4 nm for 8 dips). Nonetheless, it is reasonable to expect that the same amount of coating material is applied during each dip, which means that with increasing thickness of the porous layers, the additional coating material is preferably deposited inside the porous network instead of on top.

Transmission of glass samples single-sided coated with CNC-Ag composite particles
Regular probes used in four point probe measurements for the determination of the sheet resistance of the coating penetrate the particle layer, and consequently destroy the three dimensional coating network. Therefore, this method is not suited for determining the conductivity. Instead, we measured resistances between small dots of conductive silver glue applied at distances of 8, 16, 24 and 32 mm from each other. When plotting the measured resistance R T between these contact points as function of their distance L (Fig. 11), sheet R S and contact resistance R C can be derived from the slope and axis intersection, respectively,
with W as width of the conducting path (Wolf and Tauber 1986).

a Schematic representation of the measured samples and b R T as a function of L using conducting paths of 20 mm width (squares data points for coating resulting from 6 sequential dips, triangles data points for coating resulting from 8 sequential dips)
For the coatings prepared using 6 and 8 sequential dips, the sheet resistances were determined using 10 (Online Resource 1f) and 20 mm wide conducting paths (Fig. 11). The average sheet resistance values resulting from these measurements are 31 Ω sq−1 (6 dips, t = 92.6 nm) and 3.6 Ω sq−1 (8 dips, t = 99.4 nm). The corresponding linear regressions display a coefficient of determination R 2 of 0.99, which underlines that the regression line excellently fits the experimental data points. This is not the case for the sample resulting from 4 sequential dips: using a linear regression, the sheet resistance of this sample could be determined as 5.6 × 105 Ω sq−1 (4 dips, t = 70.5 nm), albeit with a poor fit of the regression line with the experimental data points (R 2 = 0.80–0.86, Online Resource 1 g). Note that the decrease in sheet resistance cannot simply be explained by an increase in coating thickness with increasing number of dips. Although the difference in coating thickness between the layers produced in 6 and 8 sequential dips is only minor, the difference in the number of conductive CNC-Ag nanoparticles per surface area of substrate is more than 30% (vide supra).The conductivity σ of these coatings can be derived from R S and layer thickness t,
and was calculated as 5.4 × 10−1 S cm−1, 3.5 × 103 S cm−1, and 2.9 × 104 S cm−1, respectively. Latter conductivity exceeds the conductivity of the best performing cellulose composites reported to date by a factor ~30 (Shi et al. 2013).
Conclusions
We have successfully developed a conductive coating consisting of Ag-decorated CNCs, with a conductivity that exceeds the best performing cellulose composites reported to date by a factor 30. In line with our previous work on CNC-based antireflective coatings, we demonstrated that surface modified CNCs are interesting building blocks for application in high tech functional coatings, and realized the first conductive coating based on CNCs. Here, we decorated the CNC surface with Ag using a two-step metallization procedure, and realized CNC-Ag composite particles with an unprecedented high degree of metal coverage. These were dispersed in ethanol and applied to glass substrates through dip coating. The resulting particle layers were photonically sintered, yielding coatings with an electrical conductivity up to 2.9 × 104 S cm−1.
References
Anderson RE, Guan J, Ricard M, Dubey G, Su J, Lopinski G, Dorris G, Bourne O, Simard B (2010) Multifunctional single-walled carbon nanotube-cellulose composite paper. J Mater Chem 20:2400–2407
Anothumakkool B, Soni R, Bhange SN, Kurungot S (2015) Novel scalable synthesis of highly conducting and robust PEDOT paper for a high performance flexible solid supercapacitor. Energy Environ Sci 8:1339–1347
Aoki Y, Huang J, Kunitake T (2006) Electro-conductive nanotubular sheet of indium tin oxide as fabricated from the cellulose template. J Mater Chem 16:292–297
Buskens P, Mourad M, Meulendijks N, Van Ee R, Burghoorn M, Verheijen M, Van Veldhoven E (2015) Highly porous, ultra-low refractive index coatings produced through random packing of silicated cellulose nanocrystals. Coll Surf A 487:1–8
Buskens P, Burghoorn M, Mourad MCD, Vroon Z (2016) Antireflective coatings for glass and transparent polymers. Langmuir 32:6781–6793
Carlsson DO, Nyström G, Zhou Q, Berglund LA, Nyholm L, Strømme M (2012) Electroactive nanofibrillated cellulose aerogel composites with tunable structural and electrochemical properties. J Mater Chem 22:19014–19024
Da Silva Perez D, Montanari S, Vignon MR (2003) TEMPO-mediated oxidation of cellulose III. Biomacromolecules 4:1417–1425
