
Abstract
Fusarium wilt (FW) and Ascochyta blight (AB) are two important diseases of chickpea which cause 100 % yield losses under favorable conditions. With an objective to validate and/or to identify novel quantitative trait loci (QTLs) for resistance to race 1 of FW caused by Fusarium oxysporum f. sp. ciceris and AB caused by Ascochyta rabiei in chickpea, two new mapping populations (F2:3) namely ‘C 214’ (FW susceptible) × ‘WR 315’ (FW resistant) and ‘C 214’ (AB susceptible) × ‘ILC 3279’ (AB resistant) were developed. After screening 371 SSR markers on parental lines and genotyping the mapping populations with polymorphic markers, two new genetic maps comprising 57 (C 214 × WR 315) and 58 (C 214 × ILC 3279) loci were developed. Analysis of genotyping data together with phenotyping data collected on mapping population for resistance to FW in field conditions identified two novel QTLs which explained 10.4–18.8 % of phenotypic variation. Similarly, analysis of phenotyping data for resistance to seedling resistance and adult plant resistance for AB under controlled and field conditions together with genotyping data identified a total of 6 QTLs explaining up to 31.9 % of phenotypic variation. One major QTL, explaining 31.9 % phenotypic variation for AB resistance was identified in both field and controlled conditions and was also reported from different resistant lines in many earlier studies. This major QTL for AB resistance and two novel QTLs identified for FW resistance are the most promising QTLs for molecular breeding separately or pyramiding for resistance to FW and AB for chickpea improvement.
Similar content being viewed by others


Introduction
Chickpea (Cicer arietinum L.) or Garbanzo beans (Latin America) is a short-duration pulse crop and originated from Southeastern Turkey (Ladizinsky 1975). It is a self-pollinated diploid (2n = 2x = 16) crop with genome size of 738 Mb (see Varshney et al. 2013). Chickpea is cultivated in different parts of the world mainly in the Mediterranean, South Asia, North Africa, Middle East and North and Central America. It is classified as ‘desi’ and ‘kabuli’ type based on seed size, shape and color. It is a rich and cheap source of vegetarian protein and also used as green vegetable dish whereas, other food legumes such as pigeonpea (Cajanus cajan), green gram (Vigna radiata), black gram (Vigna mungo) and lentils (Lens culinaris) are essentially consumed after drying and preservation. Chickpea contains vitamins and minerals such as Ca, Mg, Zn, K, Fe, and phosphorus (http://www.whfoods.com/genpage.php?tname=nutrientprofile&dbid=68, Jukanti et al. 2012) and it is also free from anti-nutritional factors therefore, making nutritionally more valuable and increasing consumer preference for this legume. The major nutritional value of chickpea is mainly because of its superior fiber content (12.0 in g; http://www.wehealnewyork.org/healthinfo/dietaryfiber/fibercontentchart.html#c) over many cereals and consumption of even smaller amounts of chickpea improves insulin secretion and controls blood sugar levels (http://www.whfoods.com/genpage.php?tname=foodspice&dbid=58). These valuable aspects of chickpea caused an increase in its global cultivation and the overall production reached 10.0 million metric tons from 6.6 million metric tons (http://www.cgiar.org/our-research/crop-factsheets/chickpea, as on 23rd April 2013) during last 30 years. South Asia accounts for more than 75 % of the total area under chickpea cultivation and India is the world leader in chickpea production with 7.5 m tons (FAOSTAT data 2010), followed by Pakistan and Turkey. In spite of availability of chickpea cultivars with good grain yield, the crop is highly susceptible to many diseases from time to time and due to this constraint the production is largely affected in all chickpea growing areas.
Among biotic constraints affecting chickpea production, Fusarium wilt (FW) caused by Fusarium oxysporum f. sp. ciceris and Ascochyta blight (AB) caused by Ascochyta rabiei are major diseases reducing yield up to 90 %. FW is prevalent under dry and warm conditions while AB is prevalent under humid conditions (Pande et al. 2005) in the temperature ranges of 23–25 °C during heavy rains (>150 mm). AB attacks all aerial parts of the plant and it is necrotrophic in nature which kills the cells of the host plant and then feeds on it. In the case of FW, the disease is seed and soil borne, the tap root system of the plant is affected including destruction of vascular bundles leading to a disturbance in plant-water relations and ultimately plant dies by showing gradual wilting symptoms (Castro et al. 2010). The FW infected chickpea plants also exhibit drooping of petioles, rachis and leaflets as the disease progresses. Both AB and FW are prevalent across all chickpea growing regions of the world including India, however, AB mainly occurs in northwestern plains while FW is mostly restricted to central and southern parts.
In the case of FW, so far, eight races (0, 1A, 1B/C, 2, 3, 4, 5 and 6) were reported (Jiménez-Gasco and Jiménez-Diaz 2003). The genetics of resistance to five races (race 1A, race 2, race 3, race 4 and race 5) was reported by Sharma et al. (2005) while, genetics of resistance to three races (race 1B, race 1C and race 6) is yet to be studied. In addition, efforts were also made to map quantitative trait loci (QTLs)/genes and markers flanking these QTLs were reported for different Fusarium wilt races. For instance, markers flanking “Foc0” locus (OPJ20600 and TR59), Foc1 locus (TA110 and H3A12), Foc2 locus (H3A12 and TA96), Foc3 locus (TA96 and TA194), Foc4 locus (TA96 and CS27) and Foc5 locus (TA59 and TA96) determining resistance to race 0 (Cobos et al. 2005), race 1 (Gowda et al. 2009), race 2 (Gowda et al. 2009); race 3 (Sharma et al. 2004; Gowda et al. 2009); race 4 (Winter et al. 2000; Sharma et al. 2004, 2005) and race 5 (Cobos et al. 2009) respectively were reported.
