
New esters (sulfate, nicotinates, and 4-methoxycinnamate) of 18α-glycyrrhizic acid (18α-GA) were synthesized; these were D/E-trans-isomers of natural 18β-GA (GA), which is the major triterpene glycoside in the roots of Spanish licorice and Urals licorice (Glycyrrhiza glabra L. and Gl. uralensis Fisher). Changes in the stereochemistry of the GA aglycone led to significant decreases in its anti-HIV-1 activity.
Similar content being viewed by others
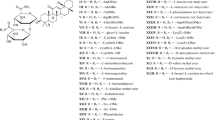


Glycyrrhizic acid (GA) is the major triterpene glycoside in the roots of the Spanish licorice (Glycyrrhiza glabra L.) and Urals licorice (Gl. uralensis Fisher) (Leguminosae) and has antiviral and immunotropic activities [1, 2]. GA and its monoammonium salt (glycyrrham) inhibit the reproduction of a number of DNA and RNA viruses (Vaccinia, Newcastle disease, vesicular stomatitis, herpes simplex type 1, influenza, etc.) in vitro [3]. GA also inhibits varicella-zoster virus [4], hepatitis A, B, and C viruses (HAV, HBV, HCV) [5–8], human cytomegalovirus [9], and the new human herpes-viruses types 6 and 7 (human herpes virus, HHV6, HHV7) [10]. Reports showing the ability of GA to inhibit Epstein-Barr virus [11] and SARS-associated coronaviruses, which produce atypical pneumonia [12], have appeared.
A number of chemically modified GA derivatives have been found to have more marked inhibitory effects on the reproduction of HIV-1 in vitro [13].
GA nicotinate (niglizin) has been patented as an HIV Inhibitor [14]. In acute HIV infection, niglizin has stronger virus-inhibiting properties than GA and glycyrrham [15]. This substance is more active than azidothymidine in models of chronic infection and has high inhibitory activity against both wild and mutant forms of HIV-1 reverse transcriptase.
Furthermore, GA nicotinate is of medicinal interest as a hepatoprotector and anti-inflammatory substance [16, 17]. There is interest in synthesizing compounds with different lipophilicities based on the stereoisomer 18α-GA (I), which is the D/E-trans-isomer of GA. Compound I has undergone clinical trials in China in the treatment of hepatitis and has been found to be more active than 18β-GA in decreasing alanine and aspartate aminotransferase activities [18].
Compound I was prepared by alkaline isomerization of 18β-GA (92%) as described in [19], followed by purification as potassium salts by dissolution in acetone and precipitation of the 3K salt of I with 10% KOH/MeOH solution, conversion of 3K salts of I into monopotassium salts (II) by crystallization from glacial acetic acid, and recrystallization from aqueous ethanol. Treatment of purified salts II with the cation exchange resin KU-2-8 in the H+ form in 85% EtOH yielded acid I with a purity of 85 ± 2% (HPLC data), which was used to prepare esters (III – VI).
Treatment of I with excess SO3∙Py in dry pyridine in the dark for 48 h followed by neutralization with 1 N NaOH and treatment with cationic exchange resin KU-2-8 (H+) in 50% EtOH yielded the sodium penta-O-sulfonate of I as the di-sodium salt III (yield 56%), which was characterized by IR, 1H and 13C NMR spectra and elemental analysis data. The 13C NMR spectrum of this compound contained a characteristic signal of the free 30-COOH group at 180.0 ppm.
Acylation of I with an excess of nicotinoyl chloride hydrochloride in a mixture of pyridine and tributylamine (6:1) at 80 – 90-°C for 3 h in the presence of a 4-Å molecular sieve produced dinicotinate IV, as confirmed by elemental analysis, with a yield of 80%.


Acylation of I with an excess of nicotinoyl chloride (1:8 mmol) at 90 – 100°C for 8 h in tributylamine produced the penta-O-nicotinate of 18α-GA (V) with a yield of 60.7%. The IR spectra of nicotinates IV and V contained absorption peaks at 1550 and 1600 cm –1, due to valent oscillations of Py residues. The 1H NMR spectrum of compound V contained signals from aromatic protons at 7.4 – 9.0 ppm as a multiplet; the 13C NMR spectrum contained intense signals from aromatic C atoms at 123.7 – 153.4 ppm.