Habibi Y, Chanzy H, Vignon MR (2006) TEMPO-mediated surface oxidation of cellulose whiskers. Cellulose 13:679–687
Ifuku S, Tsuji M, Morimoto M, Saimoto H, Yano H (2009) Synthesis of silver nanoparticles templated by TEMPO-mediated oxidized bacterial cellulose nanofibers. Biomacromolecules 10:2714–2717
Jur JS, Sweet WJ III, Oldham CJ, Parsons GN (2011) Atomic layer deposition of conductive coatings on cotton, paper and synthetic fibers: conductivity analysis and functional chemical sensing using “all-fiber” capacitors. Adv Funct Mater 21:1993–2002
Kobayashi Y, Salgueriño-Maceira V, Liz-Marzán LM (2001) Deposition of silver nanoparticles on silica spheres by pretreatment steps in electroless plating. Chem Mater 13:1630–1633
Koga H, Nogi M, Komoda N, Nge TT, Sugahara T, Suganuma K (2014) Uniformly connected conductive networks on cellulose nanofiber paper for transparent paper electronics. NPG Asia Mater 6:e93
Li B, Xu W, Kronlund D, Määttänen A, Liu J, Smätt J-H, Peltonen J, Willför S, Mu X, Xu C (2015) Cellulose nanocrystals prepared via formic acid hydrolysis followed by TEMPO-mediated oxidation. Carbohydr Polym 133:605–612
Mann D, Chattopadhyay S, Pargen S, Verheijen M, Keul H, Buskens P, Möller M (2014) Glucose-functionalized polystyrene particles designed for selective deposition of silver on the surface. RSC Adv 4:62878–62881
Mann D, Nascimento-Duplat D, Keul H, Möller M, Verheijen M, Xu M, Urbach PH, Adam AJL, Buskens P (2016) The influence of particle size distribution and shell imperfections on the plasmon resonance of Au and Ag nanoshells. Plasmonics. doi:10.1007/s11468-016-0345-8
Qaiser AA, Hyland MM, Patterson DA (2011) Surface and charge transport characterization of polyaniline–cellulose acetate composite membranes. J Phys Chem B 115:1652–1661
Raqaz A, Mihranyan A, Welch K, Nyholm L, Strømme M (2009) Influence of the type of oxidant on anion exchange properties of fibrous cladophora cellulose/polypyrrole composites. J Phys Chem B 113:426–433
Saito T, Isogai A (2004) TEMPO-mediated oxidation of native cellulose. The effect of oxidation conditions on chemical and crystal structures of the water-insoluble fractions. Biomacromolecules 5:1983–1989
Schroder KA (2011) Mechanisms of photonic curing™: processing high temperature films on low temperature substrates. Nanotech 2:220–223 (CRC Press: Boca Raton, FL)
Shi Z, Phillips GO, Yang G (2013) Nanocellulose electroconductive composites. Nanoscale 5:3194–3201
Shi Z, Gao H, Feng J, Ding B, Cao X, Kuga S, Wang Y, Zhang L, Cai J (2014) In situ synthesis of robust conductive cellulose/polypyrrole composite aerogels and their potential application in nerve regeneration. Angew Chem Int Ed 53:5380–5384
Tkalya E, Ghislandi M, Thielemans W, van der Schoot P, de With G, Koning C (2013) Cellulose nanowhiskers templating in conductive polymer nanocomposites reduces electrical percolation threshold 5-fold. ACS Macro Lett 2:157–163
Weng Z, Su Y, Wang D-W, Li F, Du J, Cheng H-M (2011) Graphene–cellulose paper flexible supercapacitors. Adv Energy Mater 1:917–922
Wolf S, Tauber RN (1986) Silicon processing for the VLSI era, volume 1: process technology. Lattice Press, Sunset Beach
Xu D, Fan L, Gao L, Xiong Y, Wang Y, Ye Q, Yu A, Dai H, Yin Y, Cai J, Zhang L (2016) Micro-nanostructured polyaniline assembled in cellulose matrix via interfacial polymerization for applications in nerve regeneration. ACS Appl Mater Interfaces 8:17090–17097
Zabetakis D, Dinderman M, Schoen P (2005) Metal-coated cellulose fibers for use in composites applicable to microwave technology. Adv Mater 17:734–738
Acknowledgments
The authors affiliated to TNO gratefully acknowledge the European Commission for funding this work as part of the EU Project GREENANOFILMS. This project has received funding from the European Union Seventh Framework Program (FP7/2007-2013) under Grant Agreement No. 603519. Solliance is acknowledged for funding the high resolution TEM facility.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10570_2017_1240_MOESM1_ESM.docx
Online Resource 1a Chemical band assignment of FT-IR for non-oxidized and TEMPO oxidized CNCs, b TGA analysis of CNC-Ag after the second metallization step, c extinction spectrum of CNC-Ag composite particles after the second metallization step, d measured resistance values and corresponding temperatures during the photonic sintering, e measured transmission of CNC-Ag coatings on glass before sintering, f R T as a function of L for W = 10 mm (6 and 8 dips), g R T as a function of L for samples with 4 dips. (DOCX 287 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Meulendijks, N., Burghoorn, M., van Ee, R. et al. Electrically conductive coatings consisting of Ag-decorated cellulose nanocrystals. Cellulose 24, 2191–2204 (2017). https://doi.org/10.1007/s10570-017-1240-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-017-1240-y