In the case of AB, a number of pathotypes were reported; for instance, more than ten pathotypes by Vir and Grewal (1974); five pathotypes by Nene and Reddy (1987); three pathotypes by Udupa et al. (1998) and ten pathotypes by Ali et al. (2009). Udupa et al. (1998) reported occurrence of three pathotypes (pathotype I (less aggressive), pathotype II (aggressive) and pathotype III (most aggressive) as revealed by microsatellites and RAPD markers. Varshney et al. (2009b) characterized 64 isolates of Ascocyta rabiei using AFLP and SSR markers and reported four distinct groups based on STRUCTURE analysis. Further, Kaur et al. (2012b) characterized AB isolates and reported 10 pathotypes based on morphological variation. A large number of QTLs/genes for AB resistance and markers flanking these QTLs have been reported, for instance QTLs for resistance to AB using F2 populations (Flandez-Galvez et al. 2003; Taràn et al. 2007; Anbessa et al. 2009; Kottapalli et al. 2009; Aryamanesh et al. 2010) and recombinant inbred line populations (Millán et al. 2003; Udupa and Baum 2003; Iruela et al. 2006, 2007) were reported. Madrid et al. (2012) reported characterization of E1N4-like sequence (CaETR-1) located in QTLAR1 (Iruela et al. 2006) implicated in AB resistance in chickpea. More recently, Madrid et al. (2013) also reported development of a co-dominant marker (CaETR) based on allele sequence length polymorphism in an ethylene response gene located in QTLAR1 region.
The development of resistant cultivars is generally the preferred strategy for managing the above two diseases as host plant resistance is economical and eco-friendly. Host plant resistance also preserves the quality of the final produce. Therefore, in this direction, mapping of important QTLs/genes responsible for AB and FW resistance has also been considered an important input for present day breeding programmes based on both traditional and modern approach for instance, marker-assisted backcrossing (MABC) which aims at conversion of targeted lines with respect to one or two traits without disturbing remaining all other native traits of the line (Varshney et al. 2009b).
In the present study, molecular mapping for FW resistance was carried out with respect to race 1 (synonymous 1A, Indian isolate) which was reported from India (Haware and Nene 1982). The resistance to race 1 was shown to be controlled by three independent genes and display complete resistance in combination or incomplete (late wilting) when present alone (Upadhyaya et al. 1983; Singh et al. 1987a, b; Sharma and Muehlbauer 2007). The timing of early and/or late wilting appears to be influenced by polygenes according to Brindha and Ravikumar (2005). A limited number of QTL studies were carried out so far using a popular or high yielding genetic background in chickpea for mapping AB and FW resistance.
In view of above, the present study aims at detecting QTLs for the FW and AB, two devastating diseases of chickpea using crosses developed from widely adapted variety ‘C 214’, (a common parent for both AB and FW crosses) and genotypes, ‘WR 315’ and ‘ILC 3279’ highly resistant to FW and AB respectively. This study was also aimed at validating QTLs identified in the previous QTL studies using different parental combinations and to detect new QTLs, if any, as resistant parents used in the present investigation were used for mapping studies in the past.
Materials and methods
Plant material and DNA isolation
In the present study, two mapping populations, ‘C 214’ × ‘WR 315’ and ‘C 214’ × ‘ILC 3279’ were developed for mapping FW resistance and AB resistance respectively. ‘C 214’ is a well-adapted FW and AB susceptible (Kaur et al. 2012a) desi variety suitable for rainfed conditions (Bhardwaj et al. 2010) while ‘WR 315’ is a desi landrace from central India resistant to race 1A, race 2, race 3, race 4 and race 5 of FW (Mayer et al. 1997, Sharma et al. 2005). ‘ILC 3279’ is AB resistant kabuli landrace (Udupa et al. 1998) originated from former USSR and was also used as a source of AB resistance in previous studies (Udupa and Baum 2003; Iruela et al. 2006, 2007).
DNA was isolated from 188 F2 plants of each mapping population as per Cuc et al. (2008) and DNA was normalized to 5 ng/μl.
Screening for FW resistance under field conditions
The F2:3 families were sown in the wilt-sick plot at Patancheru, India during crop season 2011–2012 in two replications using randomized complete block design (RCBD). Experimental plots were 4 m long, row to row spacing of 60 cm and spacing of 10 cm between plants. Observations for disease incidence were recorded at 60 days after sowing (DAS). The wilt incidence was measured on the basis of a formula (% wilt incidence = number of wilted plants/total number of plants × 100).
Screening for AB resistance
The AB populations were screened for both seedling resistance (SR) and adult plant resistance (APR). For SR, F2:3 families were phenotyped as described in Pande et al. (2010), in plastic trays (40 × 30 × 5 cm) filled with sand and vermiculate mixture in 10:1 ratio under controlled environment facility (CEF) at ICRISAT-Patancheru. The pathogen inoculum was sprayed onto foliage of 10 days old seedlings and observations were recorded 10 days after inoculation.
