
Abstract
Purpose of Review
Despite the clinical successes of immune checkpoint blockade across multiple tumor types, many patients do not respond to these therapies or become resistant after an initial response. This underscores the need to improve our understanding of the molecular determinants of response to guide more personalized and rational utilization of these therapies. Here, we describe available biomarkers of checkpoint blockade activity by classifying them into four major categories: tumor-intrinsic, immune microenvironmental, host-related, and dynamic factors.
Recent Findings
The clinical experience accumulated thus far with checkpoint blockade now offers the opportunity to comprehensively study the molecular and immune features associated with response. This is yielding a growing set of biomarkers whose integration will be key to more accurately predict clinical outcome.
Summary
We propose a model for systematic assessment of available baseline and dynamic biomarkers in relationship with patients’ outcomes. This will improve our understanding of the tumor-immune interactions and dynamics that predict a clinical response and will provide key information to develop more personalized and effective treatment strategies.
Similar content being viewed by others

Introduction
The recognition that CTLA-4 and PD-1 are key regulators of T cell responses led to the hypothesis that blocking these molecules could be a viable option to activate immune responses against cancer [1, 2]. CTLA-4 and PD-1 are both up-regulated on the T cell surface upon T cell receptor (TCR) engagement and CD28 co-stimulation. In addition, CTLA-4 is constitutively overexpressed on regulatory T cells. CTLA-4 and PD-1 respond to different ligands involved in different phases of the immune response [3]. CTLA-4 outcompetes with CD28 for binding to CD80 and CD86 on professional antigen-presenting cells (APCs), which are mainly located in T cell priming sites (i.e., secondary lymphoid organs). PD-1 binds to its ligands, PD-L1 and PD-L2, which are typically up-regulated on inflamed tissues in response to IFN-γ stimulation to prevent unwanted immune attack and maintain tissue homeostasis. The CTLA-4 and PD-1 downstream signaling pathways can both mitigate TCR activation cascade by recruiting and activating protein phosphatases SHP-1 and/or SHP-2. Because of the central role of CTLA-4 and PD-1 in controlling respectively T cell priming and effector function, these molecules have been defined as immune checkpoints. In light of the localization of these immune checkpoints on the T cell surface, a logical approach to block their function was the use of antagonist antibodies. This immunotherapeutic strategy has revealed unprecedented successes in the treatment of multiple cancer types, leading to a renewed confidence of the scientific community in this approach, which has been thus considered again as a viable strategy for cancer treatment. CTLA-4 blockade with the fully human IgG1 antibody ipilimumab for the first time showed the possibility to extend survival in metastatic melanoma patients [4, 5••] and received the US Food and Drug administration (FDA) approval for the treatment of this disease in 2011. PD-1 pathway blockade has extended these clinical successes to additional tumor types. Blocking PD-1 with the antagonist antibody nivolumab or pembrolizumab has been approved for the treatment of non-small cell lung cancer (NSCLC, squamous and non-squamous carcinoma), Merkel cell carcinoma, head and neck squamous cell carcinoma, urothelial and kidney cancers, microsatellite instability (MSI)-high cancers (such as MSI-high colorectal cancer), refractory HL, hepatocellular carcinoma, and gastric cancer [6]. Similarly, PD-L1-blocking antibodies atezolizumab, avelumab, and durvalumab have become part of standard of care for patients with advanced NSCLC, urothelial carcinoma, or Merkel cell carcinoma [6]. Clinical responses to these agents are generally durable across tumor type; however, they occur in a minority of patients, with few exceptions in HL, desmoplastic melanoma, Merkel cell carcinoma, and MSI-high cancers that respond to checkpoint blockade therapy in ≥ 50% of cases [6]. Understanding the determinants of response and resistance to checkpoint blockade is therefore key for more successful utilization of these agents. The experience accumulated thus far with checkpoint blockade now offers the opportunity to comprehensively study the molecular and immune features associated with response. This is yielding a growing set of biomarkers whose integration will be key to more accurately predict clinical outcome. Given that checkpoint blockade is becoming the foundation of multiple-combination anti-cancer programs, the capacity to identify patients more likely to respond to immunotherapy and anticipate their outcome is extremely critical. Here, we provide an overview of the main biomarkers of response to checkpoint blockade identified so far. Importantly, most of these factors are not merely indicators of clinical outcome, but often play a mechanistic role in determining response and, as such, can also constitute mechanisms of responsiveness/refractoriness. We classify these biomarkers into four major categories, depending on their origin (tumor-intrinsic, microenvironmental, and host-related biomarkers) and relevance during treatment (dynamic biomarkers). We then discuss the importance of systematically assessing baseline and dynamic biomarkers for guiding proper therapeutic decisions.
