
Abstract
Hepatitis B is a DNA virus affecting hundreds of millions of individuals worldwide. As the clinical sequelae of cirrhosis and hepatocellular cancer are increasingly recognized to be related to viral levels, the impetus increases to offer treatment to those previously not treated. With the development of more robust antivirals with reasonable safety profiles, long-term treatment is becoming more common. The oral nucleos(t)ide analogs have become the preferred first-line therapies for most genotypes of hepatitis B. Five are now available, all with different potencies and resistance profiles. Long-term data spanning several years are now available for most compounds in this arena. This article focuses on the common natural variants and those secondary to nucleos(t)ide therapy, as well as diagnostic methods to detect resistance.
Similar content being viewed by others

Introduction
The hepatitis B virus (HBV) is a DNA virus of the Hepadnaviridae family, similar to viruses that infect several avian and mammalian species. Eight genotypes exist with several subtypes defined by insertions or deletions of nucleotides, but with less than 10% overall variation among them [1]. Many viral genomic variants occur in nature, and many more occur under the pressure of therapy, namely direct-acting antivirals. Given the nature of the HBV genome, variants produced under selection pressure are archived indefinitely, which allows for a broadening of the quasispecies pool and subsequent cross-resistance to other drugs of similar structure. This article discusses the mechanisms of HBV viral resistance and the clinical implications.
Hepatitis B Virology
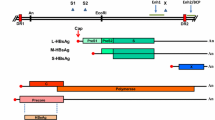
The HBV genome is comprised of two linear DNA strands that partially overlap and are thus partially double stranded, circular, and relaxed in configuration (rcDNA). The overall length is 3200 nucleotides with the DNA polymerase bound at the minus strand open 5′ end. There are four open reading frames (ORFs; Pre-S/S, Pre-C/C, Pol, and X), which overlap and are responsible for the synthesis of several sets of proteins [2].
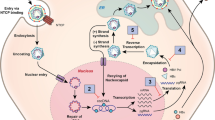
The replication of HBV is partially understood. The virus binds to the hepatocyte via a surface protein and enters the cell most likely through an endosome, although a definitive receptor has not been elucidated in human HBV [3]. Following viral entry into the cell, the HBV genome is delivered to the nucleus of the cell and forms a covalently closed circular (ccc DNA) configuration, which then acts as a transcription template for viral RNAs. Conversion of the rcDNA occurs via removal of the viral reverse transcriptase from the 5′ end of the minus strand, and removal of a short RNA segment from the 5′ end of the positive strand. These viral RNA gene segments are the two surface mRNAs, precore and pregenomic RNAs, and an X mRNA. The pregenomic messenger RNA (mRNA) leads to synthesis of the nucleocapsid unit and the viral reverse transcriptase, which attaches to the 5′ end of its own mRNA template, allowing for DNA synthesis. The cccDNA genome is produced intracellularly and remains throughout the life of the chronically infected host, allowing for viral genomic replication for decades after the initial infection.
Several major protein products are produced by HBV. The polymerase is necessary for viral replication, whereas the core gene encodes the capsid protein, or hepatitis B core antigen (HBcAg). The surface proteins are required for the shell of the virus, and the X gene product is required for in vivo infection in woodchucks; its role in human HBV is less clear, although most researchers focus on its potential carcinogenicity role [4, 5].
Naturally Occurring Mutations
Viral replication is estimated to be 1011 virions per day, with an error rate of 10−5 [6]. Both precore and core mutations are common, particularly in some geographic regions, and are associated with some genotypes more than others [7•]. The precore is a transition mutation, most commonly in the 1896 codon position (G1896A) and leads to a stop codon, resulting in decreased hepatitis B early antigen (HBeAg) production. Genotypes A and F rarely have precore mutations. The corresponding nucleotide of the stem-loop structure to position 1896 is position 1858, which is a cytidine in these genotypes, and the preferred pairing of cytidine is with a guanine at position 1896. Conversely, in genotypes where precore mutations are common, the 1858 position is often a thymidine, which is more stable when paired with adenine, hence the frequency of the precore mutation in genotypes B, C, and D [6].
