
Abstract
In 2004, H9N2 influenza A viruses were isolated from pigs with respiratory syndrome in commercial swine farms in Henan province, China. Antigenic and genetic characterization were performed for seven swine H9N2 influenza viruses. The hemagglutinin antigenicity of swine H9N2 viruses was similar to those of avian H9N2 viruses of A/duck/Hong Kong/Y280/1997 (Dk/HK/Y280/97)-like sublineage prevalent in China. It is noteworthy that the neuraminidase of these isolates had no deletions in the stalk, which was seldom observed in those viruses of Dk/HK/Y280/97-like sublineage. Genetic analysis revealed that all seven isolates had an -R-S-S-R- motif at the HA cleavage site, which was the same as those of Dk/HK/Y280/97-like viruses established in avian population in China. Phylogenetic analyses showed that the seven swine H9N2 viruses were completely derived from avian influenza viruses of Dk/HK/Y280/97-like sublineage. The present results indicated that avian-to-pig interspecies transmission of H9N2 viruses continued to exist in China through 2004; therefore, surveillance of swine influenza should be given a high priority.
Similar content being viewed by others


Introduction
Influenza A viruses are classified into 16 hemagglutinin (HA) and 9 neuraminidase (NA) subtypes based on antigenic differences [1, 2], of which H9N2 subtype virus is a conspicuous member of the influenza family, because it can not infect only chickens, ducks, and pigs, but also humans [3–6]. In China, the H9N2 virus was first isolated from a chicken in 1992 in Guangdong province [7] and is now the most prevalent subtype of influenza virus in poultry in China. Pigs are permissive to both avian and human influenza A viruses because the cells of their respiratory tract express both the sialic acid-α2,3-galactose (SAα2,3Gal) receptors preferred by avian influenza viruses and the sialic acid-α2,6-galactose (SAα2,6Gal) receptors preferred by human influenza viruses [8]. As such, pigs are proposed to be intermediate hosts for adaptation of avian influenza viruses to replicate in mammals [9]. Surveillance of swine H9N2 influenza viruses as possible sources of pandemic influenza should be given a high priority.
In 2004, surveillance of H1N1, H3N2, and H9N2 subtypes was conducted in 13 swine farms in Henan province, China. Laboratory diagnosis and virus isolation demonstrated that H9N2 influenza viruses were broadly distributed among these swine farms. Furthermore, 15 swine H9N2 viruses were isolated from 361 tracheal swabs. In order to elucidate the antigenic and genetic characteristics of these viruses and the relationships of the swine H9N2 viruses with avian influenza viruses, we analyzed seven H9N2 swine influenza virus isolates from 11 pig farms antigenically and genetically.
Materials and methods
Viruses
Nasal swabs and lung tissue samples were collected from the diseased and/or dead swine. Initial isolation of the viruses from the samples was performed in cultures of Madin-Darby canine kidney cells in the present of 2.5 μg/ml trypsin. Supernatant fluids were harvested for sequential passage and used as stock for sequence analysis. Subtype identification of these viruses were determined by standard hemagglutination inhibition (HI) tests and NA inhibition tests with a panel of reference antisera recommended by the World Health Organization (http://www.who.Int/csr/resources/publications/en/#influenza). Reference avian influenza viruses were employed for HI analysis; A/quail/Hong Kong/G1/1997 (H9N2) (Qa/HK/G1/97), A/chicken/Hong Kong/G9/1997 (H9N2) (Ck/HK/G9/97), and A/turkey/Wisconsin/1966 (H9N2) (Ty/WI/66) were kindly provided by Dr. H. Kida, Graduate School of Veterinary Medicine, Hokkaido University. The seven virus isolates obtained in this study were named as follows: A/swine/Henan/2/2004 (Sw/HN/2/04), A/swine/Henan/3/2004 (Sw/HN/3/04), A/swine/Henan/4/2004 (Sw/HN/4/04), A/swine/Henan/5/2004 (Sw/HN/5/04), A/swine/Henan/6/2004 (Sw/HN/6/04), and A/swine/Henan/7/2004 (Sw/HN/7/04).