Tumor-Intrinsic Biomarkers
Tumor-intrinsic biomarkers refer to tumor-specific factors that alter the capability of tumor cells of being recognized by immune cells or responding to immune stimuli. Aligned with the fact that tumor elimination induced by checkpoint blockade is primarily mediated by T cells, the major tumor-intrinsic factors associated with response to checkpoint blockade are:
-
Tumor mutation burden, in terms of non-self-peptide load generated by mutations
-
Tumor PD-L1 expression, as a measure of tumor sensitivity to PD-1 pathway blockade or more generally a feature of adaptive resistance to an already-generated immune response
-
IFN-γ responsiveness and tumor antigen presentation
Mutation and Neoantigen Tumor Load
We and others have reported that tumor somatic mutational burden [non-synonymous mutations and insertion/deletion (indel) variants] is associated with clinical efficacy of checkpoint blockade in advanced melanoma, NSCLC [7•, 8•, 9, 10, 11•]. The hypothesis is that, during its development, the tumor acquires mutations, some of which create immunogenic neoantigens that can promote activation of specific cytotoxic T cell responses, in particular when immune checkpoints are blocked. However, the more the tumor diverges from self (due to mutations), the more it needs to evade immune surveillance to survive and progress. Reducing immune constraints for (neo)antigen recognition with checkpoint blockade may render tumor cells more vulnerable to a specific T cell attack. Based on this principle, either total tumor mutational burden (clonal + passenger mutations), or clonal mutations (shared by all tumor cells), or the amount of predicted immunogenic neopeptides (capable of binding to patient-specific HLA molecules) have been intensively studied in association with response to checkpoint blockade. Snyder et al. performed whole exome sequencing on tumor tissue biopsies collected from advanced melanoma patients either before or after treatment with ipilimumab and showed that patients with tumors harboring more than 100 mutations had a significantly longer survival [7•] (Table 1). Similarly, Van Allen et al. analyzed mutational burden in tumor biopsies from melanoma patients before treatment with ipilimumab and found that patients experiencing clinical benefit were enriched for tumors with relatively higher numbers of non-synonymous mutations and neoantigens predicted to bind to patient-matched HLA class I molecules [9] (Table 1). Similar results were observed in melanoma and NSCLC patients treated with PD-1 blockade monotherapy [8•, 10] and more recently in NSCLC patients treated with combination PD-1 and CTLA-4 blockade [12] (Table 1). In general, total tumor mutational burden was found to correlate with the amount of potentially immunogenic predicted neoantigens [9, 12, 13]. These results helped explain the observation that across all human cancer types, those with relatively high numbers of genetic somatic mutations are generally more responsive to checkpoint blockade [14••]. These findings led to the hypothesis that tumors harboring mutations in mismatch-repair (MMR) genes, having a reduced capacity to correct DNA replication errors, could be optimal targets for immunotherapy with checkpoint blockade. One of the most common outcomes of MMR deficiency is microsatellite instability (MSI). According to this hypothesis, MSI-high solid tumors were found to be particularly responsive to PD-1 blockade [15••, 16], which was thus granted a specific indication for malignant diseases with these genetic abnormalities independent of the tissue of origin. Aligned with this concept, mutations in genes that control genomic stability, such as BRCA2 [17] and p53 [18], were found more frequently altered in responding tumors in melanoma and NSCLC patients treated with checkpoint blockade [10, 12, 19], further highlighting an association between mutations in genes controlling DNA stability/repair, tumor somatic mutation load, and responsiveness to immunotherapy. As an additional modality to enrich for tumors more likely able to respond to checkpoint blockade, quantification of indels has been proposed based on the rationale that indels, generating frameshift mutations, can produce higher numbers of immunogenic neoantigens [11•]. Interestingly, all checkpoint-blockade-approved tumor types have significantly higher frameshift neoantigens compared to malignancies for which these therapies have not been approved [11•]. This is particularly relevant for renal cell carcinoma, which is characterized by a low burden of somatic mutations, but, having on average elevated proportions of coding indels [11•], may result immunogenic and can respond to checkpoint blockade [20]. Given the impact of somatic tumor mutational load on response to checkpoint blockade, facilitating assessment and quantification of this parameter is desirable to implement its utilization and improve patients’ management. Toward this goal, it has recently been demonstrated that tumor exonic mutation burden can be faithfully estimated by targeted next-generation sequencing of most common oncogenes, such as those included in MSK-IMPACT (468 genes) or FoundationOne (315 genes) panel tests, which are routinely employed for tumor molecular diagnosis [12].