Transcription of the precore and pregenomic RNAs is controlled by the basal core promoter (BCP). Two mutations described in this gene segment (A1762T, G1764A) are associated with decreased HBeAg production. Because of its location in the X ORF, two associated codon mutations of the X protein can also occur (L130M, V1311); however, binding of the hepatocyte transcription factor, HNF-1, can lead to pregenomic RNA transcription and production of HBeAg, even in the absence of wild-type HBV [8]. This sometimes leads to the presence of both HBeAg and hepatitis B early antibody (HBeAb) in the same patient.
Independent of viral load, mutations occurring naturally in the HBV genome lead to variable risks for hepatocellular carcinoma (HCC). In a retrospective review of 2762 Asian patients with largely HBeAg-negative disease, a multivariable-adjusted HR of developing HCC was 1.76 (95% CI , 1.19–2.61) for genotype C versus genotype B and 1.73 (95% CI, 1.13–2.67) for BCP A1762T/G1764A versus wild type. Risk was highest among participants infected with genotype C HBV, wild type for the precore 1896 variant and mutant for the BCP 1762/1764 variant (adjusted HR=2.99; 95% CI, 1.57–5.70, P < 0.001) [9].
The surface ORF encodes for three proteins, S (small), M (middle), and L (large). Of these, S is the major surface protein and is also part of the M and L components. It has both a hydrophobic and hydrophilic region, the latter being exposed on the surface of virions and subviral particles. This segment includes the highly conserved a determinant, which is recognized by protective antibodies. Flanked by subdeterminants d or y, and w or r, and associated with polymorphisms at amino acid positions 122 and 160, it defines the four major serologic subtypes that have associations with specific HBV genotypes. Point mutations in this a determinant lead to escape mutants from neutralizing antibodies. This finding is well described in transplant recipients receiving HBIG [10].
Mechanisms of Antiviral Drug Resistance
As described above, the HBV genome is susceptible to naturally occurring variants, most of which do not have adequate replication fitness to be detectable with standard commercially available assays. The high frequency of variants is in large part because of the lack of proofreading during reverse transcriptase, and the large replication space available in the liver to allow for the existence of a broad range of quasispecies. The apparent inefficiency of the virus in replication, however, also allows it to overcome immune pressure of the host, and hence it can escape immune suppression. The mechanism is most notable in the precore mutation, which reactivates years or decades after natural suppression of the wild-type HBeAg-positive virus by the host.
Hence, it is not surprising that pharmacologic pressure leads to selection of these natural variants as the wild-type virus is suppressed. The pharmacologic agent itself also determines the likelihood of drug resistance based on its potency and genetic barrier to mutations. Finally, other factors such as host compliance, adequate dosing, and drug metabolism will affect the rate of resistance.
Terminology of Drug Resistance
The definition of resistance is not as apparent as one might assume. The inability to suppress virus by more than 10-fold in the first 3 months of treatment in the setting of proper dosing is not considered resistance, but rather primary antiviral failure or nonresponse. Virus that is suppressed, but increases by more than 10-fold above nadir on two consecutive samples at least 1 month apart, is considered secondary treatment failure. This outcome can be the result of noncompliance, or most commonly resistance [11].
Other commonly used terms are genotypic mutations or polymorphisms, genotypic resistance, and phenotypic resistance. A genotypic mutation or polymorphism is an amino acid change not associated with antiviral resistance in vitro. Genetic resistance refers to viral populations with amino acid changes in the polymerase region that are associated with some degree of phenotypic resistance to an antiviral agent. Phenotypic resistance is determined by in vitro assessment of drug resistance to viral populations, and can often be quantified. Cross-resistance is a term indicating that drugs of a similar class will have similar resistance characteristics, which can be confirmed by phenotypic assays. Mutations can be archived as cccDNA and persist indefinitely, and can be passed on to others [12].