Acylation of I with an excess of 4-methoxycinnamoyl chloride (1:8 mmol) in a mixture of pyridine and tributylamine (1:1) at 60 – 70°C for 12 h produced the penta-O(4-methoxy)cinnamate derivative (VI) (yield 65.6%), whose structure was confirmed by IR, UV, and NMR spectroscopy.
The cytotoxicities of I and its derivatives were greater than that of GA in cultures of transplantable human T-lymphocytes (line MT-4) (Table 1). Changes in the stereochemistry of the GA molecule led to decreases in the anti-HIV-1 activity of the glycoside. Thus, the index of selectivity (IS) of I (with trans-linkage of the A and B rings) was 10 times lower than that of GA, with cis-linking of the A and B rings. The esters of I studied here also had little activity. Thus, the aglycone with the native structure was important in producing this type of activity.
Experimental chemical section
IR spectra were recorded on a Specord M-80 spectrophotometer in pastes with Vaseline grease. UV spectra were taken on UF-400 spectrometer in methanol. 1H and 13C NMR spectra were recorded on a Brüker AM-300 spectrometer at working frequencies of 300 and 75.5 MHz with wide-band and non-resonant proton suppression. The internal standard was tetramethylsilane. Optical activity was measured using a Perkin-Elmer 241 MC polarimeter in a 1-dm tube. Melting points were measured using a Boetius microstage.
Thin layer chromatography (TLC) was performed on Silufol (Czech Republic) or Sorbfil (ZAO Sorbpolimer) plates using chloroform-methanol (20:1) (A) and toluene-ethyl acetate (10:1) (B) solvent systems. Substance spots were detected with 20% phosphotungstic acid or 5% H2SO4 in ethanol followed by heating to 110 – 120°C for 2 – 3 min. Column chromatography (CC) was performed on KSK silica gel (fraction 50 – 150, dry classification) (ZAO Sorbpolimer). HPLC analysis was performed was performed on a Du Pont chromatography system using a Chromsil CN reverse phase column and a mobile phase (MP) consisting of phosphate buffer pH 5.17; acetonitrile, and dioxane (210:15:10 (v/v); detection with was UV light (λ 254 nm).
Tributylamine (Bu3N) was kept over KOH for one day and distilled. Pyridine (Py) was distilled over BaO and stored over a 4-Å molecular sieve. Other solvents were purified as described in [20]. Solvents were evaporated in vacuo at 50 – 60°C. Molecular sieves were calcinated at 300 – 350°C for 3 h. Elemental analysis data coincided with calculated values.
18α-Glycyrrhizic acid (I) and its monopotassium salt (II).
1. A solution of 6.6 g (7.5 mmol) of 18β-GA (88 ± 2%) in 200 ml of 2 N KOH in 50% aqueous pyridine was boiled for 16 h, cooled to 20 – 22°C, diluted with iced water to 400 ml, and acidified with concentrated hydrochloric acid to pH 2. The precipitate was collected by filtration, washed with cold water and ether, and dried. This yielded 3.6 g (53%) of a mixture of the 18α and 18β isomers of GA, of which 70 ± 2% was shown by HPLC to be the 18α isomer.
2. A solution of 3.6 g of I in 200 ml of acetone was supplemented with 10% KOH/MeOH to pH 8 – 9. The precipitate of the 3K+ salt was collected by filtration, washed with acetone and ethanol, and dried. The dry salt was recrystallized from glacial CH3COOH with heating to 90 – 95°C. The resulting precipitate was collected by filtration, washed with acetone and ether, and dried in a drying cupboard at 100 – 110°C. The yield was 3.5 g of salt II (94.7%), which was recrystallized twice from a mixture of EtOH and H2O (4:1). The yield was 2.4 g (64.9%). [α D 20] +24 ± 2° (c 0.05; 25% EtOH). The UV spectrum, λ 25%EtOHmax (lg ε), was: 249 nm (4.18). C42H61O16K.3H2O.
3. A solution of 1.6 g of salt II in 25 ml of 85% EtOH, 1.6 g of cation exchange resin KU-2-8 (H+), and 0.1 g of activated charcoal powder was mixed at 20 – 22°C for 2 h. The resin and charcoal were collected by filtration and washed with ethanol, and the filtrate was evaporated to dryness in vacuo. The yield was 1.3 g of I (86.7%). [α D 20] +25 ± 2° (c 0.12; EtOH). The UV spectrum, λ 25%EtOHmax (lg ε), was: 244.5 nm (4.03). The content of the target substance was 85 ± 2%. Published data: [α D 20] +25.5° (c 0.05; 50% EtOH).
