
Abstract
Thanks to the fast improvement of the computing power and the rapid development of the computational chemistry and biology, the computer-aided drug design techniques have been successfully applied in almost every stage of the drug discovery and development pipeline to speed up the process of research and reduce the cost and risk related to preclinical and clinical trials. Owing to the development of machine learning theory and the accumulation of pharmacological data, the artificial intelligence (AI) technology, as a powerful data mining tool, has cut a figure in various fields of the drug design, such as virtual screening, activity scoring, quantitative structure-activity relationship (QSAR) analysis, de novo drug design, and in silico evaluation of absorption, distribution, metabolism, excretion and toxicity (ADME/T) properties. Although it is still challenging to provide a physical explanation of the AI-based models, it indeed has been acting as a great power to help manipulating the drug discovery through the versatile frameworks. Recently, due to the strong generalization ability and powerful feature extraction capability, deep learning methods have been employed in predicting the molecular properties as well as generating the desired molecules, which will further promote the application of AI technologies in the field of drug design.
Similar content being viewed by others
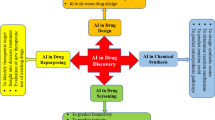

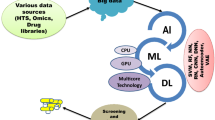
References
Abagyan, R., Totrov, M., and Kuznetsov, D. (1994). ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem 15, 488–506.
Ain, Q.U., Aleksandrova, A., Roessler, F.D., and Ballester, P.J. (2015). Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. WIREs Comput Mol Sci 5, 405–424.
Altae-Tran, H., Ramsundar, B., Pappu, A.S., and Pande, V. (2017). Low data drug discovery with one-shot learning. ACS Cent Sci 3, 283–293.
Andras, P. (2017). High-dimensional function approximation with neural networks for large volumes of data. IEEE Trans Neural Netw Learn Syst 99, 1–9.
Angermueller, C., Pärnamaa, T., Parts, L., and Stegle, O. (2016). Deep learning for computational biology. Mol Syst Biol 12, 878.
Artursson, P., and Karlsson, J. (1991). Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 175, 880–885.
Ash, S., Cline, M.A., Homer, R.W., Hurst, T., and Smith, G.B. (1997). ChemInform abstract: SYBYL line notation (SLN): a versatile language for chemical structure representation. ChemInform 28, no.
Ashburn, T.T., and Thor, K.B. (2004). Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3, 673–683.
Bai, F., Morcos, F., Cheng, R.R., Jiang, H., and Onuchic, J.N. (2016). Elucidating the druggable interface of protein-protein interactions using fragment docking and coevolutionary analysis. Proc Natl Acad Sci USA 113, E8051–E8058.
Bender, A., And, H.Y.M., Glen, R.C., and Reiling, S. (2004). Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J Chem Inf Comput Sci 44, 1708–1718.
Cabreiro, F., Au, C., Leung, K.Y., Vergara-Irigaray, N., Cocheme, H.M., Noori, T., Weinkove, D., Schuster, E., Greene, N.D., and Gems, D. (2013). Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 153, 228–239.
Cao, D.S., Xu, Q.S., Hu, Q.N., and Liang, Y.Z. (2013). ChemoPy: freely available python package for computational biology and chemoinformatics. Bioinformatics 29, 1092–1094.
Chen, B., Sheridan, R.P., Hornak, V., and Voigt, J.H. (2012). Comparison of random forest and pipeline pilot naïve bayes in prospective QSAR predictions. J Chem Inf Model 52, 792–803.
Chen, R., Li, L., and Weng, Z. (2003). ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87.
Chen, Y.C. (2015). Beware of docking! Trends Pharmacol Sci 36, 78–95.
Coley, C.W., Barzilay, R., Green, W.H., Jaakkola, T.S., and Jensen, K.F. (2017). Convolutional embedding of attributed molecular graphs for physical property prediction. J Chem Inf Model 57, 1757–1772.
Coley, C.W., Rogers, L., Green, W.H., and Jensen, K.F. (2018). SCScore: synthetic complexity learned from a reaction corpus. J Chem Inf Model 58, 252–261.
Copeland, R.A. (2010). The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Discovery 5, 305–310.
Cortes, C., Kuznetsov, V., and Mohri, M. (2014). Ensemble methods for structured prediction. Proceedings of 31st International Conference on Machine Learning 2014, 1134–1142.