Genotypic and Phenotypic Resistance to HBV Nucleos(t)ide Analogs
The mainstays of therapy for HBV are oral nucleos(t)ide analogs (NA). Most of these were developed as a result of HIV research. Indeed, the HIV polymerase is also an RNA-dependent DNA polymerase, which is inhibited by nucleoside agents. Nucleosides and nucleotides (additional phosphate group) have three structural categories: L-nucleoside analogs such as lamivudine (LVD), emtricitabine (FTC), and telbivudine (LdT); acyclic phosphonates such as adefovir (ADV) and tenofovir (TDF); and deoxyguanosine analogs such as entecavir. Figure 1 summarizes the frequency of mutations over several years of clinical trials.

Genotypic resistance through 5 years for HBV nucleoside analogs in treatment-naïve patients
Resistance to NAs was noted with LVD, the first HIV/HBV crossover compound. The polymerase gene of HBV is 344 amino acids in length and has conserved domains lettered A to F. There are eight codons in the polymerase of HBV that are associated with primary drug resistance (codons 169, 180, 181, 184, 202, 204, 236, and 250) (Table 1).
Resistance to LVD was noted in the C domain (rtM204V/I), and was initially referred to as the YMDD motif mutation. A second compensatory mutation in the upstream B domain is often present as well (rtL180M), and improves replication of the variant. Other compensatory mutations are also described that allow for improved replication fitness, in particular the rtV173L mutation, which accompanies rtL180M and rtM204V in about 10% of cases with LVD use [13•]. The reported annual incidence of resistance to LVD is as high as 32% [14]. However, perhaps the best trial in recent years using LVD as the control arm was the 1367-patient Globe trial assessing LdT. In this 2-year study, the rate of LVD resistance in the HBeAg-positive cohort was 39.5% versus 25.1% for LdT, and in the HBeAg-negative group, it was 25.9% versus 10.8%. It must be noted that these resistance figures could be predicted by the week-24 antiviral response. In patients who had complete viral suppression by week 24, the resistance in the LVD arm was only 10%, whereas it was about 5% in the LdT group by week 104 [15]. This finding clearly suggests that resistance rates can be decreased if patients who are not responding to the drug are discontinued early. A rollover study for an additional 2 years of LdT was conducted on patients who were viral negative at entry. Although viral suppression was relatively good for those who remained in the study, 11.3% and 8% developed resistance at years 3 and 4, respectively, in spite of being viral-negative at study entry [16].
Entecavir (ETV) is an analog of d′-deoxyguanosine and inhibits both negative-and positive-strand DNA synthesis. In spite of its robust antiviral activity, mutations have been described in two patterns, both of which involve the rtM204V and the rtL180M. These mutation clusters are rtI169T + rtL180M + rtM204V + rtM250V and rtL180M + rtT184G + rtS202I and rtM204V. Because so many mutations are required to lead to viral breakthrough, it is rare for treatment-naïve patients to develop resistance to ETV. The cumulative probability of resistance in HBeAg-positive and HBeAg-negative patients is 1.2% at years 5 and 6 [17, 18]. The study design precluded continuation of drug in those with high viral loads at the end of years 1 and 2, the group most likely to develop resistance. This differs from earlier trial designs using other compounds, but is more reflective of clinical practice, in which drugs that are not working are not continued as monotherapy. Conversely, in patients with preexisting LVD mutations, the cumulative 6-year resistance to ETV is 57% [18]. Given the high rate of resistance in this group, most clinicians do not use ETV in LVD-experienced patients.