For APR, both CEF as well as natural field conditions were used. F2 population consisting of 188 F2 individuals was used for APR screening in CEF at ICRISAT. Cut-twig screening technique-sand (CTST-S), which can accommodate testing more number of samples, reported by Pande et al. (2010) was adopted for screening in CEF, which is a modified non-destructive technique and was earlier referred to as cut-twig screening technique-water (CTST-W; Singh et al. 1982; Pande et al. 2005). Disease severity was recorded on 1–9 scale, where, 1 = no symptoms (highly resistant), 9 = highly susceptible (up to 100 % killing of cut twigs) (Pande et al. 2010). The experiment was conducted under complete randomized block design (RCBD). In terms of field conditions, the entries (F3 families) were planted in RCBD with two replications in PAU (test location–Ludhiana). The experimental units were one row plots of 2 m length with 10 cm spacing between plants and 40 cm between rows. Susceptible check ‘ICC 4991’ was planted after every four-test rows to provide a constant disease pressure for the AB. All plants of test entries were spray inoculated with conidial suspension derived from single spore culture of A. rabiei containing 4 × 104 conidia/ml during the flowering stage of the crop. Water sprinkling was provided through perfo-spray system to maintain the relative humidity (RH) and temperature from the following day after the inoculation for 21 days. The disease was recorded when the susceptible check shows the maximum disease severity of ‘9’ on a scale of 1–9 where 1 is highly resistant and 9 is highly susceptible.
Statistical analysis of phenotyping data
Phenotyping data for resistance to FW and AB obtained were used to compute the best linear unbiased predictors (BLUPs) of the random effect in restricted maximum likelihood (REML) variance components analysis using SAS 9.1 (SAS Institute Inc 2004) with replicates as fixed models and genotypes as random effects. BLUPs were used for QTL mapping. Analysis of variance (ANOVA) was conducted using the PROC GLM of SAS for the disease rating data. Broad-sense heritability was calculated by dividing genotypic variance of trait with that of total phenotypic trait variance \( \left( {H^{2} \, = \, \sigma^{2}_{g} /\sigma^{2}_{p} } \right) \)
Marker genotyping
The parental polymorphism was studied using a total of 371 SSR markers reported earlier (Hüttel et al. 1999; Winter et al. 1999; Lichtenzveig et al. 2005; Varshney et al. 2009a; Nayak et al. 2010; Thudi et al. 2011; Supplementary Table 1).
For marker genotyping, the PCR was setup in 5 μl reaction volume containing 5 ng template DNA, 0.2 mM dNTPs, 2 mM MgCl2, 2 pmol of forward and reverse primer, 1 U of Taq DNA polymerase (Sib enzyme) and 1× PCR buffer in GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA, USA). A common touch down PCR amplification profile as described in Gujaria et al. (2011) was adopted for PCR amplification and PCR products generated using SSR markers were analyzed on capillary electrophoresis using ABI PRISM® 3730 DNA analyzer, and allele calling was done employing GENEMAPPER (Applied Biosystems, Foster City, CA, USA).
Linkage and QTL analysis
Linkage analysis was performed by JoinMap® 3.0 program (van Ooijen and Voorrips 2001) and the linkage maps were developed on the basis of 188 F2:3 plants for both FW and AB crosses. The logarithm of odds (LOD) score for the test of linkage between marker pairs was set at 3.0 and most-likely marker orders were determined by using ‘ripple’ command. Markers that were attributed to a linkage group at a LOD grouping threshold of 3.0 were only included. Recombination frequencies were converted to genetic distances using the Kosambi mapping function (Kosambi 1944).
Both main and epistatic effect QTLs were determined in the present study using QTL Cartographer version 2.5 (Wang et al. 2005) and QTLNetwork 2.0 (Yang et al. 2005). In the case of FW, a dataset obtained from sick plot experiment was used for mapping APR. In the case of AB, two datasets obtained from controlled conditions (F2:3 families for SR and, F2 plants for APR) at ICRISAT and, a dataset from field (Test location–Ludhiana) experiment for APR were used for the analysis. The QTL analysis was performed by composite interval mapping (CIM) (Jansen and Stam 1994; Zeng 1994) in QTL Cartographer, which facilitates automatic co-factor selection by a forward/backward regression. The threshold log likelihood ratio (LOD) score was estimated with 1000 permutations to declare significance at 0.05 for the trait evaluated. Model 6 of the Zmapqtl module of QTL Cartographer was used for scanning marker intervals with a window size of 10, and LOD threshold of 2.5 was used to declare presence of QTL. The confidence interval for each QTL was set at 1-LOD support interval. The QTLs were designated with italicized symbol composed of disease name initially (for instance FW and AB to refer to Fusarium wilt and Ascochyta blight respectively), a hyphen, followed by Q, a trait name, the symbol for the chromosome in which the QTL is located, and, in cases where more than one QTL controlling a trait were detected in the same LG, they were numbered serially. For instance, the QTL name AB-Q-SR-4-1 refers to the QTL for seedling resistance to AB detected on chickpea linkage group LG 4.