18α-Glycyrrhizic acid pentasulfate sodium disodium salt (III).
A solution of 0.5 g (0.6 mmol) of I in 10 ml of dry Py was supplemented with 1.8 g (11.2 mmol) of the SO3. Py complex and stored in the dark for 48 h. NaOH solution (1 N) was added dropwise to pH 8. The mixture was evaporated and the residue was dissolved with heating in 50% EtOH and filtered. On standing, the filtrate again formed a precipitate, which was collected by filtration. The filtrate was evaporated to dryness, dissolved in 50% EtOH, and treated with cationic exchange resin KU-2-8 (H+) to pH 6. The resin was separated and washed with 50% EtOH and the filtrate was evaporated. The yield was 0.47 g (56%) of sulfonate III. The IR spectrum, ν, cm−1, was: 3600 – 3200; 1700; 1660; 1640; 1620.[α D 20] +30° (c 0.04, 50% MeOH). C42H55O31S5Na7. The 1H NMR spectrum (DMF-d6, δ, ppm), was: 0.70; 0.76; 1.01; 1.09; 1.15; 1.24; 1.39 (all s, 21 H, 7 CH3); 8.23; 8.75 (H-C-O-SO3Na). The 13C NMR spectrum was: 39.4 (C1); 26.5 (C2); 92.0 (C3); 40.4 (C4); 55.1 (C5); 43.9(C8); 61.8 (C9); 37.3 (C10); 199.3 (C11); 123.6 (C12); 171.0 (C13); 45.4 (C14); 33.9 (C17); 42.4 (C19); 44.1 (C20); 27.6 (C23); 16.0 (C24); 16.5 (C25); 18.5 (C26); 28.4 (C28); 180.0 (C30); 167.0 (C6′;C6″); 104.5; 102.5 (C1′; C1″); 83.0 (C2′); 78.0; 77.1; 76.0; 74.5; 74.0; 72.5; 72.5 (C2″;C3′;C3″;C4′;C4″; C5′;C5″).
18α-Glycyrrhizic acid di-O-nicotinate (IV).
A solution of 0.5 g (0.6 mmol) of I in 6 ml of dry Py and 1 ml of dry Bu3N was supplemented with 1 g of a 4-Å molecular sieve and mixed for 30 min; this was followed by addition of 0.86 g of (4.9 mmol) of nicotinoyl chloride hydrochloride and heating to 80 – 90°C for 3 h without allowing entry of moisture. The mix was cooled to 20 – 22°C, 6 ml of acetone was added and mixed, and the mixture was poured into cold water (60 ml). The resulting precipitate was collected by filtration, washed with water, and dried. This produced 0.6 g of crude product, which was recrystallized from aqueous ethanol. The yield was 0.52 g of IV (80%). The IR spectrum, ν, cm–1, was: 3600 – 3200 (OH); 1820 (C=O); 1745 (COOR); 1660 =O); 1600 (Py). The 1H (C11NMR spectrum (CHCl + DMSO-d36, δ, ppm), was: 0.6 – 1.1 (7 CH3, 21H); 5.4 (s, 1H, H12); 7.2; 7.6 (m, arom. H). C54H68N2O20.
18α-Glycyrrhizic acid penta-O-nicotinate (V).
A solution of 0.82 g (1 mmol) of I in 12 ml of dry Bu3N was supplemented with 4 g of a 4-Å molecular sieve and mixed for 30 min; this was followed by cooling in an ice bath and addition of 1.42 g (8 mmol) of nicotinoyl chloride hydrochloride. The mixture was heated to 90 – 100°C for 8 h without allowing entry of moisture and cooled to 20 – 22°C; 10 ml of acetone was added and the mixture was diluted with 100 ml of cold water and 5% hydrochloric acid to pH ~5. The precipitate was collected by filtration, washed with water, and dried. This produced 1.35 g of crude product, which was recrystallized from a mixture of EtOH and H2O (4:1 v/v). The yield was 0.82 g (60.7%). R f = 0.5 (B). The IR spectrum, ν, cm–1, was: 1690 (COOH); 1620, 1600, 1510 (Py). C72H77N5O21. The 1H NMR spectrum (300 MHz, CDCl3 + DMF-d6, δ, ppm), was: 0.70 – 1.35 (7 CH3, 21 H); 2.65 (s, 1H, H-9); 3.48 (s, 1H, H-3); 5.40 (s, 1H, H12); 7.40; 7.90; 7.95; 8.20; 8.24; 8.46; 8.70; 9.04 (m, arom. H,). The 13C NMR spectrum was (75.5 MHz, CDCl3 + DMF-d6, δ, ppm): 39.2 (C1); 89.0 (C3); 56.9 (C5); 44.9 (C8); 63.8 (C9); 36.5 (C10); 199.2 (C11); 123.6 (C12); 170.0 (C13); 26.6 (C16); 42.2 (C18); 43.7 (C20); 37.4 (C22); 27.6 (C23); 15.8 (C24); 16.4 (C25); 18.2 (C26); 24.4 (C27); 180.7 (C30); 103.8 (C1′;C1″); 81.5 (C2′); 76.4; 76.0 (C5′;C5″); 75.0; 74.6 (C3′;C3″); 72.4; 71.8 (C4′;C4″); 166.2; 165.0 (C6′; C6″); 153.4; 150.4; 150.2; 136.9; 126.2; 123.7 (C pyridine).