Resistance to ADV occurs primarily in the B and D domains of the polymerase, hence there is no cross-resistance with compounds exhibiting C domain mutations. The primary mutation in the D domain is the rtN236T mutation, which may also harbor other mutations such as rtP237H, rtN238T/D, rtV84M, and rtS85A, all of which can occur independently but are both less common and problematic [19]. The domain B rtA181V mutation leads to a fourfold decrease in sensitivity to ADV, and in cases of viral breakthrough is usually found in conjunction with the rtN236T mutation. In a 5-year study of HBeAg-negative patients, genotypic resistance to ADV monotherapy went from 3% at year 2 up to 29% at year 5 [20]. In the original HBeAg-positive study, because of errors in methodology, a clear picture of resistance could not be determined, but was reported as 20% at week 235 [21]. As with LdT and LVD, patients with robust viral suppression by week 48 in the case of ADV had less than 10% resistance by week 192 of therapy, compared with nearly 50% in those not suppressed by week 48 [20].
The other acyclic phosphonate compound is TDF, which has completed 3 years of an intended 8-year prospective study. The first year compared TDF with ADV and found that no resistance occurred in either arm. The ADV arm was changed to open-label TDF at the beginning of year 2, and all patients are to continue for an additional 7 years on TDF. The 3-year data were presented at an international congress, and no resistance or signature mutations to TDF have been described. An early report of rtA194T in the B–C domain border suggested possible increase in IC50 when found with the rtL180M and rtM204V mutations. However, this suggestion has not been borne out in clinical studies. In the pivotal study, patients who do not have complete viral suppression can be changed to combination TDF and FTC (emtricitabine, 200 mg) at the discretion of the investigator. Of the 51 patients in both the HBeAg-positive and HBeAg-negative studies, 34 had FTC added, and 17 remained on monotherapy. No difference was seen in viral kinetics up to week 144 after that point whether the patient was on combination therapy or remained on monotherapy with TDF, and the majority of patients had complete suppression [22, 23].
Entering the third year of therapy, 22 of 556 patients in both studies were viremic with more than 400 copies/mL. Of these 22 patients, 11 were on monotherapy with TDF and 11 were on combination TDF and FTC. Genotypic testing was done using population di-deoxy sequencing of the HBV pol/RT. Conserved site changes were noted in a total of five patients, usually at a single site, and included rtR51K, rtL180M, rtM204V, rtA181T, rtR192H, rtG152E, rtN236T, and rtR274Q. None of the conserved changes were associated with persistent viral breakthrough; none showed altered susceptibility to TDF in vitro using HepG2 cells transiently transfected with a pool of recombinant HBV plasmid DNA or mutant virus created by site-directed mutagenesis in the pCMV/HBV (genotype D) or pHY92 (genotype A) backbone. The addition of FTC did not change the viral kinetics, nor did it appear to affect conserved site changes [24].
Diagnosis of Resistance
Resistance testing is still an evolving practice and still not done consistently outside research settings. Many clinicians monitor biochemical parameters that indicate a histologic flare of disease. An increasing number of clinicians are now monitoring HBV DNA levels using polymerase chain reaction (PCR) technology. This technology is a more sensitive test to determine NA failure because biochemical breakthrough may lag behind by months, or may not occur at all. However, a rise in DNA or failure of DNA to drop or continue to decline after initial response (plateau) is not necessarily caused by resistance or NA failure. Patient compliance is a critical component to successful therapy and must be assessed. Additionally, some patients will have suboptimal response to a given drug as a result of pharmacogenomic parameters (eg, host polymorphisms) that lead to inefficient drug metabolism.
To determine resistance, ideally specific genotyping of the polymerase nucleotide sequence should be obtained at baseline and at the time of virologic breakthrough during treatment. Because the baseline data are usually not available for practical reasons, the previously described and known mutations associated with that particular drug can be determined via viral genotyping (Table 2).