Cukuroglu, E., Engin, H.B., Gursoy, A., and Keskin, O. (2014). Hot spots in protein–protein interfaces: Towards drug discovery. Prog Biophys Mol Biol 116, 165–173.
Dahl, G.E., Jaitly, N., and Salakhutdinov, R. (2014). Multi-task neural networks for QSAR predictions. Comput Sci, arXiv:1406.1231v1.
Dang, N.L., Hughes, T.B., Krishnamurthy, V., and Swamidass, S.J. (2016). A simple model predicts UGT-mediated metabolism. Bioinformatics 32, 3183–3189.
Danishuddin, and Khan, A.U. (2016). Descriptors and their selection methods in QSAR analysis: paradigm for drug design. Drug Discov Today 21, 1291–1302.
De Haes, W., Frooninckx, L., Van Assche, R., Smolders, A., Depuydt, G., Billen, J., Braeckman, B.P., Schoofs, L., and Temmerman, L. (2014). Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci USA 111, E2501–E2509.
DiMasi, J.A., Grabowski, H.G., and Hansen, R.W. (2016). Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Economics 47, 20–33.
Dobchev, D., Pillai, G., and Karelson, M. (2014). In silico machine learning methods in drug development. CTMC 14, 1913–1922.
Du, T., Liao, L., Wu, C.H., and Sun, B. (2016). Prediction of residue-residue contact matrix for protein-protein interaction with Fisher score features and deep learning. Methods 110, 97–105.
Duch, W., Swaminathan, K., and Meller, J. (2007). Artificial intelligence approaches for rational drug design and discovery. CPD 13, 1497–1508.
Dudek, A., Arodz, T., and Galvez, J. (2006). Computational methods in developing quantitative structure-activity relationships (QSAR): a review. CCHTS 9, 213–228.
Durant, J.L., Leland, B.A., Henry, D.R., and Nourse, J.G. (2003). Reoptimization of MDL keys for use in drug discovery. J Chem Inf Comput Sci 34, 1273–1280.
Duvenaud, D., Maclaurin, D., Aguilera-Iparraguirre, J., Hirzel, T., and Adams, R.P. (2015). Convolutional networks on graphs for learning molecular fingerprints. In Proceedings of the 28th International Conference on Neural Information Processing Systems, pp. 2224–2232.
Esposito, E.X., Hopfinger, A.J., and Madura, J.D. (2004). Methods for applying the quantitative structure-activity relationship paradigm. Methods Mol Biol 275, 131–214.
Falchi, F., Caporuscio, F., and Recanatini, M. (2014). Structure-based design of small-molecule protein-protein interaction modulators: the story so far. Future Medicinal Chem 6, 343–357.
Free, S.M., and Wilson, J.W. (1964). A mathematical contribution to structure-activity studies. J Med Chem 7, 395–399.
Friesner, R.A., Banks, J.L., Murphy, R.B., Halgren, T.A., Klicic, J.J., Mainz, D.T., Repasky, M.P., Knoll, E.H., Shelley, M., and Perry, J.K. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47, 1739–1749.
Ghasemi, F., Mehridehnavi, A.R., Fassihi, A., and Pérez-Sánchez, H. (2017). Deep neural network in biological activity prediction using deep belief network. Appl Soft Comput 62, doi: 10.1016/j.asoc.2017.09.040.
Goh, G.B., Hodas, N.O., Siegel, C., and Vishnu, A. (2017). SMILES2Vec: An Interpretable General-Purpose Deep Neural Network for Predicting Chemical Properties. arXiv:1712.02034v2.
Gómez-Bombarelli, R., Wei, J.N., Duvenaud, D., Hernández-Lobato, J.M., Sánchez-Lengeling, B., Sheberla, D., Aguilera-Iparraguirre, J., Hirzel, T.D., Adams, R.P., and Aspuru-Guzik, A. (2018). Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci 4, 268–276.
Goodfellow, I., Bengio, Y., and Courville, A. (2016). Deep Learning (Cambridge: The MIT Press)
Guengerich, F.P. (2011). Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab Pharmacokinetics 26, 3–14.
Hansch, C., and Fujita, T. (1964). Additions and corrections -ρ-σ-π analysis. A method for the correlation of biological activity and chemical structure. J Am Chem Soc 86, 5710.
Hartenfeller, M., and Schneider, G. (2011). De novo drug design. Methods Mol Biol 672, 299–323.
Hassan Baig, M., Ahmad, K., Roy, S., Mohammad Ashraf, J., Adil, M., Haris Siddiqui, M., Khan, S., Amjad Kamal, M., Provazník, I., and Choi, I. (2015). Computer aided drug design: success and limitations. CPD 22, 572–581.