Results and discussion
Variation and distribution of resistance to FW and AB
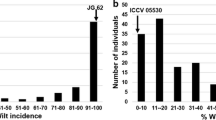
The resistant parents ‘WR 315’ (6.0 % wilt incidence) and ‘ILC 3279’ (AB resistance score of 4.0, 3.3 and 4.0) for SR (controlled conditions), APR (controlled conditions) and APR (field conditions) respectively of two different mapping populations showed greater resistance to both FW and AB respectively as compared to susceptible parent ‘C 214’ (with 54.5 % wilt incidence & AB score of 6.0, 8.4 and 7.0 for SR (controlled conditions), APR (controlled conditions) and APR (field) respectively) (Table 1). The frequency distribution of the FW and AB disease scores based on mean values is illustrated in Fig. 1. The distribution for all the traits analyzed was normal except in the case of AB for seedling resistance (controlled conditions) and the disease scores (for instance seedling resistance for AB) extended beyond the parents, suggesting transgressive segregation. Transgressive segregation for resistance (appearance of extreme individuals than the resistant parent of the mapping populations) suggested that the parents involved in the present crosses for both FW and AB possess positive alleles of different genes governing resistance. The transgressive segregation also suggests the resistance to be quantitative in nature.

Frequency distribution of disease scores for: A FW resistance under field conditions, B seedling resistance (SR) to AB under controlled conditions, C adult plant resistance (APR) to AB under control conditions, and D APR to AB under field conditions
ANOVA for disease severity revealed significant differences among families (P < 0.001) indicating high phenotypic variation. The H2 estimates were low to high (0.21 in the case of FW resistance and 0.78 in the case of AB). The high heritability value in the case of AB indicates high influence of genetic variability than environmental variability. On the other hand, the low heritability in the case of FW might be due to more influence of environment on the trait, which is evident from the normal distribution of FW resistance (Fig. 1). Components of variance (variance due to genotype and variance due to environment) estimated for the disease severities are presented in Table 2. Significant (P < 0.001) genotype effect to FW and AB in both field and controlled conditions was found. In the case of FW, the maximum observed FW survival rate in the population was 45.0 % and the minimum was 5.0 % (Fig. 1). A majority of individuals (n = 120) exhibited less survival rate (<25 %) against FW incidence. For AB resistance, in the case of both controlled and field conditions, more number of individuals (ranging from 50 to 120) coincided with moderate resistance with a score of 4.0 on 1.0–9.0 scale (Toker and Hseyin 2003). This observation is similar to results obtained by Kottapalli et al. (2009) using a F2 cross between AB susceptible parent (‘ICC 4991’) and an AB resistant parent (‘ICCV 04516’).
Genetic linkage maps
Of 371 SSR markers tested on parents of both FW and AB crosses, a total of 298 SSR markers produced scorable amplification in the case of FW parents (‘C 214’ and ‘WR 315’) which further allowed identifying a total of 62 polymorphic SSR markers between ‘C 214’ and ‘WR 315’. In the case of AB, a total of 302 SSR markers produced scorable amplification in both the parents (‘C 214’ and ‘ILC 3279’) and a set of 69 polymorphic SSR markers were identified consequently (Supplementary Table 1).
F2 populations (n = 188 each) for both FW and AB were genotyped with all the above polymorphic markers identified in polymorphism survey and used for construction of genetic maps for the respective populations. The linkage groups in the genetic maps were named following linkage group nomenclature given in Thudi et al. (2011) and Hiremath et al. (2012). In the case of ‘C 214’ × ‘WR 315’ population, 57 (91.9 %) markers could be mapped onto 8 linkage groups spanning a total map length of 347.9 cM (Fig. 2). Five markers namely CaM1650, CaM0475, TA76, CaM0436 and ICCM0076, however, remained unlinked. Similarly in the case of AB cross (‘C 214’ × ‘ILC 3279’), of 69 polymorphic markers, only 58 markers could be mapped onto 10 linkage groups with the total map length of 386.3 cM (Fig. 3). Eleven markers namely, CaM1451, TR58, TR3, ICCM0068, CaM1648, GA26, TA76 s, TA142, CaM2155, TA3 and CaM1301 remained unlinked and this may be due to lack of informative adjacent loci. The distribution of markers across linkage groups in the linkage map developed was comparable to Thudi et al. (2011). Linkage groups 1 and 5 consisted of subgroups LG 1A, LG 1B and LG 5A, LG 5B respectively and, with the addition of a few more markers, these subgroups (LG 1 and LG 5) may merge into separate groups.

Genetic linkage and QTL maps for FW resistance based on C 214 × WR 315 population of chickpea. Markers are shown on right side of the LG while map distances are shown on the left side

Genetic linkage and QTL maps for AB resistance based on C 214 × ILC 3279 population of chickpea Markers are shown on right side of the LG while map distances are shown on the left side
QTLs for FW resistance
QTL analysis was conducted with dataset obtained from wilt sick plot experiment and detected two QTLs (FW-Q-APR-6-1 and FW-Q-APR-6-2) for FW resistance on linkage group 6 in the FW cross (‘C 214’ × ‘WR 315’) (Table 3; Supplementary Fig. 1) with LOD values 8.0 and 7.6 and explained 10.4 and 18.8 % phenotypic variation respectively. Analysis with QTLNetwork based on Mixed-model based composite interval mapping (MCIM) resulted into identification of the same QTL (FW-Q-APR-6-1) flanked by SSR markers CaM1402 and CaM1101 on LG 6 explaining 16.4 % of phenotypic variation. No epistatic QTL affecting FW resistance in this population was detected. Another QTL (FW-Q-APR-6-2) detected by QTL cartographer was not detected by QTLNetwork.