Genotypic Resistance Testing
Direct DNA sequencing using population-based PCR is a common method of genotyping, but the sequencing usually has a sensitivity of about 20% of the total quasispecies pool. Amplification of clones of PCR products is more sensitive but requires multiple clonal analyses. A commercially available DNA sequencing assay is the TRUGENE™ HBV Genotyping Kit (TRUGENE HBV; Bayer HealthCare LLC, Tarrytown, NY). A more sensitive direct-sequencing method is restriction fragment-length polymorphism (RFLP) analysis, which is sensitive to 5% of the total viral pool, but requires a separate set of endonuclease reactions for each mutant [25, 26••].
The hybridization-based technologies include real-time PCR, sequencing with microchips using oligonucleotide microarrays to improve sensitivity down to 10%, and the most sensitive method within this group, matrix-assisted laser-desorption ionization time of flight mass spectrometry (MALDI-TOF MS) analysis of DNA fragments, which is sensitive to 1% of the total viral pool. Hybridization is limited by the need to have specific probes for every mutant. The available commercial assay is the line-probe assay (INNO-LiPA; Innogenetics, Ghent, Belgium), which uses a series of short oligonucleotide probes to assess for mutant sequences, with a sensitivity down to 5%. For most clinical purposes, this option is very good [26••].
The newest and most powerful technology is ultra-deep pyrosequencing (UDPS), which amplifies a DNA segment and through the use of multiple primer pairs, sequences single DNA strands via synthesis of complementary strands. It has sensitivity of 0.1% and will find mutations that have not been found with other less sensitive methods [27•, 28]. This technology may become the standard in clinical research arenas (Table 2).
Phenotypic Testing
The presence of genotypic mutations must be placed into the clinical context of how this affects therapy. The use of in vitro phenotypic analysis allows assessment of NA susceptibility of a particular mutant clone. The comparison is made to wild type and is reported as the effective concentration of the NA required to inhibit 50% of the viral target (EC50). In general, a 10-fold change is considered clinically meaningful, but changes higher than that sometimes do not lead to drug failure in vivo, and conversely, changes less than that occasionally impact drug efficacy.
Most phenotyping methods involve the use of vectors. Examples include an enzyme assay that uses polymerase expressed in insect cells using a baculovirus vector, transient transfection of recombinant HBV replication-stable clones, and a recombinant HBV/Baculovirus in hepatocyte cell lines. Permanent cell lines that express mutant virus are a more reproducible method. However, as new mutations are discovered, more cell lines need to be produced [29] (Table 2).
Prevention and Management of Resistance
The first step in preventing resistance to NAs is to select appropriate patients for treatment. Patients with a history of noncompliance or those unable to remain on drug are more likely to develop resistance through their actions. The second step is to select a drug with a high barrier to resistance and to monitor regularly to detect resistance early.
Once the decision is made to start medication in a given patient, the goal must be complete viral suppression, preferably within 1 year. In cases of viral load plateau, an alternative drug or, rarely, a second drug may be necessary. The use of combination NAs was intensively studied when the available compounds were not potent and had low barriers to resistance (LVD and ADV). This strategy worked reasonably well and decreased resistance and multidrug resistance [30]. However, with more potent single-agent compounds (eg, ETV and TDF), there is little role for combination, except in the setting of suboptimal response to one or the other. The current American Association for the Study of Liver Diseases guidelines on HBV has an algorithm for drug resistance, which suggests the addition of a second drug if resistance to LVD, LdT, or ADV is discovered [7•]. However, recent data suggest that monotherapy with TDF may be sufficient for the commonly found mutations associated with LVD, LdT, and ADV [22, 31]. Further study is required to confirm these initial impressions. For patients who develop either ETV resistance or, more commonly, lack of complete response, the addition of TDF in small series has been very successful [32]. Further study is needed to determine if the combination is necessary or if switching to TDF is adequate.