Penta-O-4-methoxycinnamate ester of 18α-glycyrrhizic acid (VI).
A solution of 0.5 g (0.6 mmol) of I in a mixture of 5 ml of dry Py and 5 ml of Bu3N was supplemented with 4 g of a 4-Å molecular sieve and mixed for 30 min; this was followed by cooling in an ice bath and addition of 0.96 g (8 mmol) of 4-methoxycinnamoyl chloride in 5 ml of dichloroethane. The reaction was mixed for 2 h at 20 – 22°C and 12 h at 60 – 70°C. The mixture was then diluted with cold water (80 ml) and acidified with 5% hydrochloric acid to pH 4. The aqueous solution was extracted with CH2Cl2 (two 50-ml portions), washed with 5% hydrochloric acid and water, dried, and evaporated. The product (1.0 g) was recrystallized from aqueous EtOH. The yield was 0.61 g of VI (65.6%) (cream-colored powder). The melting point was >260°C. [α D 20 ] +18° (c 0.04, MeOH). R f = 0.63 (A). The IR spectrum, ν, cm−1, was: 1680, 1620, 1600, 1510 (Ph). The UV spectrum, λ 25%EtOHmax (lg ε), was: 246 nm (4.86). The 1H NMR spectrum (300 MHz, CDCl3, δ, ppm), was: 0.70; 0.80; 0.90; 0.96; 1.12; 1.20; 1.32 (all s, 21 H, 7 CH3); 3.48 (s, H3, 1H); 5.58 (H12, 1 H); 6.24; 6.30; 6.64; 6.70; 6.86; 7.24; 7.42; 7.62; 7.68; 7.70; 7.72 (m, arom. H). The 13C NMR spectrum (75.5 MHz, CDCl3, δ, ppm), was: 39.1 (C1); 90.0 (C3); 40.3 (C4); 55.3 (C5; 5CH3O cinnamate); 44.9 (C8); 63.9 (C9); 36.5 (C10); 199.9 (C11); 124.0 (C12); 166.6 (C13); 43.7 (C14); 26.6 (C16); 42.3 (C18); 31.8 (C21); 37.6 (C22); 28.4 (C23); 16.5; 15.9 (C24; C25); 18.4 (C26); 100.2 (C1’); 102.4 (C1”); 81.0 (C2’); 73.5; 72.0; 71.0; 68.0; 67.5 (C4’; C4”; C3’; C3”; C2”); 77.2 (C5’; C5”); 171.6 (C6’; C6”); 161.6; 160.7; 146.6; 142.2; 129.8; 129.2; 128.2; 126.9; 115.2; 114.9; 114.3; 114.2 (arom. C). C92H102O26.
Experimental biological section
The anti-HIV activity of I and its esters III and IV was studied using the traditional model based on primary infection of MT-4 lymphoid cells with HIV (an acute HIV infection model) using strain HIV/EVK [21]. The reference agent was GA at a concentration of 100 μg/ml. The cytotoxicity of the study compounds was assessed in cultures of transplantable MT-4 human T-lymphocytes. Compounds were dissolved in DMSO and dilutions were added to the wells of 96-well plates (three for each dilution) on cell seeding. After incubation, the proportions of viable cells were measured in a Goryaev chamber after staining with trypan blue. Dose-response curves were plotted and the concentrations of compounds causing death of 50% of the cells (CD50, toxic dose) were determined.