Antisera and hemagglutination inhibition
The viruses were antigenically analyzed by HI testing by a panel of hyperimmune chicken antisera. Chicken hyperimmune sera against Qa/HK/G1/97 (Qa/HK/G1/97, representing the Qa/HK/G1/97-like sublineage) and Ck/HK/G9/97 (Ck/HK/G9/97, representing the Dk/HK/Y280/97-like sublineage) were provided by Dr H. Kida. Chicken hyperimmune sera against A/chicken/Hebei/1/1996 (H9N2) (Ck/HB/1/96, isolated by J. H. Liu, seemingly not belonging to any known H9N2 sublineage) and A/swine/Shandong/FJN/2003 (H9N2) (Sw/SD/FJN/03, representing the Sw/SD/1/03-like sublineage) were prepared in our laboratory. HI tests were performed as previously described [10].
Gene sequencing and phylogenetic analysis
Viral RNA was extracted from supernatant fluids using Trizol reagents (Gibco-BRL). Reverse transcription-PCR and gene sequencing were done, as described previously [11]. Assembly of partial sequences, translation of nucleotide sequences into amino acid sequences, and initial multiple sequence alignments were performed with the Clustal V method using MegAlign software version 1.03 (DNAStar Inc., Madison, WI). The phylogenetic relationships were estimated from the nucleotide sequences of each H9N2 swine influenza viral gene relative to selected H9N2 and H5N1 subtype influenza reference strains obtained from GenBank database (using the PHYLIP software package (http://www.ddbj.nig.ac.jp/E-mail/clustalw-e.html)). The phylogenetic tree was drawn using TREEVIEW (version 1.40, Roderic D. M. Page, 1997). The nucleotide sequences used for the phylogenetic analysis are as follows: PB2 1342–2172, PB1 1140–1762, PA 761–1215, HA 172–1185, NP 1120-1444, NA 67-1398, M 81-560, and NS 18-787.
Nucleotide sequence accession numbers
The nucleotide sequences for all H9N2 influenza viruses analyzed in the present study are available from GenBank under accession numbers DQ981535 to DQ981574 and DQ981583 to DQ981598.
Results
Antigenic analysis
In order to investigate the antigenic properties of the swine H9N2 virus isolates in 2004, HI tests were performed with a panel of anti-H9 hyperimmune sera (Table 1). Antigenic analysis demonstrated a diversity of reaction patterns generally corresponding with their phylogenetic relationships determined by sequence comparisons (Fig. 2a). All the tested H9N2 swine viruses reacted well with antisera to Ck/HK/G9/97, however, they showed middle or low reactivity to antisera raised against Ck/HB/1/96 and Sw/SD/FJN/03, indicating that these swine isolates were related to the avian viruses of Dk/HK/Y280/97-like sublineage and there were obvious antigenic variance between swine influenza virus isolates from Henan and those from Shandong [12]. Also, these Henan H9N2 swine virus isolates did not react to antisera to Qa/HK/G1/97. The present findings indicated that the HAs of swine H9N2 viruses prevalent in Henan province were similar to those of H9N2 viruses of Dk/HK/Y280/97-like sublineage prevalent in avian population in China.
Molecular analysis
In order to try to illustrate the possible determinants of interspecies transmission of H9N2 influenza viruses from avian to pigs, the deduced amino acid sequences of viral proteins were analyzed. We determined the nucleotide sequences (76-1617, 1542 bp) of the seven H9 HAs of swine influenza virus isolates in the present study. The amino acid sequences at the HA cleavage site of them possessed an -R-S-S-R- motif, which were the same as those of Dk/HK/Y280/97-like and Qa/HK/G1/97-like viruses, -R-S-S-R- established in Asia, but had amino acid difference from the -R-L-S-R- sequence of Sw/SD/1/03-like viruses (Table 2). Amino acids at the receptor-binding sites of the HA are associated with the differences in the receptor binding specificity [13]. Table 2 shows the amino acids at the positions 183, 190, 226, 227, and 228, which are putative residues for receptor binding sites of the HAs of H9N2 viruses [14]. All of the seven swine isolates possessed N, V, and Q at amino acid position 183, 190, and 226 (numbering according to H3 HA), respectively, which are the same as that of Dk/HK/Y280/97-like viruses prevalent in avian population in mainland China.
It has been considered that the glycosylation sites might affect the receptor-binding capacity of HA [15]. Analysis of the potential glycosylation sites in the HAs of the seven H9N2 virus isolates revealed seven common sites (five in HA1 and two in HA2) with the -N-X-T/S- motif (in which X may be any amino acid except proline) in the sequenced regions of HA genes. The potential glycosylation sites of the seven isolates (Asn11, 123, 200, 280, 287, 474, and 533, respectively), were conserved in the HAs of Dk/HK/Y280/97-like viruses.