It has become clear that tumor immunogenicity is not always a linear function of tumor mutational burden and neoantigen load. The quality of tumor neoantigens is a key determinant of the depth and duration of response to checkpoint blockade. Specifically, the amount of clonal (present in all tumor cells) compared to subclonal (present only in a subset of tumor cells) tumor neoantigens was found to better correlate with survival in melanoma and NSCLC patients treated respectively with ipilimumab or anti-PD-1 monotherapies [21•]. This highlights the dual impact of tumor heterogeneity on sensitivity to immunotherapy: by promoting divergence from self, it may favor tumor immune recognition, but this comes at the expense of an increased degree of diversity within the tumor cells themselves, which may favor escape form immune reactions.
Tumor PD-L1 Expression
PD-L1 expression on the tumor cell surface has a dual value: on the one hand, it indicates the presence of a specific target for checkpoint blockade, on the other it works as a surrogate biomarker of intra-tumor T cell infiltration and/or T cell responses. This is because PD-L1 expression is induced by IFN-α and IFN-γ, via JAK/STAT signaling during an ongoing immune response, presumably as a defense mechanism to ensure tissue homeostasis. In tumor cells, PD-L1 overexpression may result from the same mechanism and leads to “adaptive” immune resistance to ongoing inflammation. Accordingly, PD-L1 expression was found to be associated with poor prognosis and reduced overall survival in patients with kidney [22], esophageal [23], gastric [24], and ovarian cancers [25], underscoring a mechanism of resistance to spontaneous anti-tumor immune responses. In light of currently available PD-1 pathway blocking agents, the interpretation of PD-L1 overexpression has now changed into a sign of potential tumor vulnerability to immunotherapy. In a phase I trial with nivolumab in patients with multiple advanced solid cancers, baseline PD-L1 expression in > 5% tumor cells was associated with a higher overall response rate [26••]. In subsequent studies with PD-1 blockade, the absolute or binary predictive value of this parameter has not been entirely consistent (Table 2). The use of different assays, antibodies, and scoring methods to assess PD-L1 expression across these studies may contribute to the difficulties to reach a definitive agreement on this biomarker [39]. Currently, PD-L1 expression status is part of the therapeutic indication of pembrolizumab in metastatic NSCLC, with 1 and 50% PD-L1+ tumor cells being respectively the cutoff for second- and first-line treatment based on two studies demonstrating efficacy in these patient populations [38, 40]. In other malignancies, the predictive value of tumor PD-L1 expression has to be determined. In the CheckMate 067 phase III trial comparing checkpoint blockade monotherapy versus combination in previously untreated advanced melanoma patients, nivolumab + ipilimumab achieved a slightly improved survival outcome in patients with tumor PD-L1 expression levels < 1%, highlighting the potential benefit of using combination checkpoint blockade in these cases [37••].
PD-L1 up-regulation on tumor cells is not only a response to microenvironmental immune stimuli but can also be induced as part of tumor-intrinsic oncogenic signaling cascades, such as JAK-STAT [41, 42], Akt/mTOR [43], MYC [44], and RAS [45], by loss of function in tumor suppressor genes, such as PTEN [46], and by genetic alteration in the PDL1 gene itself, including amplification [47] and structural variations causing PDL1 3′-UTR disruption and transcript stabilization [48]. An emblematic example of these mechanisms is HL, where the recurrent chromosome 9p24 amplification leads to PDL1, PDL2, and JAK copy number increase. The genetic basis for PD-L1 overexpression in HL and the characteristically pronounced but ineffective inflammatory response in the involved lymph nodes constituted the rationale to test PD-1 blockade against this malignancy. Both nivolumab and pembrolizumab have been granted accelerated approval by the FDA for the treatment of refractory classical HL based on exceptional anti-tumor activity [49••, 50]. Of note, the anti-tumor activity of PD-1 blockade in HL seems to rely more on CD4+ than CD8+ T cell-mediated mechanisms, as most of responding tumors have dysfunctional class-I (but not class-II) MHC antigen presentation [35]. The impact of oncogene-driven/genetic-driven relative to immune-driven tumor PD-L1 overexpression has not been systematically evaluated. It is logical to think that tumors overexpressing PD-L1 because of intrinsic mechanisms of PD-L1 induction, without substantial T cell infiltration, will not be particularly sensitive to PD-1 pathway blockade.
