
Abstract
A high yield of isolated protoplasts and efficient regeneration protocols are prerequisites for successful development of somatic hybrids. In the present study, protoplast isolation and regeneration were evaluated in 12 Kalanchoë accessions belonging to nine species. The highest protoplast yield was obtained from K. blossfeldiana ‘Charming Red Meadow’ with 10.78 ± 0.51 × 105 protoplasts per gram fresh weight. We observed significant differences of protoplast yield while there was no distinct difference in viability among the accessions. Seven accessions reached the microcolony stage and four developed microcalli in medium supplemented with 1.0 mg/l 1-naphthaleneacetic acid (NAA), 0.5 mg/l 6-benzylaminopurine (BAP) and 0.5 mg/l 2,4-dichlorophenoxy acetic acid (2,4-D). Using five selected accessions we optimized the PGR (plant growth regulators) concentrations using combinations of NAA, BAP and 2,4-D. K. blossfeldiana cultivars ‘Charming Red Meadow’ and ‘Paris’ produced significantly different numbers of calli depending on the PGR concentrations. For plant regeneration, the medium was supplemented with 1 mg/l NAA and 2 mg/l BAP or 2 mg/l zeatin. Shoots were regenerated on medium supplemented with NAA and BAP for K. blossfeldiana ‘Charming Red Meadow’ and K. blossfeldiana ‘Paris’. The plants successfully developed roots on the medium supplemented with IAA. The medium containing zeatin induced root formation directly from callus in K. blossfeldiana ‘Charming Red Meadow’. Our findings have the potential to facilitate the use of Kalanchoë species in somatic hybridization breeding programs.
Key Message
The study revealed a strong genotype-dependent efficiency of colony and microcallus formation. Plants were regenerated from two Kalanchoë blossfeldiana cultivars on medium supplemented with NAA and BAP.
Similar content being viewed by others


Introduction
Kalanchoë is a widespread flowering plant genus belonging to the Crassulaceae family. It comprises around 140 species that are mainly succulent plants native to Madagascar and East and South Africa (Descoings 2003). Kalanchoë blossfeldiana is the most exploited species of the genus and it gave rise to the majority of commercial cultivars. Kalanchoë cultivars are widely used as potted plants due to plentiful flowering, longevity of individual flowers, postharvest performance, and low-care requirements (Mackenzie et al. 2018). Kalanchoë is the second most economically important potted plant in Europe with a turnover of 69 million euros in 2017 (Royal Flora Holland 2018). The genus is also increasingly utilized as cut flowers (Mackenzie et al. 2018). Breeding of new cultivars started as early as 1930s aiming at the development of compact cultivars with various flower colours. Currently, the cultivar improvement is achieved using cross-breeding and interspecific hybridization (Izumikawa et al. 2008; Kuligowska et al. 2015a), though genetic engineering has also been reported (Lütken et al. 2011). The expansion of the gene pools and generation of new Kalanchoë cultivars using genetic resources of wild species is of great interest (Mackenzie et al. 2018). Successful interspecific hybridization was accomplished within the Kalanchoë genus (Izumikawa et al. 2008; Kuligowska et al. 2015a; Huang and Chu 2017), however, development of hybrids is limited due to hybridization barriers related to sexual reproduction (Kuligowska et al. 2015b). Fusion of protoplasts from different species provides a practical breeding tool that can bypass sexual hybridization-related barriers. Nevertheless, development of an efficient protoplast-to-plant system is often a limiting step in somatic hybridization (Eeckhaut et al. 2013). Protoplast regeneration can be influenced by a number of factors, including donor plant material and genotype, culture system and media composition. In particular, the concentration and ratios of main plant growth regulators, auxins and cytokinins are of key importance (Davey et al. 2005). Thus, to obtain a high regeneration frequency, the protoplast culture conditions need to be optimized.
Previous studies of Kalanchoë in vitro cultures documented shoot regeneration using different explants and hormone combinations. Varga et al. 1988 examined the effect of auxins and cytokinins in K. blossfeldiana (indole-3-acetic acid—IAA, indole-3-butyric acid—IBA, zetin, kinetin and N6-(2-Isopentenyl)adenine—2iP) on shoot regeneration from leaf-derived callus. Khan et al. (2006) evaluated effects of BAP, NAA and PGR-free medium on direct regeneration of Kalanchoë tomentosa from shoot tips. The successful shoot regeneration from leaf and internode explants using thidiazuron (TDZ) was also reported in K. blossfeldiana (Sanikhani et al. 2006; Lütken et al. 2011). Furthermore, one study reported regeneration of plants from protoplasts of a single K. blossfeldiana cultivar (Castelblanque et al. 2010). Electric fusion was reported between isolated vacuoles and protoplasts of K. daigremontiana (Vienken et al. 1981), while neither chemical fusion nor successful somatic hybridization was obtained in Kalanchoë genus.