Analysis of the potential glycosylation sites in the NAs of the seven H9N2 virus isolates revealed eight common sites at Asn44, 61, 69, 86, 146, 200, 234, and 402, respectively, except for Sw/HN/5/04, which had an additional glycosylation site at Asn215. Most of the NAs of the Dk/HK/Y280/97-like viruses contain a deletion of three amino acid residues at position 63–65 in the stalk of the protein compared with those of Ck/HK/G9/97, which resulted in the loss of one potential glycosylation site (Asn61). However, the seven 2004 swine H9N2 virus isolates had no such deletion (Fig. 1). Although the functional relevance of the observed deletion in the NAs of the seven 2004 swine H9N2 viruses is not known, the deletion will serve as useful markers of this lineage of H9N2 viruses in China.

Deletion of amino acids in the stalk of the NAs of H9N2 viruses


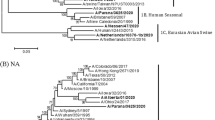
Phylogenetic trees of the eight gene segments of influenza viruses. Horizontal distances are proportional to the minimum number of nucleotide differences required to join nodes and sequences. Vertical distances are for spacing branches and labels. Viruses isolated in the present study are in bold, and H5 viruses are underlined. All other viruses are H9N2. Abbreviations used in virus designations are as follows: Ck, chicken; Dk, duck; Gf, guinea fowl; Gs, goose; Pg, pigeon; Qa, quail; Sb, shorebird; Sw, swine; Ty, turkey; WDk, wild duck; Ark, Arkansas; BJ, Beijing; CA, California; DE, Delaware; FJ, Fujian; GD, Guangdong; GX, Guangxi; GZ, Guangzhou; HB, Hebei; HK, Hong Kong; HLJ, Heilongjiang; HN, Henan; HuB, Hubei; HZ, Hangzhou; JL, Jilin; JS, Jiangsu; Kor, Korea; MDJ, Mudanjiang; NC, Nanchang; NM, Neimeng; SD, Shandong; SH, Shanghai; ST, Shantou; SZ, Shenzhen; WI, Wisconsin; Yok, Yokohama; ZJ, Zhejiang
Homology analysis of nucleotide sequences of all genes
The identity among the seven swine H9N2 isolates for each of the gene sequences by DNAStar are shown in Table 3. The homology analysis of the nucleotide sequences of eight genes of the seven 2004 swine H9N2 virus isolates compared with other available sequences in GenBank was performed (Table 3), which showed that they were wholly avian origin.
Phylogenetic analysis
The phylogenetic relationships of swine H9N2 viruses prevalent in Henan province to selected reference strains were estimated from the nucleotide sequences of each viral gene. The phylograms for all genes are shown in Fig. 2a–h. Phylogenetic analysis of the H9 HAs showed that at least five distinct sublineage H9N2 viruses have been defined in avian population in mainland China, with the prototype viruses being A/chicken/Beijing/1/1994 (Ck/BJ/1/94), A/chicken/Shanghai/F/1998 (Ck/SH/F/98), Qa/HK/G1/97, Dk/HK/Y280/97, and A/Swine/Shandong/1/2003 (Sw/SD/1/03) [12, 16, 17]. All HAs of the swine H9N2 viruses (99.7–100% homology) tested in the present study clustered into Dk/HK/Y280/97-like sublineage formed by recent avian viruses and were most closely related to the A/pigeon/Nanchang/2-0416/2000 (Pg/NC/2-0461/00 (H9N2)), with above 98% identity. Two H9N2 viruses isolated by Peiris [14], A/swine/Hong Kong/2106/1998 (Sw/HK/2106/98) and A/swine/Hong Kong/3297/1998 (Sw/HK/3297/98) also appeared closely related to contemporary avian viruses of the Dk/HK/Y280/97-like sublineage. The other swine H9N2 isolates in 1998 (A/swine/Hong Kong/9/1998 (Sw/HK/9/98) and A/swine/Hong Kong/10/1998 (Sw/HK/10/98)) were more closely related to A/chicken/Guangdong/SS/1994 (Ck/GD/SS/94) and A/chicken/Guangdong/11/1997 (Ck/GD/11/97) and exhibited an outgroup relationship to Dk/HK/Y280/97 and Sw/SD/1/03-like sublineage. Since the swine H9N2 isolates do not form a single lineage, the results imply that multiple introductions from avian hosts to pigs occurred. The phylogeny of the NA genes paralleled to that of the HA genes, in which the seven swine isolates still belonged to Dk/HK/Y280/97-like sublineage. The sequence similarity among seven swine H9 virus isolates and A/chicken/Jiangsu/1/2000 (Ck/JS/1/00 (H9N2)) ranged from 98.4% to 99.4% nucleotide identity.