IFN-γ Responsiveness and Antigen Presentation
Tumor expression of the IFN-γ receptor signaling cascade and antigen presentation machinery are important factors determining response to immunotherapy. IFN-γ is produced mainly by natural killer (NK) cells or activated antigen-specific T cells. When sensed by tumor cells, IFN-γ promotes direct anti-tumor effects by limiting tumor growth and survival and indirect immune-mediated anti-tumor activity by favoring tumor immune recognition via the JAK/STAT pathway, leading to up-regulation of inducible proteasome subunits, transporters associated with antigen processing (TAP), class I and class II MHC molecules, and T cell-recruiting chemokines [51, 52]. Given the harmful consequences of IFN-γ-mediated signaling, PD-L1 is concomitantly induced to control for excessive unwanted T cell responses against host damaged tissues. In the setting of a tumor, this mechanism can favor immune escape and indicates adaptive immune resistance. Signs of adaptive immune resistance (i.e., PD-L1 overexpression in association with elevated IFN-γ and immune infiltrates) in turn may indicate sensitivity to immunotherapy with checkpoint blockade, in particular PD-1/PD-L1 blockade. However, under the selective pressure of strong anti-tumor immune responses, the tumor can evolve to lose the capacity to respond to IFN-γ by acquisition/selection of loss-of-function mutations or epigenetic inactivation of JAK1/JAK2 genes, becoming insensitive to immunotherapy [53, 54]. Of note, JAK1 loss-of-function precludes signaling from both type-I [IFN-α, IFN-β] and type-II interferons (IFN-γ), while JAK2 loss-of-function primarily abolishes responsiveness to IFN-γ [55]. The tumor can also directly lose the capacity of being recognized by immune cells by acquisition/selection of loss-of-function mutations in the antigen presentation machinery, with the most common genetic alterations occurring in the beta 2-microglobulin (β2M) gene [56, 57•, 58]. β2M serves as a structural protein to properly assemble class I MHC on the cell surface. The tumor evolutionary advantage of losing β2M stems from the possibility to inactivate all class-I HLA alleles through the loss of a single gene (β2M). Tumor loss of IFN responsiveness and antigen presentation machinery may result from a natural immunoediting process that can be accelerated during treatment with immunotherapy leading to refractoriness. Importantly, CRISPR-Cas9-genome-editing-based screening of genes affecting the capacity of melanoma cell lines to be recognized and killed by CD8+ T cells objectively demonstrated that loss of genes involved in IFN-γ signaling and/or antigen presentation is a top mechanism leading to immune resistance in vitro and in vivo [59, 60]. Inactivating mutations of class I HLA and β2M genes are significantly enriched in immunogenic tumors characterized by high mutation burden (i.e., MSI-high tumors) and correlate with T cell infiltration and cytolytic activity, highlighting an active mechanism of immune escape under the pressure of on-going anti-tumor T cell responses [13, 61, 62]. In addition, analysis of loss-of-function mutations in the type I IFN response pathway in TCGA datasets of skin cutaneous melanoma, breast invasive carcinoma, and prostate carcinoma underscored an association between the presence of these alterations and poor clinical outcome [55, 63]. Acquisition of loss-of-function mutations in the IFN response pathway and antigen presentation machinery during treatment with checkpoint blockade have been also observed and reported to occur in association with the development of resistance in particular in melanoma and lung cancer patients [56, 57•, 58].
An interesting exception to this model is HL, which largely loses expression of class-I MHC and β2M [35] and can still respond to PD-1 blockade. Recent work showed the correlation between expression of class-II MHC and clinical response of HL patients to PD-1 blockade and the predictive potential of class-II MHC expression for prolonged survival in these patients, indicating the involvement of CD4+ T cells in the anti-lymphoma activity of anti-PD-1 [35].