In the present study we present a modified protocol for isolation of mesophyll protoplasts from nine Kalanchoë species. The results reveal the regeneration ability of several Kalanchoë species and the role of plant growth regulators in protoplast regeneration.
Materials and methods
Plant material
Nine Kalanchoë species were used in the present experiments: K. aromatica, three K. blossfeldiana cultivars: ‘Charming Red Meadow’, ‘Charlie’ and ‘Paris’, K. gracilipes, K. marnieriana, K. miniata, K. pinnata wild type (WT) and a root oncogenic loci (rol)—gene transformed type (unpublished), K. pumila, K. streptantha and K. rotundifolia. The plant cuttings were established in peat soil (NPK 14-7-15 0.650 kg per m3 + micro 50 g per m3, soil type: ‘Pindstrup Substrate No. 1’, Pindstrup Mosebrug A/S, Ryomgård, Denmark) in 11 cm diameter pots and grown in a greenhouse with a day/night temperature of 24 ± 4 °C/18 ± 4 °C and a photoperiod of 16 h. Additional illumination of 260 μmol s−1 m−2 supplied by Philips Master SON-T PIA Green Power 400 W, was provided when natural light irradiance was below 180 μmol s−1 m−2. Ebb-flow irrigation was performed with water containing fertilizer (Pioneer NPK Macro 14-3-23 + Mg combined with Pioneer Micro; pH 5.5; EC = 1.3, Azelis, Antwerp, Belgium) once a week.
Axenic plant cultures
Stem segments with axillary buds were used as in vitro explant source. The plant stems were collected from 1- to 2-month-old plants and surface sterilized according to Lütken et al. (2011). Explants of 3 ± 0.2 cm were cultured on MS medium (Murashige and Skoog 1962) containing 30 g/l sucrose, 0.5 g/l 2-(N-morpholino)ethanesulfonic acid (MES), 0.66 g/l CaCl2, 0.08 g/l Ethylenediamino-N,N′-bis(2-hydroxy-phenyl)acetic acid iron (EDDHA-Fe), 1 ml/l Atamon™ (sodium benzoate), 100 mg/l timentin, 0.05 mg/l IAA and 3 g/l gelrite. The pH was adjusted to 5.8 prior to autoclaving. All cultures were maintained in a growth chamber at 24 °C with a 16 h photoperiod and a light intensity of 150 μmol s−1 m−2. Plants were subcultured monthly/every second month depending on the cultivar, and maintained as a source of leaf material for protoplast isolation.
Protoplast isolation
Protoplasts were isolated from young fully expanded leaves of in vitro plants. One gram of leaf tissue was cut into 1–2 mm stripes in 10 ml of washing medium (WM: 1.65 g/l MS macro elements, 106 g/l mannitol, 7 g/l glycine, 1.5 g/l CaCl2, 0.5 g/l MES, pH 5.8, autoclaved) in 5 cm Petri dishes. After 30 min incubation, the WM solution was replaced with filter-sterilized (0.2 μm, Whatman, International Ltd., Maidstone, Kent, UK) enzymatic solution containing 0.5% (w/v) Cellulase Onozuka R-10 (Duchefa Biochemie, Haarlem, The Netherlands) and 0.1% (w/v) Driselase (Sigma-Aldrich, St. Louis, US) dissolved in WM. Digestion was performed at room temperature in darkness with shaking at 40 rpm for 16-18 h. The protoplast suspension was filtered through 4-layer sterile cheesecloth and centrifuged at 100×g for 10 min. The protoplast pellet was suspended in 10 ml flotation medium (FM: 205 g/l sucrose, 0.64 g/l MES, pH 6.0, filter-sterilized). Protoplasts were purified by density gradient centrifugation at 50×g for 10 min, after layering 1 ml of modified W5 (Menczel et al. 1981) (0.46 g/l sucrose, 18.38 g/l CaCl2·2H2O, 0.37 g/l KCl, 9.076 g/l NaCl, pH 5.8, autoclaved) on the FM. The collected protoplasts were suspended in 10 ml W5 and centrifuged at 100×g for 10 min to collect protoplasts.
Yield and viability assessment
The purified protoplasts were counted using a Fuchs-Rosenthal haemocytometer chamber. The yield was expressed as number of protoplasts per gram fresh weight (gfw). The protoplast viability was determined by fluorescein diacetate (FDA) staining according to Widholm (1972). For viability assessment, 0.5 μl FDA solution (25 mg FDA dissolved in 5 ml acetone, Sigma-Aldrich, St. Louis, USA) was added to 100 μl protoplast suspension. Protoplasts were observed under a fluorescence microscope (Leica DM2000 LED, Leica, Germany; excitation filter BP 450–490 nm, dichromatic mirror: 510 nm and emission filter: LP 515 nm) equipped with a digital camera (Leica DFC420, Leica, Germany). The protoplast viability was determined as the ratio between the green fluorescent protoplasts and the total number of observed protoplasts. The mean protoplast yield and viability were calculated based on two independent isolations per genotype. At least three microscopic fields per isolation with minimum 50 protoplasts were screened.