Heller, S.R., McNaught, A., Pletnev, I., Stein, S., and Tchekhovskoi, D. (2015). InChI, the IUPAC international chemical identifier. J Cheminform 7, 23.
Higueruelo, A.P., Jubb, H., and Blundell, T.L. (2013). Protein-protein interactions as druggable targets: recent technological advances. Curr Opin Pharmacol 13, 791–796.
Huang, S.Y., and Zou, X. (2010). Inclusion of solvation and entropy in the knowledge-based scoring function for protein-ligand interactions. J Chem Inf Model 50, 262–273.
Huang, S.Y., Grinter, S.Z., and Zou, X. (2010). Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys Chem Chem Phys 12, 12899–12908.
Hubatsch, I., Ragnarsson, E.G.E., and Artursson, P. (2007). Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc 2, 2111–2119.
Jimenez, J., Skalic, M., Martinez-Rosell, G., and De Fabritiis, G. (2018). KDEEP: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J Chem Inf Model 58, 287–296.
Jin, W., Barzilay, R., and Jaakkola, T. (2018). Junction tree variational autoencoder for molecular graph generation. arXiv:1802.04364v2.
Kadurin, A., Aliper, A., Kazennov, A., Mamoshina, P., Vanhaelen, Q., Khrabrov, K., and Zhavoronkov, A. (2017)a. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8, 10883.
Kadurin, A., Nikolenko, S., Khrabrov, K., Aliper, A., and Zhavoronkov, A. (2017)b. druGAN: An advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol Pharm 14, 3098–3104.
Kearnes, S., Goldman, B., and Pande, V. (2016)a. Modeling industrial ADMET data with multitask networks. arXiv:1606.08793v3.
Kearnes, S., Mccloskey, K., Berndl, M., Pande, V., and Riley, P. (2016)b. Molecular graph convolutions: moving beyond fingerprints. J Comput Aid Mol Design 30, 1–14.
Khamis, M.A., Gomaa, W., and Ahmed, W.F. (2015). Machine learning in computational docking. Artif Intell Med 63, 135–152.
Kim, K.H., Kim, N.D., and Seong, B.L. (2010). Pharmacophore-based virtual screening: a review of recent applications. Expert Opin Drug Discov 5, 205–222.
Kinnings, S.L., Liu, N., Tonge, P.J., Jackson, R.M., Xie, L., and Bourne, P. E. (2011). A machine learning-based method to improve docking scoring functions and its application to drug repurposing. J Chem Inf Model 51, 408–419.
Klaeger, S., Heinzlmeir, S., Wilhelm, M., Polzer, H., Vick, B., Koenig, P. A., Reinecke, M., Ruprecht, B., Petzoldt, S., Meng, C., et al. (2017). The target landscape of clinical kinase drugs. Science 358, eaan4368.
Labbé, C.M., Kuenemann, M.A., Zarzycka, B., Vriend, G., Nicolaes, G.A. F., Lagorce, D., Miteva, M.A., Villoutreix, B.O., and Sperandio, O. (2016). iPPI-DB: an online database of modulators of protein-protein interactions. Nucleic Acids Res 44, D542–D547.
Lavecchia, A., and Giovanni, C. (2013). Virtual screening strategies in drug discovery: a critical review. CMC 20, 2839–2860.
LeCun, Y., Bengio, Y., and Hinton, G. (2015). Deep learning. Nature 521, 436–444.
Leelananda, S.P., and Lindert, S. (2016). Computational methods in drug discovery. Beilstein J Org Chem 12, 2694–2718.
Li, H., Hou, J., Adhikari, B., Lyu, Q., and Cheng, J. (2017). Deep learning methods for protein torsion angle prediction. BMC BioInf 18, 417.
Liew, C.Y., Ma, X.H., Liu, X., and Yap, C.W. (2009). SVM model for virtual screening of Lck inhibitors. J Chem Inf Model 49, 877.
Lombardo, F., and Jing, Y. (2016). In silico prediction of volume of distribution in humans. Extensive data set and the exploration of linear and nonlinear methods coupled with molecular interaction fields descriptors. J Chem Inf Model 56, 2042–2052.
Lombardo, F., Obach, R.S., Varma, M.V., Stringer, R., and Berellini, G. (2014). Clearance mechanism assignment and total clearance prediction in human based upon in silico models. J Med Chem 57, 4397–4405.