Together, these findings underscore the importance of assessing the functional status of the IFN response pathway and antigen presentation machinery in tumor cells at baseline and during treatment with checkpoint blockade to anticipate clinical outcome and guide proper therapeutic decisions. In this context, particular attention should be given to those cases showing loss of heterozygosity in JAK1/2 and β2M, because, under a strong immune selective pressure, these abnormalities can predispose to the acquisition of inactivating mutations in the second allele resulting in complete loss of gene function, which will render the tumor refractory to immunotherapy [56, 63]. To overcome resistance conferred by deficiencies in the antigen presentation process, the use of NK-stimulatory or adoptive therapies represents a rational approach. Preclinical evidence supports the relevance of potentiating NK surveillance against β2M-deficient tumors [56], and checkpoint blockade in combination with antibodies blocking NK inhibitory receptors (such as inhibitory KIR) is actively investigated in patients with solid and hematologic malignancies (NCT01592370, NCT03335540, NCT03341936, NCT03203876, NCT01714739) [64].
Microenvironmental Biomarkers
Throughout its development, the tumor establishes interactions within its environment, with the immune system being the most dynamic component. Tumor-immune actions and reactions occur in an ongoing dialog that shapes tumor evolution toward immune escape [65]. Depending on the stage of tumor evolution with respect to immune escape, this dialog may be more or less coherent. For example, elevated mutational burden and neoantigen load in tumors with intact antigen presentation machinery may very likely associate with abundant T cell infiltration. This in turn can activate immune suppressive mechanisms, including expression of inhibitory factors (i.e., PD-L1) and recruitment of immunosuppressive cells [Tregs, myeloid-derived suppressor cells (MDSCs), M2 macrophages] as a physiologic feedback loop to preserve tissue homeostasis. A similar situation can indicate potential responsiveness to immunotherapy. However, tumor and microenvironmental immune features are not always “in phase” in these bidirectional interactions [66, 67] and predicting the outcome of immunotherapy based only on one of these two sides may not necessarily be successful. Assessment of microenvironmental parameters, in particular immune cells, is thus as important as evaluation of tumor-intrinsic biomarkers. However, characterization of immune infiltrate is more challenging than quantification of a single tumor marker or genetic features because of immune processes being very dynamic and changing over time. Another limitation is technical and resides in the amount and quality of tumor tissue required and difficulties in the analysis standardization.
All immune cell types can infiltrate a tumor, including innate cells (macrophages, dendritic cells, mast cells, NK cells) and B and T cells at different stages of maturation/activation and different subsets of CD4+ T cells [T helper 1 (Th1) cells, Th2 cells, Th17 cells, Tregs, T follicular helper (TFH) cells]. These immune cells can be present in the tumor core and/or invasive margins. Both composition and localization of tumor immune infiltrate can influence the outcome of cancer patients [68]. Before the advent of immunotherapy, several studies clearly showed the association between long survival and high intra-tumor density of CD3+ T cells and cytotoxic and memory CD8+ T cells in patients with different types of cancer undergoing surgery [69]. Based on this evidence, quantitation of memory CD45RO+ and cytotoxic CD8+ T cells in the tumor core and invasive margins was proposed as a new scoring system to predict survival [70]. The so-called immune score was specifically established to predict disease recurrence after surgery in patients with local colorectal cancer based on the evidence that a baseline low immune score significantly enriched for patients that developed recurrent disease after surgery [70,71,72, 73•]. Application of this concept to predict response to checkpoint blockade has shown promising results [74•]. Comparison of CD8+, CD4+, PD-1+, and PD-L1+ cell population densities within the tumor and the invasive margin in pre-treatment biopsies from advanced melanoma patients treated with pembrolizumab revealed that invasive margin CD8+ T cell density has the strongest predictive potential of response to PD-1 blockade [74•] (Table 3). Interestingly, in these patients, the presence of tumor-associated CD8+ T cells correlated with both improved clinical outcome and PD-1 and PD-L1 expression in the tumor [74•]. This indicates the importance of pre-existing tumor-associated CD8+ T cells as well as an active PD-1/PD-L1 pathway inhibiting these T cells in dictating sensitivity to PD-1 blockade.