The QTLs for FW resistance identified in the present study were not reported so far and therefore these QTLs may be considered novel. Identification of these novel QTLs for FW race 1 as compared to some earlier studies on QTL mapping (Gowda et al. 2009; Sharma et al. 2004) involving WR 315 as resistant parent may be explained due to differences in the experimental conditions employed in our study and the earlier studies. It is important to note that FW screening was carried out under lab conditions in the earlier studies while FW screening was undertaken under field conditions in the present study. Therefore for breeding applications, the identified QTLs in the present study seem to be the most promising QTLs for FW race 1. Furthermore, as the QTL region also included four bacterial artificial chromosome end (BAC-end) derived SSR markers (CaM1402, CaM1101, CaM1125 and CaM0594), integration of BACs for these corresponding markers into physical map of chickpea (http://probes.pw.usda.gov:8080/chickpea/) may facilitate the isolation of genes that are responsible for FW resistance.
QTLs for resistance to AB
QTL analysis for resistance to AB was conducted for both SR and APR. For SR, QTL analysis detected two QTLs (AB-Q-SR-4-1 and AB-Q-SR-4-2) on LG 4 (Table 3; Fig. 3). These two QTLs were detected with 8.8 and 2.9 LOD values and explained 31.9 and 10.3 % phenotypic variation respectively. AB-Q-SR-4-1 was flanked by SSR markers STMS11 and TA130 while, AB-Q-SR-4-2 was flanked by H4G11 and CaM2049 markers (Fig. 3). QTLNetwork identified the same QTL (AB-Q-SR-4-1) (Table 3) explaining 21.0 % phenotypic variation. Furthermore, this QTL was also involved in epistatic interaction and this QTL region has its own effect and also involved in interaction with other loci with an epistatic effect of 0.23 (Fig. 4).

A snapshot showing significant interaction of loci (in red). Circles denote QTLs with additive effect whereas squares represent QTLs with dominance effect
This QTL could be the same QTL identified in several earlier studies (Flandez-Galvez et al. 2003; Udupa and Baum 2003; Tekeoglu 2004; Cho et al. 2004; Iruela et al. 2006). This assumption was supported by the presence of common marker, (either TA130 or GAA47) in the corresponding QTL regions. It is important to note that the resistant parent (ILC 3279) used in the present study was also used in the QTL studies of Udupa and Baum (2003); Iruela et al. (2006, 2007). In the case of other QTL studies (Flandez-Galvez et al. 2003; Tekeoglu 2004; Cho et al. 2004), a different resistance source was used for mapping AB resistance. By taking all these studies together, the QTL (AB-Q-SR-4-1) identified in the present study seems to be a very promising QTL region for SR resistance to AB in chickpea breeding as it was identified across different genetic backgrounds as well as locations.
For APR to AB, a total of four QTLs were identified in QTL Cartographer analysis using two datasets (ICRISAT-Patancheru and PAU-Ludhiana). Of four QTLs, two QTLs (AB-Q-APR-6-1 and AB-Q-APR-6-2) were detected on LG 6 (Fig. 3) using dataset from controlled conditions (Table 3). These QTLs detected with LOD values 4.8 and 5.1, explained 2.2 and 11.5 % of phenotypic variation, respectively. QTLNetwork detected AB-Q-APR-6-2 with 10.8 % phenotypic variation. Taràn et al. (2007) reported a QTL on LG 6 in the vicinity of TA106 marker which in fact, flanked QTL (AB-Q-APR-6-2) in the present study on LG 6 and small differences in the QTL region in terms of precise marker order from this study with that of Taràn et al. (2007) may be due to differences in meiotic recombinations, methodology and linkage testing softwares.
Another dataset based on field screening at Ludhiana detected two QTLs, AB-Q-APR-4-1 and AB-Q-APR-5B on LG 4 and LG 5B, respectively. AB-Q-APR-4-1 explained 26.4 % variation with a LOD of 4.3 and, AB-Q-APR-5B explained 1.5 % of phenotypic variation with LOD value 3.1 (Table 3). AB-Q-APR-4-1 QTL was also detected by QTLNetwork with 6.7 % phenotypic variation. The detection of same genomic region/QTL for both SR (AB-Q-SR-4-1) and APR (AB-Q-APR-4-1) on LG 4 flanked by STMS11 and TA130 might be due to presence of common genes conditioning resistance to AB at different plant growth stages.
Comparison of QTL analysis for SR and APR provided at least one QTL that was identified for both, SR (AB-Q-SR-4-1) as well as APR detected under both field (APR) and controlled conditions (lab screening for SR). Detection of the same resistance QTL for SR and APR may support the presence of common genes conditioning AB resistance at different developmental stages of the plant. In fact, Rajesh et al. (2008) predicted candidate genes for AB resistance in the same genomic region using sequence information from corresponding BAC clones and recently, Madrid et al. (2012) reported characterization of a functional sequence (EIN4-like sequence (CaETR-1) located under the QTL (AR1) (Iruela et al. 2006) and its involvement in AB resistance. The above QTL (AR1) is the same or similar to AB-Q-SR-4-1 identified in the present study for AB resistance. The other QTL (AB-Q-SR-4-2) identified in the present study was reported earlier by Lichtenzveig et al. (2006). This study however added one additional marker (CaM2049), derived from end sequence of BAC (Thudi et al. 2011), for this QTL region. Integration of the BAC corresponding to this SSR marker into physical map may provide additional BACs covering this region and eventually isolation of resistance genes to AB. In a separate study integration of the genetic and physical map of chickpea indicated that the SSR marker was mapped on to the BAC contig (ctg2052). In fact, Zhang et al. (2010) also reported one BAC/BIBAC contig (ctg3270) containing or closely linked to the same QTL. Nevertheless, the availability of draft genome sequence of chickpea and the resequencing data on ninety chickpea lines (Varshney et al. 2013) will enable identification of genes and superior alleles underlying in the novel QTLs regions identified for FW and AB resistance.