The results of the previous surveillance and released data in GenBank revealed that H9N2 influenza viruses were cocirculating with the H5N1 influenza viruses during the recent past, raising the possibility of genetic exchange among these viruses [6, 12, 16, 18–21]. Phylogenetic analysis of the internal genes of the seven swine H9N2 virus isolates showed that these isolates were not closely related to H5N1 viruses (Fig. 2c–h). Taking these results together, the seven 2004 swine H9N2 virus isolates belong to Dk/HK/Y280/97-like sublineage and are the result of transmission of avian H9N2 viruses to pigs in toto.
Discussion
In China, H9N2 influenza viruses were first isolated from chickens in Guangdong province in 1992. Subsequently, sporadic and endemic outbreaks of the H9N2 influenza have often occurred in many provinces from Northern to Southern China [5]. Hitherto, H9N2 influenza virus infection has frequently occurred in pig population in China. Seropositivity to H9 viruses has been reported among pigs in China [22]. Peiris et al. were the first to identify that cocirculation of avian H9N2 and contemporary human H3N2 viruses in pigs had occurred in southeastern China and predicted that the cocirculation of H9 and H3 viruses in pigs would provide an opportunity for genetic reassortment leading to the emergence of viruses with pandemic potential [14]. It is noteworthy that reassortant viruses between avian H5N1 and H9N2 viruses had caused pig diseases and death in Shandong province [6, 12]. Since pigs can serve as intermediate hosts for adaptation of avian influenza viruses to infect mammals, swine influenza infections have been the focus of increasing attention.
In 2004, a characteristic respiratory syndrome in pigs was again observed on many pig farms in Henan province. The H9N2 infections in the pigs were diagnosed by virus isolation and serological tests. Although these viruses caused apparent clinical disease in pigs, genetic analysis showed that these viruses had the characteristic of low pathogenic avian influenza virus. The low pathogenic motif of -R-S-S-R- at the cleavage site of HA1 and HA2 was consistent with those of most H9N2 influenza viruses prevalent in the avian population in mainland China, but had an amino acid different from those of Shandong isolates in 2003, -R-L-S-R-. It is not clear whether this amino acid sequence motif at the cleavage site is related to the infection, pathogenicity and tissue tropism, although it is similar to the motif (-R-X-R/K-R-, in which X may be any amino acid) required for highly pathogenic viruses of the H5 and H7 subtypes. One nucleotide substitution could change the -R-X-S-R- motif to -R-X-R-R- motif, and Guo [4] suggested that the H9N2 viruses prevalent in China are potentially capable of becoming highly pathogenic viruses.
The host range of influenza viruses is associated with different amino acids within and around the HA receptor-binding pocket [23]. In the present study, all swine isolates possessed amino acids N, V, and Q within the receptor binding sites of HA at positions 183, 190, and 226, respectively. Glutamine at position 226 is typical of the sequences found in avian viruses [24]. The H3 HA possessing Q-226 binds to the SAα2,3Gal receptors [23]. It has also been shown that avian H9N2 viruses possessing Q-226 preferentially bind to the SAα2,3Gal receptors. In conjunction with amino acid residue at 226, V at position 190 is reported to have high binding affinity for SAα2,6Gal receptors [25]. The direct binding assays [26, 27] have shown that these Henan swine virus isolates displayed not only a high affinity for the 3′SL-PAA-biotin, but also a weak affinity for the 6′SL-PAA-biotin (data not shown). Therefore, it is predicted that the seven swine H9N2 virus isolates have the potential to bind to SAa2,6Gal receptors found on human cells. This could pose a significant pandemic threat to the human population immunologically naive to the H9 antigen.
The sequence and length of the stalk region are known to vary among and within NA subtypes [28]. The NA genes of most of Dk/HK/Y280/97-like viruses had 3 amino acid deletions at residues 63–65 in the stalk. However, no deletion was observed in the Henan swine isolates. Guo et al. [4] considered that acquisition of the small deletion in the H9N2 NA may be related to changes in characteristics of the HA. Glycosylation of HA and stalk-length of NA combine to regulate the growth of influenza virus [29]. Further research is needed on the correlations between deletions in the NAs of H9N2 viruses and viral pathogenicity, the affinity of receptor binding, and the mechanism on virus crossing interspecies transmission.