Lotfi Shahreza, M., Ghadiri, N., Mousavi, S.R., Varshosaz, J., and Green, J. R. (2017). A review of network-based approaches to drug repositioning. Brief Bioinform, doi: 10.1093/bib/bbx017.
Luo, Y., Zhao, X., Zhou, J., Yang, J., Zhang, Y., Kuang, W., Peng, J., Chen, L., and Zeng, J. (2017). A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun 8, 573.
Lusci, A., Pollastri, G., and Baldi, P. (2013). Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model 53, 1563–1575.
Ma, J., Sheridan, R.P., Liaw, A., Dahl, G.E., and Svetnik, V. (2015). Deep neural nets as a method for quantitative structure-activity relationships. J Chem Inf Model 55, 263–274.
Ma, X., Jia, J., Zhu, F., Xue, Y., Li, Z., and Chen, Y. (2009). Comparative analysis of machine learning methods in ligand-based virtual screening of large compound libraries. CCHTS 12, 344–357.
Maheshwari, S., and Brylinski, M. (2016). Template-based identification of protein-protein interfaces using eFindSitePPI. Methods 93, 64–71.
Martin-Montalvo, A., Mercken, E.M., Mitchell, S.J., Palacios, H.H., Mote, P.L., Scheibye-Knudsen, M., Gomes, A.P., Ward, T.M., Minor, R.K., Blouin, M.J., et al. (2013). Metformin improves healthspan and lifespan in mice. Nat Commun 4, 2192.
Mason, J.S. (2007). Introduction to the volume and overview of computer-assisted drug design in the drug discovery process. In Taylor, J.B., and Triggle, D.J., ed. Comprehensive Medicinal Chemistry II (Elsevier), pp. 1–11.
Matlock, M.K., Hughes, T.B., and Swamidass, S.J. (2015). XenoSite server: a web-available site of metabolism prediction tool. Bioinformatics 31, 1136–1137.
Mauri, A., Consonni, V., Pavan, M., and Todeschini, R. (2006). DRAGON software: An easy approach to molecular descriptor calculations. Match Commun Math Comput Chem 56, 237–248.
Mayr, A., Klambauer, G., Unterthiner, T., and Hochreiter, S. (2016). DeepTox: toxicity prediction using deep learning. Front Environ Sci, https://doi.org/10.3389/fenvs.2015.00080.
Melville, J., Burke, E., and Hirst, J. (2009). Machine learning in virtual screening. CCHTS 12, 332–343.
Mnih, V., Kavukcuoglu, K., Silver, D., Rusu, A.A., Veness, J., Bellemare, M.G., Graves, A., Riedmiller, M., Fidjeland, A.K., Ostrovski, G., et al. (2015). Human-level control through deep reinforcement learning. Nature 518, 529–533.
Mullard, A. (2017). The drug-maker’s guide to the galaxy. Nature 549, 445–447.
Myint, K.Z., and Xie, X.Q. (2010). Recent advances in fragment-based QSAR and multi-dimensional QSAR methods. IJMS 11, 3846–3866.
Ning, X., and Karypis, G. (2011). In silico structure-activity-relationship (SAR) models from machine learning: a review. Drug Dev Res 72, 138–146.
O’Boyle, N.M., and Hutchison, G.R. (2008). Cinfony—combining Open Source cheminformatics toolkits behind a common interface. Chem Cent J 2, 1–10.
OECD. (2014). Guidance document on the validation of (quantitative) structure-activity relationship [(Q)Sar] models. 69, 1–154.
Olivecrona, M., Blaschke, T., Engkvist, O., and Chen, H. (2017). Molecular de-novo design through deep reinforcement learning. J Cheminform 9, 48.
Pan, S.J., and Yang, Q. (2010). A survey on transfer learning. IEEE Trans Knowl Data Eng 22, 1345–1359.
Pereira, J.C., Caffarena, E.R., and Dos Santos, C.N. (2016). Boosting docking-based virtual screening with deep learning. J Chem Inf Model 56, 2495.
Pu, Y., Wang, W., Henao, R., Chen, L., Gan, Z., Li, C., and Carin, L. (2017). Adversarial symmetric variational autoencoder. arXiv:1711.04915v2.
Ramsundar, B., Liu, B., Wu, Z., Verras, A., Tudor, M., Sheridan, R.P., and Pande, V. (2017). Is multitask deep learning practical for pharma? J Chem Inf Model 57, 2068–2076.