At this time, few indications exist to perform baseline drug resistance testing before initiating therapy if the patient is treatment-naïve. Currently available assays are not sensitive enough and the results usually would not alter management. However, once a patient is started on treatment, careful assessment of DNA levels every 3 months is a must until viral negativity is achieved. Using drugs with a high barrier to resistance is preferred, and with these compounds most patients should achieve undetectable levels within 1 year. For those still viremic at week 48, but who continue to show viral decline, continued therapy with a single agent is indicated. For those who have a viral plateau, resistance testing is suggested, along with assurance of patient compliance. The addition of a second agent, or in some cases, switching to another agent may be necessary to achieve viral suppression. Once virus is undetectable, laboratory monitoring of DNA can be performed every 6 months, particularly for compliant patients on ETV or TDF. For patients who are treatment-experienced with a nucleoside agent such as lamivudine, baseline monitoring may be indicated, but because of the lack of sensitivity of most assays, even existing mutations that are archived may not be detected. On-treatment monitoring is imperative in this group to detect existing mutations that emerge as wild-type virus is suppressed. Medication adjustment can be made accordingly in those cases.
Conclusions
Resistance to nucleoside agents targeting hepatitis B is an important public health issue in terms of pharmacoeconomics and the potential for multidrug-resistant HBV that is passed to treatment-naïve individuals. Because the current antiviral agents have only one target, it is logical to restrict clinical use to only the most potent agents with the lowest risk for resistance. For the oral agents, this is tenofovir and entecavir. Both have reasonable safety profiles and low resistance in treatment-naïve patients. Ideally, new targets of drug therapy will allow for true combination studies to maximize viral suppression and inhibit the development of resistance. In lieu of this, however, the responsible use of current single agents, perhaps the use of two agents in selected settings, and careful monitoring for compliance and viral breakthrough are necessary to minimize viral resistance in HBV infection.
References
Papers of particular interest, published recently, have been highlighted as:• Of importance•• Of major importance
Myers R, Clark C, Khan A, et al.: Genotyping hepatitis B virus from whole-and sub-genomic fragments using position-specific scoring matrices in HBV STAR. J Gen Virol 2006, 87:1459–1464.
Schaefer S: Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J Gastroenterol 2007, 13:14–21.
DeMeyer S, Gong JZ, Suwandhi W, et al.: Organ and species specificity of hepatitis B virus (HBV) infection: a review of literature with a special reference to preferential attachment of HBV to human hepatocytes. J Viral Hepatol 1997, 4:145–153.
Zoulim F, Saputelli J, Seeger C: Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol 1994, 68:2026–2030.
Takada S, Tsuchida N, Kobayashi M, et al.: Disruption of the function of tumor-suppressor gene p53 by the hepatitis B virus X protein and hepatocarcinogenesis. J Cancer Res Clin Oncol 1995, 121:593–601.
Gunther S, Fischer L, Pult I, et al.: Naturally occurring variants of hepatitis B virus. Adv Virus Res 1999, 52:25–137.
• Lok AS, McMahon BJ: Chronic hepatitis B: update 2009. Hepatology 2009, 50:661–662. This article is a very comprehensive overview of hepatitis B treatment guidelines.
Zheng Y, Li J, Ou JH: Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J Virol 2004, 78:6908–6914.
Yang HI, Yeh SH, Chen PJ, et al.: Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst 2008, 100:1134–1143.
Grottola A, Buttafoco P, Del Buono MG, et al.: Pretransplantation pre-S2 and S protein heterogeneity predisposes to hepatitis B virus recurrence after liver transplantation. Liver Transpl 2002, 8:443–448.
Locarnini S, Hatzakis A, Heathcote J, et al.: Management of antiviral resistance in patients with chronic hepatitis B. Antivir Ther 2004, 9:679–693.
Zhou T, Saputelli J, Aldrich CE, et al.: Emergence of drug-resistant populations of woodchuck hepatitis virus in woodchucks treated with the antiviral nucleoside lamivudine. Antimicrob Agents Chemother 1999, 43:1947–1954.
Delaney WE IV, Yang H, Westland CE, et al.: The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J Virol 2003, 77:11833–11841.
Lai CL, Dienstag J, Schiff E, et al.: Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis 2003, 36:687–696.