Protoplast culture
Protoplasts were suspended in culture medium 1 (M1—Table 1) and cultured at a concentration of 105 protoplasts ml−1 in 1 ml or 2.5 ml depending on the experiment. Cultures were refreshed weekly by adding 0.4 volume of fresh medium with lower mannitol concentration (M2—Table 1). After 4 weeks, the culture medium was replaced by the same volume of medium 3 (M3—Table 1). In M3, the cultures were first cultured for 2 weeks in dark, followed by 2 weeks of 16 h photoperiod. Cultures were inspected regularly under a microscope (Leica DM2000 LED, Leica, Germany) for cell division and microcolony formation. The number of microcalli (1–2 mm diameter) was determined and they were transferred using tweezers to 5 cm Petri dishes containing solid medium 4 (M4—Table 1). The developed shoots were separated from organogenic callus and cultured on solid medium 5 (M5—Table 1). The media composition, culture conditions and culture duration are shown in Table 1. PGR concentrations differed between experiment 1 and 2 and are shown in Table 2 and 3.
Experiment 1: Protoplast yield and viability screening
Nine Kalanchoë species (12 accessions) described above were used. The protoplast isolation, yield and viability assessment were performed as described above using 0.5 g leaf material per isolation. The protoplast cultures were established with 2.5 ml of culture per 5 cm Petri dish. The media were as described in Table 1 and they were supplemented with PGR according to Table 2. The protoplast cultures were observed daily during the first week and once a week during the remaining culture period. Three independent isolations were carried out for each plant accession. The mean values and standard deviations were calculated. Analysis of variance (ANOVA) and Tukey’s honestly significant difference (HSD) tests were performed using SPSS 9.1 (SPSS Institute, Chicago, USA) to assess the effect of the treatments.
Experiment 2: Optimization of PGR concentrations
To determine the effect of plant PGR on protoplast regeneration, protoplast cultures with 12 different PGR concentrations and one control (without PGR) were established using five Kalanchoë accessions: K. blossfeldiana ‘Charming Red Meadow’, ‘Charlie’ and ‘Paris’, K. marnieriana and K. pinnata WT. The protocols for protoplast isolation and yield assessment were as described above. The quantity of leaf material for protoplast isolation was determined based on the result of experiment 1 to yield 39 × 105 protoplasts. The protoplast cultures were set up in 35 mm Petri dishes with 1 ml of protoplast culture at a density of 105 protoplasts ml−1. The cultures were performed under the conditions described in Table 1. The media were supplemented with PGR concentrations according to Table 3. Each treatment consisted of 3 technical replicates (Petri dish) and was repeated twice. After 5 weeks in solid medium M4, if no shoot or root formation was observed, the calli were maintained on M4 for an additional 4 weeks. Analysis of variance (ANOVA) and Tukey’s honestly significant difference (HSD) tests were performed using SPSS 9.1 (SPSS Institute, Chicago, USA) to assess the effect of the treatments.
Results
Experiment 1: Protoplast yield and viability screening
The leaves of in vitro plants were used for protoplast isolation when they had developed two new leaf pairs. The time required to reach this developmental stage was between 4 and 6 weeks depending on the accessions (Supplementary Fig. 1). Viable protoplasts were isolated from all 12 accessions. They were spherical, with a diameter between 10 and 70 μm. Different protoplast types were observed following isolation (Fig. 1a-I–IV). The protoplast yield varied significantly among examined plants. The lowest yield was obtained for K. rotundifolia (0.52 ± 0.09 × 105 protoplasts per gfw), while the highest yield was obtained for K. blossfeldiana ‘Charming Red Meadow’ (10.78 ± 0.51 × 105 protoplasts per gfw) (Fig. 2). The protoplast viability did not differ significantly among accessions (Fig. 2).