While the presence of cytolytic immune cells in the tumor is generally a biomarker of potentially favorable outcome to immunotherapy (Table 3), the presence of immunosuppressive cells may have a dual meaning. In fact, immune inhibitory cell subsets can be recruited as a consequence of inflammation and ongoing immune response. In this case, treatment with checkpoint blockade can be particularly valuable. For example, high baseline intra-tumor Tregs was found to be associated with an improved clinical outcome to CTLA-4 blockade [75] (Table 3). This may be due to the capacity of anti-CTLA-4 to directly inhibit one of the major mechanisms of Treg suppression (CTLA-4) in a tumor that recruits Tregs as a key immune escape mechanism. The presence of alternative immunosuppressive cell subsets at baseline may instead indicate reduced sensitivity to CTLA-4 blockade. As an example, elevated frequencies of circulating MDSCs have been shown to inversely correlate with response to ipilimumab in advanced melanoma patients [77] (Table 3). If checkpoint blockade cannot directly inhibit the immunosuppressive populations that accumulate into an inflamed tumor, combinations with agents targeting these regulatory subsets may be effective. Pre-clinical studies in mouse tumor models have shown that the presence of MDSC or tumor-associated macrophages can be efficiently counteracted by inhibition of PI3Kγ or CSF-1R respectively in combination with checkpoint blockade leading to improved anti-tumor effects [80, 81].
Host Biomarkers
In addition to tumor- and immune-related factors, certain features of the host may also impact on the response to checkpoint blockade. Recent studies have highlighted that the composition of the gut microbiota can influence the outcome of multiple types of cancer therapies, including checkpoint blockade [82, 83]. Seminal observations in mouse preclinical models opened the way for investigating the impact of gut microbiota in response to immunotherapy in cancer patients. Anti-tumor activity of checkpoint blockade was found to be significantly reduced in germ-free mice or mice treated with broad-spectrum antibiotics [84•, 85•]. Checkpoint blockade also deeply altered the composition of the gut microbiota and in different ways in responding compared to non-responding animals, and oral supplementation of the favorable microbiota could restore the therapeutic efficacy of checkpoint blockade [84•, 85•]. Analysis of anti-PD-1 clinical benefit in the relationship with the consumption of antibiotics for routine indications showed significantly shorter survival in cancer patients that were treated with antibiotics shortly before, during, or shortly after PD-1 blockade [86], highlighting the importance of the host microbiome in the response to immunotherapy in cancer patients. Multiple studies in melanoma patients have also recently described an association between the baseline composition of gut microbiota and improved activity of PD-1 or CTLA-4 blockade [87,88,89,90]. Importantly, this association vanishes if oral microbiota is interrogated in place of gut microbiota [89]. Even though different microbial species may be associated with response to checkpoint blockade therapy, there seems to be a trend toward an enrichment in Firmicutes in responders and Bacteroidetes in non-responders [87, 88]. Importantly, these analyses have demonstrated the potential of monitoring particular microbial species in the gut to predict clinical outcome of checkpoint blockade. These results are corroborated by mechanistic studies in mice showing improved tumor control and immune activity of checkpoint blockade upon fecal microbiota transplantation from responders. The presence of beneficial gut microbiota is also associated with increased T cell activation in the gut and peripheral blood and tumor and with reductions in immunosuppressive cells (Tregs and MDSCs). Initial observations suggest that the same taxa that are associated with checkpoint blockade clinical activity may also be associated with toxicities, in particular colitis [87, 88, 91]. This further strengthens the importance of further assessing gut microbiota in patients undergoing immunotherapy to anticipate both response and toxicity.
An additional host factor, Fcγ receptor polymorphisms, which alter the binding affinity for antibody Fc regions, have been studied in association with the clinical outcome of checkpoint blockade therapy, in particular ipilimumab. Previous studies with depleting, Fc competent, IgG1 antibodies as cancer treatments, including anti-CD20 rituximab, anti-HER2 trastuzumab, and anti-EGFR cetuximab, have highlighted the potential impact of host Fcγ receptor polymorphisms on anti-tumor activity [92]. In particular, single-nucleotide polymorphisms leading to FCGR2A (CD32) H131R and FCGR3A (CD16) V158F amino acid changes have been shown to increase binding affinity for IgG1 antibodies and this can improve antibody effector functions [92].