At least six different genotypes of H9N2 influenza viruses have been recognized in southeastern China [19]. Phylogenetic analysis of the H9 HAs showed that five distinct H9N2 virus sublineages were maintained in the avian population in mainland China (Fig. 2a). Consistent with the antigenic variation, the seven swine H9N2 viruses analyzed in the study belonged to the Dk/HK/Y280/97-like sublineage. All eight genes were closely related to those of recent avian H9 viruses, indicating that repeated interspecies transmission events had occurred from avian to pigs. Up to date, swine H9N2 viruses isolated from Hong Kong [14], Shandong [12], and Henan (in the present study) do not form a single lineage, implying that multiple interspecies transmission events from avian hosts to pigs occurred in nature. It has been proposed that at least 2% of blood donors tested were positive for H9 antibody in southern China [3, 30–32], suggesting that human infection with H9N2 occurred sporadically in this area. Since H9N2 virus appears to have the potential to cross the species barrier to humans more efficiently than the current H5N1 virus by the predicted affinity for SAα2,6Gal receptors [20, 32–35], it is prudent that we enhance surveillance of swine H9N2 influenza viruses as part of overall pandemic preparedness efforts.
References
R.G. Webster, W.J. Bean, O.T. Gorman, T.M. Chambers, Y. Kawaoka, Microbiol. Rev. 56, 152–179 (1992)
R.A. Fouchier, V. Munster, A. Wallensten, T.M. Bestebroer, S. Herfst, D. Smith, G.F. Rimmelzwaan, B. Olsen, A.D. Osterhaus, J. Virol. 79, 2814–2822 (2005)
M. Peiris, K.Y. Yuen, C.W. Leung, K.H. Chan, P.L. Ip, R.W. Lai, W.K. Orr, K.F. Shortridge, Lancet 354, 916–917 (1999)
Y.J. Guo, S. Krauss, D.A. Senne, I.P. Mo, K.S. Lo, X.P. Xiong, M. Norwood, K.F. Shortridge, R.G. Webster, Y. Guan, Virology 267, 279–288 (2000)
J. Liu, K. Okazaki, H. Ozaki, Y. Sakoda, Q. Wu, F. Chen, H. Kida, Avian Pathol. 32, 551–560 (2003)
C. Xu, W. Fan, R. Wei, H. Zhao, Microbes Infect. 6, 919–925 (2004)
B.L. Chen, A.J. Zhang, W.B. Chen, Chin. J. Vet. Med. 10, 3–5 (1994)
T. Ito, J.N. Couceiro, S. Kelm, L.G. Baum, S. Krauss, M.R. Castrucci, I. Donatelli, H. Kida, J.C. Paulson, R.G. Webster, Y. Kawaoka, J. Virol. 72, 7367–7373 (1998)
L. Campitelli, I. Donatelli, E. Foni, M.R. Castrucci, C. Fabiani, Y. Kawaoka, S. Krauss, R.G. Webster, Virology 232, 310–318 (1997)
A.P. Kendal, M.S. Pereira, J.J. Skehel, Concepts and procedures for laboratory-based influenza surveillance (Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA, 1982)
L.L. Shu, Y.P. Lin, S.M. Wright, K.F. Shortridge, R.G. Webster, Virology 202, 825–833 (1994)
Y.L. Cong, J. Pu, Q.F. Liu, S. Wang, G.Z. Zhang, X.L. Zhang, W.X. Fan, E.G. Brown, J.H. Liu, J. Gen. Virol. 88, 2035–2041 (2007)
W. Weis, J.H. Brown, S. Cusack, J.C. Paulson, J.J. Skehel, D.C. Wiley, Nature 333, 426–431 (1988)
J.S.M. Peiris, Y. Guan, D. Markwell, P. Ghose, R.G. Webster, K.F. Shortridge, J. Virol. 75, 9679–9686 (2001)
M. Ohuchi, R. Ohuchi, A. Feldmann, H.D. Klenk, J. Virol. 71, 8377–8384 (1997)