In summary, the present study reports two major and novel QTLs for resistance to FW and one major QTL for resistance (both SR and APR) to AB. These QTL regions are linked with some BAC-end sequence derived SSR markers. While QTLs for FW are identified based on field data, these QTLs are most promising for deployment in chickpea breeding. In case of AB resistance, one QTL that was detected under lab conditions as well as field conditions is the same QTL identified in several earlier studies. Therefore the QTLs identified and markers linked with the promising QTLs are useful resource for genomics-assisted breeding for resistance to FW and AB.
References
Ali AR, Iqbal SHM, Iqbal U, Ghafoor A, Akram A (2009) Pathogenic diversity in Ascochyta rabiei (Pass.) Lib., of chickpea. Pak J Bot 41:413–419
Anbessa Y, Taràn B, Warkentin TD, Tullu A, Vandenberg A (2009) Genetic analyses and conservation of QTL for Ascochyta blight resistance in chickpea (Cicer arietinum L.). Theor Appl Genet 4:757–765
Aryamanesh N, Nelson MN, Yan G, Clarke HJ, Siddique KHM (2010) Mapping a major gene for growth habit and QTLs for Ascochyta blight resistance and flowering time in a population between chickpea and Cicer reticulatum. Euphytica 173:307–319
Bhardwaj R, Sandhu JS, Kaur L, Gupta SK, Gaur PM, Varshney RK (2010) Genetics of Ascochyta blight resistance in chickpea. Euphytica 171:337–343
Brindha S, Ravikumar RL (2005) Inheritance of wilt resistance in chickpea: a molecular marker analysis. Curr Sci 88:701–702
Castro P, Pistón F, Madrid E, Millàn T, Gil J, Rubio J (2010) Development of chickpea near-isogenic lines for Fusarium wilt. Theor Appl Genet 121:1519–1526
Cho S, Chen W, Muehlbauer FJ (2004) Pathotype-specific genetic factors in chickpea (Cicer arietinum L.) for quantitative resistance to Ascochyta blight. Theor Appl Genet 109:733–739
Cobos MJ, Fernàndez MJ, Rubio J, Kharrat M, Moreno MT, Gil J, Millàn T (2005) A linkage map of chickpea (Cicer arietinum L.) based on populations from Kabuli × Desi crosses: location of genes for resistance to Fusarium wilt race 0. Theor Appl Genet 110:1347–1353
Cobos MJ, Winter P, Kharrat M, Cubero JI, Gil J, Millan T, Rubio J (2009) Genetic analysis of agronomic traits in a wide cross of chickpea. Field Crops Res 111:130–136
Cuc LM, Mace E, Crouch J, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea L.). BMC Plant Biol 8:55
FAOSTAT data (2010) http://faostat.fao.org/site/339/default.aspx. Accessed 12 Nov 2012
Flandez-Galvez H, Ades PK, Ford R, Pang ECK, Tayler PWJ (2003) QTL analysis for Ascochyta blight resistance in an intraspecific population of chickpea (Cicer arietinum L.). Theor Appl Genet 107:1257–1265
Gowda SJM, Radhika P, Kadoo NY, Mhase LB, Gupta VS (2009) Molecular mapping of wilt resistance genes in chickpea. Mol Breed 24:177–183
Gujaria N, Kumar A, Dauthal P, Hiremath P, Bhanu Prakash A, Farmer A, Bhide M, Shah T, Gaur PM, Upadhyaya HD, Bhatia S, Cook DR, May GD, Varshney RK (2011) Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.). Theor Appl Genet 122:1577–1589
Haware MP, Nene YL (1982) Symptomless carriers of the chickpea wilt Fusarium. Plant Dis 66:809–810
Hiremath P, Kumar JA, Penmetsa RV, Farmer A, Schlueter JA, Chamarthi SK, Whaley AM, Carrasquilla-Garcia N, Gaur PM, Upadhyaya HD, Kavi Kishor PB, Shah TM, Cook DR, Varshney RK (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10:716–732
Hüttel B, Winter P, Weising K, Choumane W, Weigand F (1999) Sequence-tagged microsatellite markers for chickpea (Cicer arietinum L.). Genome 42:210–217
Iruela M, Rubio J, Barro F, Cubero JI, Millán T, Gil J (2006) Detection of two quantitative trait loci for resistance to Ascochyta blight in an intra-specific cross of chickpea (Cicer arietinum L.): development of SCAR markers associated with resistance. Theor Appl Genet 112:278–287
Iruela M, Castro P, Rubio J, Cubero JI, Jacinto C, Millán T, Gil J (2007) Validation of a QTL for resistance to Ascochyta blight linked to resistance to Fusarium wilt race 5 in chickpea (Cicer arietinum L.). Eur J Plant Pathol 119:29–37
Jansen RC, Stam P (1994) High resolution mapping of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Jiménez-Gasco MM, Jiménez-Diaz RM (2003) Development of a specific polymerase chain reaction-based assay for the identification of Fusarium oxysporum f. sp. ciceris and its pathogenic races 0, 1A, 5 and 6. Phytopathology 3:200–209