Repasky, M.P., Shelley, M., and Friesner, R.A. (2007). Flexible Ligand Docking with Glide (John Wiley & Sons, Inc.).
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J Chem Inf Model 50, 742–754.
Sahoo, S., Adhikari, C., Kuanar, M., and Mishra, B. (2016). A short review of the generation of molecular descriptors and their applications in quantitative structure property/activity relationships. CAD 12, 181–205.
Sanchez-Lengeling, B., Outeiral, C., Guimaraes, G.L., and Aspuru-Guzik, A. (2017). Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). ChemRxiv Preprint.
Santos, R., Ursu, O., Gaulton, A., Bento, A.P., Donadi, R.S., Bologa, C.G., Karlsson, A., Al-Lazikani, B., Hersey, A., Oprea, T.I., et al. (2017). A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16, 19–34.
Schaarschmidt, J., Monastyrskyy, B., Kryshtafovych, A., and Bonvin, A. (2017). Assessment of contact predictions in CASP12: co-evolution and deep learning coming of age. Proteins 86, https://doi.org/10.1002/prot.25407.
Schmidhuber, J. (2015). Deep learning in neural networks: An overview. Neural Networks 61, 85–117.
Schneider, G., Funatsu, K., Okuno, Y., and Winkler, D. (2017). De novo drug design—Ye olde scoring problem revisited. Mol Inform 36, https://doi.org/10.1002/minf.201681031.
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R., and Wolfson, H.J. (2005). PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33, W363–W367.
Scott, D.E., Bayly, A.R., Abell, C., and Skidmore, J. (2016). Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov 15, 533–550.
Segler, M.H.S., Kogej, T., Tyrchan, C., and Waller, M.P. (2018). Generating focussed molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci 4, 120–131.
Sheridan, R.P. (2013). Time-split cross-validation as a method for estimating the goodness of prospective prediction.. J Chem Inf Model 53, 783–790.
Shin, W.H., Christoffer, C.W., and Kihara, D. (2017). In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 131, 22–32.
Shoemaker, R.H. (2006). The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 6, 813–823.
Sim, D.S.M. (2015)a. Drug Distribution (Springer International Publishing).
Sim, D.S.M. (2015)b. Drug elimination. In Chan, Y., Ng, K., and Sim, D., ed. Pharmacological Basis of Acute Care (Springer, Cham), pp. 37–47.
Smith, E.G., and Wiswesser, W.J. (1975). The Wiswesser Line-Formula Chemical Notation (New York: McGraw-Hill).
Spencer, M., Eickholt, J., and Jianlin Cheng, J. (2015). A deep learning network approach to ab initio protein secondary structure prediction. IEEE/ACM Trans Comput Biol Bioinf 12, 103–112.
Subramanian, G., Ramsundar, B., Pande, V., and Denny, R.A. (2016). Computational modeling of β-secretase 1 (BACE-1) inhibitors using ligand based approaches. J Chem Inf Model 56, 1936–1949.
Sushko, I., Salmina, E., Potemkin, V.A., Poda, G., and Tetko, I.V. (2012). ToxAlerts: a web server of structural alerts for toxic chemicals and compounds with potential adverse reactions. J Chem Inf Model 52, 2310–2316.
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., Simonovic, M., Roth, A., Santos, A., Tsafou, K.P., et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–D452.
Talele, T., Khedkar, S., and Rigby, A. (2010). Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. CTMC 10, 127–141.
Tian, S., Li, Y., Wang, J., Zhang, J., and Hou, T. (2011). ADME evaluation in drug discovery. 9. Prediction of oral bioavailability in humans based on molecular properties and structural fingerprints. Mol Pharm 8, 841–851.
Tishby, N., and Zaslavsky, N. (2015). Deep learning and the information bottleneck principle. Paper presented at: Information Theory Workshop, arXiv:1503.02406v1.
Todeschini, R., and Consonni, V. (2009). Molecular Descriptors for Chemoinformatics (Wiley-VCH).
Turner, J.R. (2010). New Drug Development (Springer New York).
Unterthiner, T., Mayr, A., Klambauer, G., Steijaert, M., Ceulemans, H., Wegner, J.K., and Hochreiter, S. (2014). Deep learning as an opportunity in virtual screening. Paper presented at: The Workshop on Deep Learning & Representation Learning.
Urban, G., Subrahmanya, N., and Baldi, P. (2018). Inner and outer recursive neural networks for chemoinformatics applications. J Chem Inf Model 58, 207–211.