Y. Guan, K.F. Shortridge, S. Krauss, P.S. Chin, K.C. Dyrting, T.M. Ellis, R.G. Webster, M. Peiris, J. Virol. 74, 9372–9380 (2000)
J.H. Lu, X.F. Liu, W.X. Shao, Y.L. Liu, D.P. Wei, H.Q. Liu, Virus Genes 31, 163–169 (2005)
Y. Guan, K.F. Shortridge, S. Krauss, R.G. Webster, Proc. Natl. Acad. Sci. USA 96, 9363–9367 (1999)
Y.K. Choi, H. Ozaki, R.J. Webby, R.G. Webster, J.S. Peiris, L. Poon, C. Butt, Y.H. Leung, Y. Guan, J. Virol. 78, 8609–8614 (2004)
K.S. Li, K.M. Xu, J.S. Peiris, L.L. Poon, K.Z. Yu, K.Y. Yuen, K.F. Shortridge, R.G. Webster, Y. Guan, J. Virol. 77, 6988–6994 (2003)
Y.P. Lin, M. Shaw, V. Gregory, K. Cameron, W. Lim, A. Klimov, K. Subbarao, Y. Guan, S. Krauss, K. Shortridge, R. Webster, N. Cox, A. Hay, PNAS 97, 9654–9658 (2000)
A. Ninomiya, A. Takada, K. Okazaki, K.F. Shortridge, H. Kida, Vet. Microbiol. 88, 107–114 (2002)
G.N. Rogers, J.C. Paulson, R.S. Daniels, J.J. Skehel, I.A. Wilson, D.C. Wiley, Nature 304, 76–78 (1983)
M. Matrosovich, A. Tuzikov, N. Bovin, A. Gambaryan, A. Klimov, M.R. Castrucci, I. Donatelli, Y. Kawaoka, J. Virol. 74, 8502–8512 (2000)
M.N. Matrosovich, S. Krauss, R.G. Webster, Virology 281, 156–162 (2001)
A.S. Gambaryan, M.N. Matrosovich, J. Virol. Meth. 39, 111–123 (1992)
W.X. Wu, M.A. Gillian, Virology 325, 340–350 (2004)
K. Subbarao, A. Klimov, J. Katz, H. Regnery, W. Lim, H. Hall, M. Perdue, D. Swayne, C. Bender, J. Huang, M. Hemphill, T. Rowe, M. Shaw, X. Xu, K. Fukuda, N. Cox, Science 279, 393–396 (1998)
S.J. Baigent, J.W. McCauley, Virus Res. 79, 177–185 (2001)
Y.J. Guo, J.G. Li, X.W. Cheng, M. Wang, Y. Zou, C.H. Li, F.C. Cai, H.Y. Liao, Y. Zhang, J.F. Guo, R.M. Huang, D. Bei, Chin. J. Exp. Clin. Virol. 13, 105–108 (1999)
K.G. Nicholson, J.M. Wood, M. Zambon, Lancet 362, 1733–1745 (2003)
K.M. Butt, G.J. Smith, H. Chen, L.J. Zhang, Y.H. Leung, K.M. Xu, W. Lim, R.G. Webster, K.Y. Yuen, J.S. Peiris, Y. Guan, J. Clin. Microbiol. 43, 5760–5767 (2005)
Y. Ha, D.J. Stevens, J.J. Skehel, D.C. Wiey, Proc. Natl. Acad. Sci. USA 98, 11181–11186 (2001)
N.V. Kaverin, I.A. Rudneva, N.A. Ilyushina, A.S. Lipatov, S. Krauss, R.G. Webster, J. Virol. 78, 240–249 (2004)
M.N. Matrosovich, T.Y. Matrosovich, T. Gray, N.A. Roberts, H.D. Klenk, Proc. Natl. Acad. Sci. USA 101, 4620–4624 (2004)
Acknowledgments
The study was supported by the National Natural Scientific Foundation (30599431, 30471282), National Basic Research Program (973) (2005CB523003) and Grant-in-Aid for Scientific Research from the Ministry of Education (NCET-05-0123). EGB is funded by the Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Yan-Long Cong, Chun-Feng Wang and Chun-Mei Yan have contributed equally to this work.
(i) All the authors have agreed to its submission and are responsible for its contents and (ii) all the authors have agreed that Yanlong Cong may act on their behalf regarding any subsequent processing of the paper.
Rights and permissions
About this article
Cite this article
Cong, YL., Wang, CF., Yan, CM. et al. Swine infection with H9N2 influenza viruses in China in 2004. Virus Genes 36, 461–469 (2008). https://doi.org/10.1007/s11262-008-0227-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-008-0227-z