Jukanti AK, Gaur PM, Gowda CLL, Chibbar RN (2012) Chickpea: nutritional properties and its benefits. Br J Nutr 108:S11–S26
Kaur L, Singh VP, Gupta VP (2012a) Peroxidase: a marker for Ascochyta blight resistance in chickpea. Arch Phytopathol Plant Protect 45:42–46
Kaur L, Singh VP, Sandhu JS (2012b) Characterization of Ascochyta rabiei isolates and evaluation of genotypic stability in chickpea. Arch Phytopathol Plant Protect 45:83–89
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kottapalli P, Gaur PM, Katiyar SK, Crouch JH, Buhariwalla HK, Pande S, Gali KK (2009) Mapping and validation of QTLs for resistance to an Indian isolate of Ascochyta blight pathogen in chickpea. Euphytica 165:79–88
Ladizinsky G (1975) A new Cicer from Turkey. Notes Roy Bot Gard. Edinburgh 34:201–202
Lichtenzveig J, Scheuring C, Dodge J, Abbo S, Zhang HB (2005) Construction of BAC and BIBAC libraries and their applications for generation of SSR markers for genome analysis of chickpea, Cicer arietinum L. Theor Appl Genet 110:492–510
Lichtenzveig J, Bonfil DJ, Zhang HB, Shtienberg D, Abbo S (2006) Mapping quantitative trait loci in chickpea associated with time to flowering and resistance to Didymella rabiei the causal agent of Ascochyta blight. Theor Appl Genet 113:1357–1369
Madrid E, Rajesh PN, Rubio J, Gil J, Millán T, Chen W (2012) Characterization and genetic analysis of an EIN4-like sequence (CaETR-1) located in QTL (AR1) implicated in Ascochyta blight resistance in chickpea. Plant Cell Rep 31:1033–1042
Madrid E, Chen W, Rajesh PN, Castro P, Millán T, Gil J (2013) Allele-specific amplification for the detection of Ascochyta blight resistance in chickpea. Euphytica 189:183–190
Mayer MS, Tullu A, Simon CJ, Kumar J, Kaiser WJ, Kraft JM, Muehlbauer FJ (1997) Development of a DNA marker for Fusarium wilt resistance in chickpea. Crop Sci 37:1625–1629
Millán T, Rubio J, Iruela M, Daly K, Cubero JI, Gil J (2003) Markers associated with Ascochyta blight resistance in chickpea and their potential in marker-assisted selection. Field Crops Res 84:373–384
Nayak SN, Zhu H, Varghese N, Datta S, Choi HK, Horres R, Jüngling R, Singh J, Kavi Kishore PB, Sivaramakrihnan S, Hoisington DA, Kahl G, Winter P, Cook DR, Varshney RK (2010) Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120:1415–1441
Nene YL, Reddy MV (1987) Chickpea diseases and their control. In: Saxena MC, Singh KB (eds) The Chickpea. CABI, Oxon, pp 233–270
Pande S, Siddique KHM, Kishore GK, Baya B, Gaur PM, Gowda CLL, Bretag T, Crouch JH (2005) Ascochyta blight of chickpea: biology, pathogenicity, and disease management. Aus J Agric Res 56:317–332
Pande S, Sharma M, Gaur PM, Gowda CLL (2010) Host plant resistance to Ascochyta blight of chickpea. Information Bulletin No. 82. Patancheru 502 324. International Crops Research Institute for the Semi-Arid Tropics, Andhra Pradesh, p 40
Rajesh PN, Bleness MO, Roe BA, Muehlbauer FJ (2008) Analysis of genome organization, composition and microsynteny using 500 kb BAC sequences in chickpea. Theor Appl Genet 117:449–458
SAS Institute Inc. (2004) SAS/STAT 9.1 User’s Guide, Cary, NC: SAS Institute Inc
Sharma KD, Muehlbauer FJ (2007) Fusarium wilt of chickpea: physiological specialization, genetics of resistance and resistance gene tagging. Euphytica 157:1–14
Sharma KD, Winter P, Kahl G, Muehlbauer FJ (2004) Molecular mapping of Fusarium oxysporum f. sp. ciceris race 3 resistance gene in chickpea. Theor Appl Genet 108:1243–1248
Sharma KD, Chen W, Muehlbauer FJ (2005) Genetics of chickpea resistance to five races of Fusarium wilt and a concise set of race differentials for Fusarium oxysporum f. sp. ciceris. Plant Dis 89:385–390
Singh G, Kapoor S, Singh K (1982) Screening chickpea for gray mold resistance. Int Chickpea Newsl 7:13–14
Singh H, Kumar J, Haware MP, Smithson JB (1987a) Genetics of resistance to Fusarium wilt in chickpeas. In: Day PR, Jellis GJ (eds) Genetics and plant pathogenesis. Blackwell, Oxford, pp 339–342
Singh H, Kumar J, Smithson JB, Haware MP (1987b) Complementation between genes for resistance to race 1 of Fusarium oxysporum f. sp. ciceris in chickpea. Plant Pathol 36:539–543
Taràn B, Warkentin TD, Tullu A, Vanderberg A (2007) Genetic mapping of Ascochyta blight resistance in chickpea (Cicer arietinum) using a simple sequence repeat linkage map. Genome 50:26–34
Tekeoglu M (2004) QTL analysis of Ascochyta blight resistance in chickpea. Turk J Agric For 28:183–187