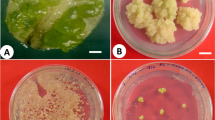
Plant regeneration from protoplasts of K. blossfeldiana ‘Charming Red Meadow’ a different types of isolated protoplast a-I: small protoplast without chloroplasts; a-II: dense protoplast with chloroplasts concentrated in the middle of the cell; a-III: dense protoplast with chloroplasts distributed homogenously throughout the cytoplasm; a-IV: vacuolated protoplast with chloroplasts concentrated in one zone of the cell; b protoplast division at 3 days after culture initiation; c microcolonies 2 weeks after culture initiation; d microcolonies 4 weeks after culture initiation; e green microcallus 8 weeks after culture initiation; f yellow microcallus 8 weeks after culture initiation; g shoot regenerated on the callus at 17 weeks; h regenerated plants at 22 weeks; scale bars: a, b 25 µm; c, d 50 µm; e–h 1 mm

Protoplast yield and viability of 12 Kalanchoë accessions in experiment 1; bars represent mean ± SD, n = 3; yield values followed by different letters are significantly different (p ≤ 0.05) according to Tukey’s honestly significant difference test; gfw: gram fresh weight; K. b.: K. blossfeldiana; ‘Ch. R. Meadow’: ‘Charming Red Meadow’
Protoplasts of all accessions regenerated cell walls in vitro within 24 h. The first cell divisions (Table 4) were observed from 3 days to 7 days in all accessions. Microcolonies developed after 2–4 weeks from protoplasts of all K. blossfeldiana cultivars, K. marnieriana, K. pinnata WT, K. rotundifolia and K. streptantha (Table 4). After 8 weeks of culture, visible microcalli of 1–2 mm were obtained from K. blossfeldiana ‘Charming Red Meadow’ and ‘Paris’, K. pinnata WT and K. rotundifolia (Table 4). K. rotundifolia had the highest calli yield, followed by K. blossfeldiana ‘Charming Red Meadow’, K. pinnata WT and K. blossfeldiana ‘Paris’ (Table 4). The calli were further transferred to the solid medium (Table 1 and 2). However, after 3 weeks they showed no growth, thus the culture was terminated.
Experiment 2: Optimization of PGR concentrations
Based on the results from experiment 1, the plants with high protoplast yield: K. blossfeldiana ‘Charming Red Meadow’, ‘Paris’, ‘Charlie’, K. pinnata WT and K. marnieriana were included in the optimization of culture conditions for regeneration. In experiment 2, the highest yield was observed for K. pinnata WT at 7.57 ± 0.86 x 105 protoplasts per gfw. K. marnieriana had the lowest yield at 2.56 ± 0.60 × 105 protoplasts per gfw (Fig. 3).

Protoplast yield in experiment 2; bars represent mean ± SD, n = 3; bars followed by different letters are significantly different (p ≤ 0.05) according to Tukey’s honestly significant difference test; gfw: gram fresh weight; K. b.: K. blossfeldiana; ‘Ch. R. Meadow’: ‘Charming Red Meadow’
The culture medium in experiment 2 was supplemented with a tenfold lower NAA concentration than in experiment 1, and different concentrations of BAP and 2, 4-D (Table 3). All protoplasts cultured on medium without PGR died within 3 days. In the cultures supplemented with PGR, protoplasts of K. pinnata WT and K. marnieriana rebuilt a cell wall and underwent first divisions, but did not develop further. K. blossfeldiana ‘Charlie’ protoplasts formed microcolonies, but they stopped their development in the fifth week of culture after transfer to 16 h photoperiod. K. blossfeldiana ‘Charming Red Meadow’ and ‘Paris’ protoplasts exhibited stable mitosis and developed microcalli that were transferred to solid medium after 8 weeks (Fig. 1b–f). The K. blossfeldiana ‘Charming Red Meadow’ calli obtained in both repetitions of experiment 2 followed a similar pattern. The lowest number of calli was obtained in media supplemented with 0.05 mg/l BAP, and the highest number of calli was obtained in media supplemented with 0.5 mg/l BAP (Fig. 4a and b). The results of two-way ANOVA (Table 5) showed that the BAP concentration had a significant influence on the calli number. Moreover, the interaction between the effect of BAP and the effect of 2,4-D was statistically significant. The effect of 2,4-D was significant only in repetition 1 (Table 5).

Number of calli after 8 weeks according to the PGR concentrations in experiment 2 a and b repetition 1 and 2 for K. blossfeldiana ‘Charming Red Meadow’; c and d repetition 1 and 2 for K. blossfeldiana ‘Paris’; bars represent mean ± SD, n = 3; bars followed by different letters are significantly different (p ≤ 0.05) according to Tukey’s honestly significant difference test
The K. blossfeldiana ‘Paris’ protoplast derived calli number differed between repetition 1 and 2. In repetition 1, the lowest calli numbers were obtained in media supplemented with 0.05 mg/l BAP. Repetition 2 showed higher calli numbers. The results of two-way ANOVA revealed that the concentration of BAP had significant influence on the calli numbers in both repetitions. In repetition 1, there was a significant interaction between the effect of BAP and 2,4-D on the calli number while 2,4-D had no significant effect. In repetition 2 however, 2,4-D had significant influence on the calli numbers, but no interaction was observed between the effect of BAP and 2,4-D (Table 5).