Vakser, I.A. (2014). Protein-protein docking: from interaction to interactome. BioPhys J 107, 1785–1793.
Valkov, E., Sharpe, T., Marsh, M., Greive, S., and Hyvonen, M. (2012). Targeting protein-protein interactions and fragment-based drug discovery. Top Curr Chem 317, 145–179.
Vinyals, O., Blundell, C., Lillicrap, T., Kavukcuoglu, K., and Wierstra, D. (2016). Matching networks for one shot learning. Papers published at the Neural Information Processing Systems Conference.
Vohora, D., and Singh, G. (2017). Pharmaceutical Medicine and Translational Clinical Research (Academic Press).
Voosen, P. (2017). The AI detectives. Science 357, 22–27.
Wallach, I., Dzamba, M., and Heifets, A. (2015). AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. Mathematische Zeitschrift 47, 34–46.
Wang, C., and Zhang, Y. (2017). Improving scoring-docking-screening powers of protein-ligand scoring functions using random forest. J Comput Chem 38, 169–177.
Wang, J., Luo, C., Shan, C., You, Q., Lu, J., Elf, S., Zhou, Y., Wen, Y., Vinkenborg, J.L., Fan, J., et al. (2015). Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat Chem 7, 968–979.
Wang, N.N., Dong, J., Deng, Y.H., Zhu, M.F., Wen, M., Yao, Z.J., Lu, A.P., Wang, J.B., and Cao, D.S. (2016). ADME properties evaluation in drug discovery: prediction of Caco-2 cell permeability using a combination of NSGA-II and boosting. J Chem Inf Model 56, 763–773.
Wang, S., Sun, S., Li, Z., Zhang, R., and Xu, J. (2017). Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol 13, e1005324.
Wang, W., Yang, S., Zhang, X., and Li, J. (2014). Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 30, 2923–2930.
Weininger, D. (2011). Simplified Molecular Input Line Entry Specification.
Willett, P. (2006). Similarity-based virtual screening using 2D fingerprints. Drug Discovery Today 11, 1046–1053.
Wójcikowski, M., Zielenkiewicz, P., and Siedlecki, P. (2015). Open Drug Discovery Toolkit (ODDT): a new open-source player in the drug discovery field. J Cheminform 7, 26.
Wu, Z., Ramsundar, B., Feinberg, E.N., Gomes, J., Geniesse, C., Pappu, A. S., Leswing, K., and Pande, V. (2017). MoleculeNet: A benchmark for molecular machine learning. arXiv:1703.00564v2.
Xing, J., Lu, W., Liu, R., Wang, Y., Xie, Y., Zhang, H., Shi, Z., Jiang, H., Liu, Y.C., Chen, K., et al. (2017). Machine-learning-assisted approach for discovering novel inhibitors targeting bromodomain-containing protein 4. J Chem Inf Model 57, 1677–1690.
Xu, Y., Pei, J., and Lai, L. (2017). Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model 57, 2672–2685.
Xue, L.C., Dobbs, D., Bonvin, A.M.J.J., and Honavar, V. (2015). Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett 589, 3516–3526.
Yamanishi, Y., Araki, M., Gutteridge, A., Honda, W., and Kanehisa, M. (2008). Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 24, i232–i240.
Yap, C.W. (2011). PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32, 1466–1474.
Zaretzki, J., Matlock, M., and Swamidass, S.J. (2013). XenoSite: accurately predicting CYP-mediated sites of metabolism with neural networks. J Chem Inf Model 53, 3373–3383.
Zhang, Q.C., Petrey, D., Norel, R., and Honig, B.H. (2010). Protein interface conservation across structure space. Proc Natl Acad Sci USA 107, 10896–10901.
Zsoldos, Z., Reid, D., Simon, A., Sadjad, S.B., and Johnson, A.P. (2007). eHiTS: a new fast, exhaustive flexible ligand docking system. J Mol Graphics Model 26, 198–212.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21210003 and 81230076 to H.J., 81773634 to M.Z. and 81430084 to K.C.), the “Personalized Medicines—Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12050201 to M.Z.), National Key Research & Development Plan (2016YFC1201003 to M.Z.), and the National Basic Research Program (2015CB910304 to X.L.).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhong, F., Xing, J., Li, X. et al. Artificial intelligence in drug design. Sci. China Life Sci. 61, 1191–1204 (2018). https://doi.org/10.1007/s11427-018-9342-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11427-018-9342-2