Liaw YF, Gane E, Leung N, et al.: 2-Year GLOBE trial results: telbivudine is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology 2009, 136:486–495.
Wang Y, Thongsawal S, Gane E, et al.: Efficacy and safety outcomes after 4 years of telbivudine treatment in patients with chronic hepatitis B (CHB). Hepatology 2009, 50(Suppl 4):A482.
Tenney DJ, Pokomowski KA, Rose RE, et al.: Entecavir at 5 years shows long-term maintenance of high genetic barrier to hepatitis B virus resistance. Hepatol Int 2008, 2:302–303.
Tenney DJ, Pokorowski KA, Rose RE, et al.: Entecavir maintains a high genetic barrier to HBV resistance through 6 years in naive patients. J Hepatol 2009, 50(Suppl 1):S10.
Bartholomeusz A, Locarnini SA: Antiviral drug resistance: clinical consequences and molecular aspects. Semin Liver Dis 2006, 26:162–170.
Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al.: Long-term therapy with adefovir dipivoxil for HBeAg negative chronic hepatitis B for up to 5 years. Gastroenterology 2006, 131:1743–1751.
Marcellin P, Chang TT, Lim SG, et al.: Long-term efficacy and safety of adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. Hepatology 2008, 48:750–758.
Marcellin P, Buti M, Krastev Z, et al.: Three years of tenofovir disoproxil fumarate (TDF) treatment in HBeAg-negative patients with chronic hepatitis B (Study 102), preliminary analysis. Hepatology 2009, 50(Suppl 4):A481.
Heathcote E, Gane EJ, De Man RA, et al.: Three years of tenofovir disoproxil (TDF) treatment in HBeAg-positive patients (HBeAg+) with chronic hepatitis B (Study 103), preliminary analysis. Hepatology 2009, 50(Suppl 4):A483.
Snow-Lampart A, Chappell B, Curtis M, et al.: Resistance surveillance for up to 144 weeks in HBeAg + and HBeAg-hepatitis B patients treated with tenofovir DF showed no relationship between virologic breakthrough and emergence of genotypic changes in HBV polymerase. Hepatology 2009, 50(Suppl 4):A480.
Sablon E, Shapiro F: Advances in molecular diagnosis of HBV infection and drug resistance. Int J Med Sci 2005, 2:8–16.
•• Zoulim F, Locarnini S: Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009, 137:1593–1608. This article provides an exceptional review of mechanisms of resistance and prevalence of current mutations.
• Shendure J, Ji H: Next-generation DNA sequencing. Nat Biotechnol 2008, 26:1135–1145. This article is a good overview of emerging diagnostics.
Margeridon-Thermet S, Shulman NS, Ahmed A: Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J Infect Dis 2009, 199:1275–1285.
Zoulim F: In vitro models for studying hepatitis B virus drug resistance. Semin Liver Dis 2006, 26:171–180.
Lampertico P, Viganò M, Manenti E, et al.: Low resistance to adefovir combined with lamivudine: a 3-year study of 145 lamivudine-resistant hepatitis B patients. Gastroenterology 2007, 133:1445–1451.
Kitrinos K, Myrick F, Curtis M, et al.: In vitro tenofovir sensitivity of HBV populations from clinical specimens containing rtA181T/V and/or rtN236T. Hepatology 2009, 50(Suppl 4):A434.
Petersen J, Lutgehetmann M, Zoulim F, et al.: Entecavir and tenofovir combination therapy in chronic hepatitis B: rescue therapy in patients with advanced fibrosis and multiple previous treatment failures. Results from an international multicenter cohort study. Hepatology 2009, 50(Suppl 4):A405.
Disclosure
No potential conflict of interest relevant to this article was reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Poordad, F., Chee, G.M. Viral Resistance in Hepatitis B: Prevalence and Management. Curr Gastroenterol Rep 12, 62–69 (2010). https://doi.org/10.1007/s11894-009-0088-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-009-0088-1