For plant regeneration, all calli were transferred to solid medium containing the same PGR as in the initial cultures. The total and average numbers of calli as well as the percentage of green and yellow calli (Fig. 1e and f) after 8 and 13 weeks of culture are shown in Supplementary Table 1 and 2. After 13 weeks of culture, the calli reached 0.5–0.8 cm in diameter. The biggest (at least 7 mm) calli were cut and all calli were divided between two media supplemented with either NAA and BAP, or NAA and zeatin (Supplementary Table 3 and 4). After 17 weeks of culture of K. blossfeldiana ‘Charming Red Meadow’, roots were regenerated on the medium supplemented with NAA and zeatin, from initial PGR treatment F (0.1 mg/l NAA, 0.1 mg/l BAP and 0.1 mg/l 2,4-D) in repetition 1 and 2, and from treatment L (0.1 mg/l NAA, 0.5 mg/l BAP and 1.0 mg/l 2,4-D) in repetition 2 (Table 6). These roots failed to regenerate into plants after further transfer. Shoots (Fig. 1g) were obtained from green calli of K. blossfeldiana ‘Charming Red Meadow’ on the medium containing NAA and BAP from initial PGR treatment F (0.1 mg/l NAA, 0.1 mg/l BAP and 0.1 mg/l 2,4-D) in repetition 1, and treatment L (0.1 mg/l NAA, 0.5 mg/l BAP and 1.0 mg/l 2,4-D) in repetition 2 (Table 6). All shoots successfully developed into plants (Fig. 1h). The callus regeneration from K. blossfeldiana ‘Paris’ yielded shoots only in repetition 2 on medium containing NAA and BAP from initial treatment L (0.1 mg/l NAA, 0.5 mg/l BAP and 1.0 mg/l 2,4-D) (Table 6). The plants successfully developed roots on the medium supplemented with IAA.
Discussion
Constant development of new decorative cultivars is a prerequisite for breeders and producers to be competitive within floriculture. Interspecific hybrids are important breeding material that can provide novel traits for commercial ornamental plants (Kuligowska et al. 2016). They can be obtained through sexual hybridization or somatic hybridization. The second method provides tools to avoid hybridization barriers related to sexual reproduction and can allow development of hybrids from more distantly related species (Eeckhaut et al. 2013; Kuligowska et al. 2016). The ability to isolate high numbers of healthy protoplasts enables a successful protoplast culture and development of an efficient protoplast-to-plant system (Rahmani et al. 2016). However, its establishment is influenced by many factors, among others: genotype, physiological status and growing conditions of the protoplast source tissue, protoplast isolation and medium composition (Davey et al. 2005; Eeckhaut et al. 2013).
In the present study, we successfully isolated protoplasts from 12 Kalanchoë accessions. A previous study of protoplast cultures in Kalanchoë reported a maximal yield of 6.0 × 105 protoplasts per gfw from K. blossfeldiana ‘Tenorio’ (Castelblanque et al. 2010). However, our preliminary studies did not produce satisfactory results following the described isolation protocol (data not shown). Using a newly developed isolation protocol based on Duquenne et al. (2007), four out of 12 Kalanchoë accessions showed a yield higher than 7.0 x 105 protoplasts per gfw, with the highest yield of 10.78 ± 0.51 × 105 protoplasts per gfw obtained in K. blossfeldiana ‘Charming Red Meadow’. Compared to Castelblanque et al. (2010) we were able to increase the duration of enzymatic digestion from 4 h to overnight by increasing the osmotic pressure of enzyme solution (change of mannitol concentration) and modifying enzyme concentrations. Despite a great increase of protoplast yield in some of the tested accessions, several plant accessions yielded significantly less protoplasts compared to the protoplast yield obtained by Castelblanque. Similar to our findings, Maćkowska et al. (2014) found that commercial plants exhibit higher protoplast yields than wild species. A plausible reason for differences in protoplast yields in our study is the leaf morphology of the tested species. High protoplast yields were generally obtained from plants with large, flat and thin leaves (K. blossfeldiana, K. pinnata). The low protoplast yields were observed in plants with small curly leaves or leaves covered with hair or wax (K. aromatica, K. pumila, K. rotundifolia). Moreover, the low protoplast yield was due to incomplete tissue digestion, suggesting that leaf morphology, at least partly disabled leaf tissue penetration by the enzymes. This assumption can be supported by the lack of differences in protoplast viability among different plant accessions. Decreased protoplast yield observed in experiment 2 (started in December 2017) compared to values obtained in experiment 1 (started in September 2017) can be attributed to seasonal variation (Keskitalo 2001; Maćkowska et al. 2014). The physiological status of the source tissue is important for successful protoplast isolation (Davey et al. 2005; Lord and Gunawardena 2010). In the present study, plants with fast in vitro growth generally yielded more protoplasts. Plants between 4 and 6 weeks were suitable for protoplast isolation. In contrast, a previous study with K. blossfeldiana ‘Tenorio’ used 8-week-old plants for protoplast isolation (Castelblanque et al. 2010).