The anti-HIV activity of the compounds was estimated by infecting MT-4 cells (2 × 106 cells/ml) with HIV/EVK at a multiplicity of infection of 0.2 – 0.5 infectious units/cell. After virus adsorption for 1 h at 37°C, infected and control cells (without virus) were diluted with culture growth medium to a concentration of 5 × 10 cells/ml and added to the wells of 96-well culture plates. The appropriate wells were then supplemented with solutions of the study compounds (three for each dilution). The final concentrations of study compounds in cell suspensions ranged from 0.1 to 100 μg/ml.
The inhibitory effects of compounds were assessed on day 4 of culture by measuring the quantity of viral antigen - virus-specific p24 protein – using an immunoenzyme method. In addition, the proportions of viable cells were measured after staining with trypan blue by counting in a Goryaev chamber. The experimental data were used to plot dose-response curves and to identify the quantitative characteristics of inhibition in terms of the ID50, i.e., the concentration of compounds suppressing virus production by 50% or providing 50% protection of cells from death due to infection, the ID90, i.e., the concentration of compounds suppressing virus production by 90% or providing 90% protection of cells from death due to infection, and the IS, i.e., the index of selectivity, which was the ratio of the toxic dose CD50 to the effective dose ID50.
This study was supported by the Russian Foundation for Basic Research (Grant No. 08-03-13512 ofi-ts).
References
G. A. Tolstikov, L. A. Baltina, V. P. Grankina, et al., Licorice: Biodiversity, Chemistry, and Use in Medicine [in Russian], Geo Academic Press, Novosibirsk (2007).
G. A. Tolstikov, L. A. Baltina, É. É. Schul’ts, and A. G. Pokrovskii, Bioorgan. Khim., 23, 691–709 (1997).
R. Pompei, O. Flore, M. Marcialis, A. Pani, B. Loddo, Nature, 281, 689–690 (1979).
M. Baba, Sh. Shigeta, Antiviral Res., 7, 99–107 (1987).
T. Okamoto, K. Kojino, O. Hino, Jpn. J. Pharmacol., 87, 177–180 (2001).
J.-M. Crance, F. Leveque, E. Biziagos, et al., Antiviral. Res., 23, 63–76 (1994).
H. Sato, W. Goto, J.-I. Yamamura, et al., Antiviral. Res., 30, 171–177 (1996).
Y. Arase, K. Ikeda, N. Murashima, et al., Cancer, 79, 1494–1500 (1997).
K. Numazaki, N. Nagata, T. Sato, S. Chiba, J. Leukocyte Biol., 5, 24–28 (1994).
C. Ceremelli, M. Portolani, B. Cotombari, et al., Phytochem. Res., 10, 527–528 (1996).
J.-C. Lin, Antiviral Res., 59, 41–47 (2003).
J. Cinatl, B. Morgenstern, G. Bauer, et al., Lancet, 361, 2045–2046 (2003).
S. Saito, T. Furumoto, A. Hosono, et al., Eur. J. Med. Chem., 31, 365–381 (1996).
A. G. Pokrovskii, O. A. Plyasunova, L. A. Baltina, et al., USSR patent 1804848 (1993).
O. A. Plyasunova, T. V. Il’ina, Ya. Yu. Kiseleva, et al., Vestn. Ros. Akad. Med. Nauk, 11, 42–46 (2004).
Kh. M. Nasyrov, D. N. Lazareva, Farmakol. Toksikol., No. 1, 84–88 (1984).
L. S. Gromakova, Author’s Abstract of Master’s Thesis in Medical Sciences [in Russian], Kazan’ (1995).
S. A. Shibata, J. Pharm. Soc. Jpn., 120, 849–862 (2000).
L. A. Baltina, N. G. Serdyuk, O. B. Flekhter, et al., Khim.-Farm. Zh., 30(10),8–11 (1996).
A. Gordon, R. Ford, The Chemist’s Companion [Russian translation], Mir, Moscow (1976).
O. A. Plyasunova, I. N. Egoricheva, N. V. Fedyuk, et al., Vopr. Onkol.,No.5–6,235–238 (1992).
Author information
Authors and Affiliations
Additional information
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 44, No. 6, pp. 15 – 18, June, 2010.
Rights and permissions
About this article
Cite this article
Baltina, L.A., Stolyarova, O.V., Baltina, L.A. et al. Synthesis and antiviral activity of 18α-glycyrrhizic acid and its esters. Pharm Chem J 44, 299–302 (2010). https://doi.org/10.1007/s11094-010-0454-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-010-0454-1