Thudi M, Bohra A, Nayak SN, Varghese N, Shah TM, Penmetsa RV, Thirunavukkarasu N, Gudipati S, Gaur PM, Kulwal PL, Upadhyaya HD, KaviKishor PB, Winter P, Kahl G, Town CD, Kilian A, Cook DR, Varshney RK (2011) Novel SSR markers from BAC-End sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). PLoS One 6:e27275
Toker C, Hseyin A (2003) Selection of chickpea (Cicer arietinum L.) genotypes for resistance to Ascochyta blight [Ascochyta rabiei (Pass.) Labr.], yield and yield criteria. Turk J Agric For 27:277–283
Udupa SM, Baum M (2003) Genetic dissection of pathotype-specific resistance to Ascochyta blight disease in chickpea (Cicer arietinum L.) using microsatellite markers. Theor Appl Genet 106:1196–1202
Udupa SM, Weigand F, Saxena MC, Kahl G (1998) Genotyping with RAPD and microsatellite markers resolves pathotype diversity in the Ascochyta blight pathogen of chickpea. Theor Appl Genet 97:299–307
Upadhyaya HD, Smithson JB, Haware MP, Kumar J (1983) Resistance to wilt in chickpea. II: further evidence for two genes for resistance to race 1. Euphytica 32:749–755
van Ooijen JW, Voorrips RE (2001) JoinMap® 3, software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao Y, Srinivasan R, Gaur PM, Siddique KHM, Town CD, Hoisington DA (2009a) A comprehensive resource of drought-and salinity responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.). BMC Genomics 10:523
Varshney RK, Pande S, Kannan S, Thudi M, Sharma M, Gaur PM, Hoisington DA (2009b) Assessment and comparison of AFLP and SSR based molecular genetic diversity in Indian isolates of Ascochyta rabiei, a causal agent of Ascochyta blight in chickpea (Cicer arietinum L.). Mycol Prog 8:87–97
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Taràn B, Millán T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CL, Singh NP, Lichtenzveig J, Gali KK, Rubio J, Nadarajan N, Dolezel J, Bansal KC, Xu X, Edwards D, Zhang G, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246
Vir S, Grewal JS (1974) Physiological specialization in Ascochyta rabiei, the causal organism of gram blight. Indian Phytopathol 27:265–266
Wang S, Basten CJ, Zeng ZB (2005) Windows QTL Cartographer 2.5. department of statistics, North Carolina State University, Raleigh, NC (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm). Accessed 1 Dec 2011
Winter P, Pfaff T, Udupa SM, Huttel B, Sharma PC, Sahi S, Arreguin-Espinoza R, Weigand F, Muehlbauer FJ, Kahl G (1999) Characterization and mapping of sequence tagged microsatellite sites in the chickpea (Cicer arietinum L.) genome. Mol Gen Genet 262:90–101
Winter P, Benko-Iseppon AM, Hüttel B, Ratnaparkhe M, Tullu A, Sonnante G, Pfaff T, Tekeoglu M, Santra D, Sant VJ, Rajesh PN, Kahl G, Muehlbauer FJ (2000) A linkage map of the chickpea (Cicer arietinum L.) genome based on recombinant inbred lines from a C. arietinum × C. reticulatum cross: localization of resistance genes for Fusarium wilt races 4 and 5. Theor Appl Genet 101:1155–1163
Yang J, Hu CC, Ye XZ, Zhu J (2005) QTLNetwork 2.0. Institute of Bioinformatics, Zhejiang University, Hangzhou, China http://ibi.zju.edu.cn/software/qtlnetwork
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang X, Scheuring CF, Zhang M, Dong JJ, Zhang Y, Huang JJ, Lee MK, Abbo S, Sherman A, Shtienberg D, Chen W, Muehlbauer F, Zhang HB (2010) A BAC/BIBAC-based physical map of chickpea Cicer arietinum L. BMC Genomics 11:501
Acknowledgments
The authors acknowledge support from Vinay Kumar Bairapaka, Rameshwar Telangre for their technical help. The present study was supported under the Accelerated Crop Improvement Programme (ACIP) from the Department of Biotechnology (DBT), Government of India and Tropical Legume I of Bill and Melinda Gates Foundation (BMGF) supported through CGIAR Generation Challenge Progamme (GCP). Thanks are also due to suggestions of Professor PK Gupta, Chair, and other members of ACIP Task Force of DBT for their useful suggestions during the course of the study. This work has been under taken as part of the CGIAR Research Program on GrainLegumes. ICRISAT is a member of CGIAR Consortium.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Sabbavarapu, M.M., Sharma, M., Chamarthi, S.K. et al. Molecular mapping of QTLs for resistance to Fusarium wilt (race 1) and Ascochyta blight in chickpea (Cicer arietinum L.). Euphytica 193, 121–133 (2013). https://doi.org/10.1007/s10681-013-0959-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-013-0959-2