Development of an efficient protoplast regeneration method relies on multiple parameters related to the culture conditions in several consecutive steps. Optimization of in vitro culture requires empirical testing of different parameters such as media composition, PGR supplementation or cultivation conditions (Davey et al. 2005). In the present study, we used medium according to Kao and Michayluk (1975) for protoplast culture. In a preliminary study using K. blossfeldiana ‘Charming Red Meadow’ and K. pinnata WT we compared KAO medium and MS-based medium according to Castelblanque et al. (2010). We only observed protoplast divisions using the first medium (data not shown). Auxins and cytokinins are the major growth regulators of sustained protoplast growth (Davey et al. 2005). In experiment 1, we used the PGR supplementation described by Castelblanque et al. (2010). The protoplast proliferation and regeneration showed genotype specific differences, as previously observed in other plant species (Meyer et al. 2009; Maćkowska et al. 2014; Bruznican et al. 2017). Even though all tested plants underwent the first protoplast divisions and we observed microcolony and even microcallus formation in some accessions, we were not able to regenerate plants from the obtained callus. We hypothesized that the relatively high auxin concentration can contribute to callus recalcitrancy. Thus, in experiment 2 the NAA concentration was lowered to 0.1 mg/l, and we tested wider ranges of 2,4-D (from 0.05 to 1.0 mg/l) and BAP (from 0.05 to 0.5 mg/l) concentrations. Compared to experiment 1, in experiment 2 we generally obtained more calli for both K. blossfeldiana ‘Charming Red Meadow’ and ‘Paris’. These results indicate that a lower NAA concentration had a positive effect on callus formation in K. blossfeldiana. However, in K. pinnata WT, calli were obtained only in experiment 1. Thus, it is likely that this species requires high NAA concentration for callus formation. In the experiment 2, we observed the differences in number of calli between two repetitions. However, we observed highly significant BAP effect on calli yield in both repetitions, while 2,4-D effect and PGR interaction effect were not consistent. This can be due to the above mentioned physiological status of the source tissue. Moreover, it is likely that higher number of technical repeats are needed for clear assessment of PGR effects.
For plant regeneration from protoplast-derived callus, we used media supplemented with combinations of either NAA and BAP, or NAA and zeatin. NAA and BAP induce shoot regeneration from protoplast cultures of K. blossfeldiana (Castelblanque et al. 2010) and Robinia pseudoacacia (Kanwar et al. 2009). Zeatin is frequently combined with NAA and used in protoplast regeneration protocols for shoot induction e.g. in Solanum (Borgato et al. 2007) and Albizia julibrissin (Rahmani et al. 2016). Moreover, zeatin has a positive effect on shoot regeneration from leaf-derived callus of K. blossfeldiana (Varga et al. 1988). However, in the present study contrary to the expected results of the of shoot development, medium supplemented with zeatin promoted root development in K. blossfeldiana ‘Charming Red Meadow’. The promotive effect of zeatin on root development was previously described in in vitro cultures of Vaccinium vitis-idaea and Fragaria ananassa (Debnath 2005, 2006). NAA and BAP were able to induce healthy shoots from both K. blossfeldiana cultivars and further plants were regenerated.
In the future research further efforts are needed to accomplish the increase in the shoot regeneration efficiency. The optimal medium supplementation with PGRs for shoot regeneration from protoplast-derived callus varies depending on the plant species and even genotypes (Davey et al. 2005; Rahmani et al. 2016). Sanikhani et al. (2006) and Lütken et al. (2011) reported successful shoot regeneration in Kalanchoë blossfeldiana by TDZ. Furthermore, shoot differentiation was induced from protoplast-derived callus in the presence TDZ in Ulmus americana (Jones et al. 2015). Thus, testing a wider range of PGRs including TDZ for shoot induction in Kalanchoë should be considered. Previously reported protoplast-to-plant system for Kalanchoë mesophyll protoplasts (Castelblanque et al. 2010) as well as this study describe plant regenration only via shoot organogenesis. Chao et al. (2004) described somatic embryogenesis induction in K. blossfeldiana on MS medium supplemented with NAA, BAP and activated carbon. Development of efficient regeneration system through indirect somatic embryogenesis could provide an alternative route to obtain high shoot regeneration efficiency in Kalanchoë. Sucrose is a regular choice of carbon source in the most protoplast cultivation media (Davey et al. 2005). However, a change in the carbon source from sucrose to glucose or maltose could promote shoot regeneration for protoplast-derived cells as demonstrated in case of Lilium and rice (Godo et al. 1996; Jain et al. 1995).