Ipilimumab is an IgG1 antibody with functional Fc region, and it can activate Fcγ downstream effector functions, including antibody-dependent cellular cytotoxicity (ADCC). In contrast to tumor-targeting antibody therapeutics, ipilimumab targets CTLA-4, which is expressed on immune cells. CTLA-4 is expressed at very high levels on Tregs, and treatment with anti-CTLA-4 antibodies has been shown to downregulate tumor-infiltrating Tregs via ADCC in preclinical mouse tumor models [93, 94]. Similarly, ipilimumab was found to effectively engage CD16 on human “non-classical” CD16+ monocytes ex vivo leading to ADCC-mediated Treg lysis, and in melanoma patients, higher baseline frequencies of circulating non-classical monocytes were associated with response to ipilimumab [95]. Although the effect of ipilimumab on Tregs in cancer patients in vivo has not yielded conclusive results [95, 96], these findings raised the possibility that Fcγ receptor polymorphisms may impact on the clinical activity of ipilimumab. Initial studies in melanoma patients treated with ipilimumab showed no correlation between Fcγ receptor polymorphisms and clinical outcome [75]. More recently, incorporation of CD16 V158F polymorphism information into multiple regression analysis of survival was found to significantly improve prediction of clinical benefit in melanoma patients [97]. Among patients with high mutational load or high intra-tumor CD8 expression in two independent patient cohorts, the presence of CD16 V158F polymorphism correlated with significantly extended overall survival [97]. These findings support further investigation of Fcγ receptor polymorphisms and Treg depletion in the anti-tumor activity of CTLA-4 blockade in patients. A recently initiated clinical trial testing a modified version of ipilimumab with more potent depleting functions will contribute to clarify the relevance of these factors (NCT03110307).
The class-I HLA (HLA-I) genotype is emerging as an additional host genetic factor that can influence the outcome of checkpoint blockade. HLA-I molecules are both polygenic—being encoded by three genes (HLA-A, HLA-B, and HLA-C)—and highly polymorphic, with over 10,000 distinct alleles described thus far [98]. The combination of multiple genes and alleles ensures enough diversification of HLA molecules to bind and present a wide range of peptides for T cell recognition and activation. It is logical to think that HLA zygosity can impact on the efficiency of antigen presentation and immune reactions. In the setting of cancer patients treated with checkpoint blockade, reduced HLA-I zygosity, decreasing the number and diversity of tumor-associated peptides possibly being presented, may negatively impact on anti-tumor activity. Initial evidence supporting this hypothesis has been recently documented by a retrospective analysis of 1535 patients with advanced solid cancers, including melanoma and NSCLC, treated with checkpoint blockade, where homozygosity at one HLA-I locus (A, or B, or C) was associated with reduced overall survival [99]. Interestingly, HLA-I zygosity in combination with mutation burden appeared to more accurately predict clinical benefit, with homozygosity in at least one HLA-I locus and < 113 mutations/exome enriching for patients with significantly shorter survival [99]. The potential usefulness of this biomarker needs to be confirmed in additional series of patients with longer follow-up.
Dynamic Biomarkers
Baseline tumor-intrinsic and immune microenvironmental factors are all valuable parameters helping to estimate the outcome of immunotherapy. However, many cases do not present with all favorable/negative features in these multifactorial categories and predicting the relative contribution of favorable and detrimental features in the overall response may be difficult. Monitoring dynamic biomarkers during treatment may add value in the prediction of a clinical result. Among the most frequently reported dynamic biomarkers of ipilimumab, increases in peripheral blood absolute lymphocyte counts (ALC), absolute eosinophil count (AEC) [100,101,102], and activated Ki67+ [103] and/or ICOS+ T cell frequencies [104, 105] have been described in melanoma patients. Of note, on-treatment increases in ALC and AEC [100,101,102], ICOS+ T cell frequencies [104, 105], and decreases in circulating neutrophil/lymphocyte ratios [106] and circulating MDSCs [77] have all been shown to associate with a positive clinical outcome to ipilimumab.
Changes in circulating T cells have been also found to be associated with activity of PD-1 blockade. Rapid increases in proliferating Ki67+PD-1+CD8+ T cells have been observed after anti-PD-1 treatment in melanoma patients, indicating re-invigoration of previously exhausted (PD-1+) CD8+ T cells [107]. Importantly, the ratio between Ki67+CD8+ T cells and tumor burden correlated with survival in these patients [107]. We have recently reported that PD-1 blockade can reduce peripheral frequencies of a non-conventional immunosuppressive TFH-like PD-1hiFoxp3−CD4+ T cell subset in association with clinical benefit in melanoma patients, highlighting the impact of CD4+ T cells in the response to anti-PD-1 [108]. Interestingly, these immunosuppressive TFH-like cells can be induced by CTLA-4 blockade, whereas addition of anti-PD-1 to anti-CTLA-4 treatment helps maintain stable levels of these cells [108]. Given the biologic relationship between TFH and B cells and in light of preliminary evidence of the poor prognostic value of TFH-associated genes in B-cell lymphoma patients treated with anti-CD20 combination immunotherapy [78] (Table 3), this immunosuppressive TFH-like cell subset deserves particular attention in the setting of B-cell malignancies.