In conclusion, our work presents an efficient protocol of protoplast isolation from fast growing Kalanchoë species. The results demonstrate that protoplast yield, but not viability can be influenced by the genotype of donor plant and their growth in vitro. The PGR concentration used during protoplast culture is critical for callus development. However, the efficiency of colony and microcalli formation is strongly genotype-dependent. Despite the high number of obtained calli, the shoot regeneration rate was low. Further research is needed to optimize this final step of a protoplast-to-plant regeneration system in Kalanchoë.
Abbreviations
- 2,4-D:
-
2,4-Dichlorophenoxy acetic acid
- BAP:
-
6-Benzylaminopurine
- FDA:
-
Fluorescein diacetate
- gfw:
-
Gram fresh weight
- IAA:
-
Indole-3-acetic acid
- MES:
-
2-(N-Morpholino) ethane sulfonic acid
- MS:
-
Murashige and Skoog
- NAA:
-
1-Naphthaleneacetic acid
- PGR:
-
Plant growth regulators
References
Borgato L, Pisani F, Furini A (2007) Plant regeneration from leaf protoplasts of Solanum virginianum L. (Solanaceae). Plant Cell Tissue Org 88(3):247–252
Bruznican S, Eeckhaut T, Van Huylenbroeck J, De Clercq H, Geelen D (2017) Regeneration of cell suspension derived Apium graveolens L. protoplasts. Plant Cell Tissue Org 131(1):163–174
Castelblanque L, García-Sogo B, Pineda B, Moreno V (2010) Efficient plant regeneration from protoplasts of Kalanchoe blossfeldiana via organogenesis. Plant Cell Tissue Org 100(1):107–112
Chao C, Guilan W, Limin T, Ruisheng C (2004) Embryoid induction and regeneration in callus of Kalanchoe blossfeldiana. Acta Hortic Sin 31(2):249–252
Davey MR, Anthony P, Power JB, Lowe KC (2005) Plant protoplasts: status and biotechnological perspectives. Biotechnol Adv 23(2):131–171
Debnath SC (2005) Micropropagation of lingonberry: influence of genotype, explant orientation, and overcoming TDZ-induced inhibition of shoot elongation using zeatin. HortScience 40(1):185–188
Debnath SC (2006) Zeatin overcomes thidiazuron-induced inhibition of shoot elongation and promotes rooting in strawberry culture in vitro. J Hortic Sci Biotechnol 81(3):349–354
Descoings B (2003) Kalanchoe. In: Eggli U (ed) Illustrated handbook of succulent plants: Crassulaceae. Springer, Berlin, pp 143–181
Duquenne B, Eeckhaut T, Werbrouck S, Van Huylenbroeck J (2007) Effect of enzyme concentrations on protoplast isolation and protoplast culture of Spathiphyllum and Anthurium. Plant Cell Tissue Org 91(2):165–173. https://doi.org/10.1007/s11240-007-9226-3
Eeckhaut T, Lakshmanan PS, Deryckere D, Van Bockstaele E, Van Huylenbroeck J (2013) Progress in plant protoplast research. Planta 238(6):991–1003
Godo T, Matsui K, Kida T, Mii M (1996) Effect of sugar type on the efficiency of plant regeneration from protoplasts isolated from shoot tip-derived meristematic nodular cell clumps of Lilium x formolongi hort. Plant Cell Rep 15(6):401–404
Huang C-H, Chu C-Y (2017) Inheritance of leaf and flower morphologies in Kalanchoe spp. Euphytica 213(1):4
Izumikawa Y, Takei S, Nakamura I, Mii M (2008) Production and characterization of inter-sectional hybrids between Kalanchoe spathulata and K. laxiflora (= Bryophyllum crenatum). Euphytica 163(1):123–130
Jain RK, Khehra GS, Lee SH, Blackball NW, Marchant R, Davey MR, Power JB, Cocking EC, Gosal SS (1995) An improved procedure for plant regeneration from indica and japonica rice protoplasts. Plant Cell Rep 14:515–519
Jones AMP, Shukla MR, Biswas GCG, Saxena PK (2015) Protoplast-to-plant regeneration of American elm (Ulmus americana). Protoplasma 252(3):925–931
Kanwar K, Bhardwaj A, Deepika R (2009) Efficient regeneration of plantlets from callus and mesophyll derived protoplasts of Robinia pseudoacacia L. Plant Cell Tissue Org 96(1):95–103. https://doi.org/10.1007/s11240-008-9465-y
Kao K, Michayluk M (1975) Nutritional requirements for growth of Vicia hajastana cells and protoplasts at a very low population density in liquid media. Planta 126(2):105–110
Keskitalo M (2001) Can protoplast production from in vitro cultured shoots of Tanacetum vary during the season? Agric Food Sci Finl 10:145–151
Khan S, Naz S, Ali K, Zaidi S (2006) Direct organogenesis of Kalanchoe tomentosa (Crassulaceae) from shoot-tips. Pak J Bot 38(4):977
Kuligowska K, Lütken H, Christensen B, Müller R (2015a) Quantitative and qualitative characterization of novel features of Kalanchoë interspecific hybrids. Euphytica 205(3):927–940
Kuligowska K, Lütken H, Christensen B, Skovgaard I, Linde M, Winkelmann T, Müller R (2015b) Evaluation of reproductive barriers contributes to the development of novel interspecific hybrids in the Kalanchoë genus. BMC Plant Biol 15(1):15
Kuligowska K, Lütken H, Müller R (2016) Towards development of new ornamental plants: status and progress in wide hybridization. Planta 244(1):1–17
Lord CEN & Gunawardena AHLAN (2010) Isolation of leaf protoplasts from the submerged aquatic monocot Aponogeton madagascariensis. Am J Plant Sci Biotechnol 4(2):6–11
Lütken H, Laura M, Borghi C, Ørgaard M, Allavena A, Rasmussen SK (2011) Expression of KxhKN4 and KxhKN5 genes in Kalanchoë blossfeldiana ‘Molly’ results in novel compact plant phenotypes: towards a cisgenesis alternative to growth retardants. Plant Cell Rep 30(12):2267–2279. https://doi.org/10.1007/s00299-011-1132-9
Mackenzie KK, Lütken H, Coelho LL, Kaaber MD, Hegelund JN, Müller R (2018) Kalanchoë. In: Van Huylenbroeck J (ed) Ornamental crops. Handbook of plant breeding, vol 11. Springer, Cham, pp 453–479
Maćkowska K, Jarosz A, Grzebelus E (2014) Plant regeneration from leaf-derived protoplasts within the Daucus genus: effect of different conditions in alginate embedding and phytosulfokine application. Plant Cell Tissue Org 117(2):241–252. https://doi.org/10.1007/s11240-014-0436-1
Menczel L, Nagy F, Kiss ZR, Maliga P (1981) Streptomycin resistant and sensitive somatic hybrids of Nicotiana tabacum + Nicotiana knightiana: correlation of resistance to N. tabacum plastids. Theor Appl Genet 59(3):191–195
Meyer L, Serek M, Winkelmann T (2009) Protoplast isolation and plant regeneration of different genotypes of Petunia and Calibrachoa. Plant Cell Tissue Org 99(1):27–34
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15(3):473–497
Rahmani M-S, Pijut PM, Shabanian N (2016) Protoplast isolation and genetically true-to-type plant regeneration from leaf- and callus-derived protoplasts of Albizia julibrissin. Plant Cell Tissue Org 127(2):475–488
Royal Flora Holland (2018) Annual report 2017. https://www.royalfloraholland.com/en. Accessed 18 Sept 2018
Sanikhani M, Frello S, Serek M (2006) TDZ induces shoot regeneration in various Kalanchoe blossfeldiana Poelln. cultivars in the absence of auxin. Plant Cell Tissue Org 85(1):75–82
Schenk RU, Hildebrandt A (1972) Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Can J Bot 50(1):199–204
Varga A, Thoma L, Bruinsma J (1988) Effects of auxins and cytokinins on epigenetic instability of callus-propagated Kalanchoe blossfeldiana Poelln. Plant Cell Tissue Org 15(3):223–231
Vienken J, Ganser R, Hampp R, Zimmermann U (1981) Electric field-induced fusion of isolated vacuoles and protoplasts of different developmental and metabolic provenience. Physiol Plant 53:64–70
Widholm JM (1972) The use of fluorescein diacetate and phenosafranine for determining viability of cultured plant cells. Stain Technol 47(4):189–194
Acknowledgments
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. This project was a part of the innovation consortium “BioFactory” funded by the Danish Agency for Science, Technology and Innovation. The authors would like to thank Knud Jepsen A/S, Hinnerup, Denmark, for providing plant material.
Author information
Authors and Affiliations
Contributions
JC, KKM, TE, RM, HL: study conception and design; JC: Acquisition of data; JC, KKM: Analysis and interpretation of data; JC, KKM: Drafting of manuscript; JC, KKM, TE, RM, HL: Critical revisions.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Ming-Tsair Chan.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cui, J., Kuligowska Mackenzie, K., Eeckhaut, T. et al. Protoplast isolation and culture from Kalanchoë species: optimization of plant growth regulator concentration for efficient callus production. Plant Cell Tiss Organ Cult 138, 287–297 (2019). https://doi.org/10.1007/s11240-019-01624-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-019-01624-4