T cell repertoire diversity and clonality assessed by next-generation deep sequencing of the complementarity-determining region 3 (CDR3) from rearranged TCR can be used to monitor induction of T cell responses. In metastatic castration-resistant prostate cancer and melanoma patients, CTLA-4 blockade with ipilimumab or tremelimumab was found to increase peripheral TCR repertoire diversity with no specific expansion of clonal T cell populations [109, 110]. The predictive/prognostic significance of this effect has to be validated in larger studies. Initial results have shown that patients with longer survival tend to less frequently lose baseline high-frequency clones upon CTLA-4 blockade, pointing to the importance of pre-existing T cell responses in the anti-tumor activity of anti-CTLA-4 [109]. This is in agreement with the association between broad baseline TCR repertoires and improved outcome of CTLA-4 blockade [111]. Monitoring changes in peripheral TCR repertoires during immunotherapy can also provide important information to anticipate toxicities. Reduction in peripheral TCR evenness was found to parallel severe organ infiltrations with mononuclear cells in mice treated with anti-4-1BB alone or in combination with anti-PD-1 [112]. Furthermore, in patients with metastatic castration-resistant prostate cancer or melanoma treated with ipilimumab or tremelimumab, increases in peripheral TCR repertoire diversity were found to be associated with the development of immune-related adverse events [110, 113].
As opposed to peripheral blood dynamic biomarkers that are more easily detectable but do not necessarily associate with clinical outcome, changes in intra-tumor immune cells may more reliably indicate an ongoing immune response and therapeutic effect. For these analyses, the limitation is the requirement of longitudinal tumor biopsies, which cannot always systematically be collected. Results from small studies with checkpoint blockade treatment in solid and hematologic cancer patients have frequently highlighted an association between clinical responses and increases in tumor-infiltrating T cell density and/or activation, including enhanced expression of IFN-γ-induced and Th1-associated genes, which are indicative of ongoing anti-tumor immune responses [75, 76, 78, 114, 115]. Effects of checkpoint blockade on intra-tumor TCR repertoire diversity in association with anti-tumor activity have also started to be investigated. Initial analyses in melanoma patients treated with nivolumab have shown that clinical responses are associated with a decrease in evenness of TCR representation in the repertoire of tumor-infiltrating T cells during therapy, indicating relative expansion of specific clonal T cell populations in responding tumors [115]. Interestingly, in responding patients, this effect correlated with elimination of clonal tumor mutations, consistent with therapy-induced T cell responses against relevant truncal neoantigens [115]. Of note, intra-tumor TCR clonality upon treatment with nivolumab increased more efficiently in patients with HLA-I heterozygosity, suggesting that HLA diversity may influence activation and expansion of relevant T cell clones [99]. The association between reduction in TCR evenness and anti-tumor effects of immunotherapy has been further confirmed in preclinical studies with B16-melanoma bearing mice treated with a panel of immunomodulatory antibodies, including anti-PD-1 [112].
Conclusions and Perspectives
The availability of an increasing number of patients treated with checkpoint blockade now offers a real opportunity to define robust biomarkers of biologic and therapeutic activity. This process has been significantly accelerated by the possibility to comprehensively interrogate tumor genome and transcriptome via next-generation sequencing techniques. Thus far, analysis of tumor-restricted factors with these methods has been the most successful approach toward the identification of biomarkers of response to checkpoint blockade. The degree of tumor foreignness, quantified by tumor somatic mutational burden and/or neoantigen load, has shown to impact on the clinical outcome as long as tumor cells have the capacity to correctly present antigens. Tumor expression of PD-L1 provides an additional indication of potential susceptibility to checkpoint blockade, in particular when it is driven by IFN-γ as a result of T cell infiltration. This highlights the relevance of microenvironmental factors in the prediction of responses. Depending on their assortment, tumor-infiltrating immune cells can provide both stimulatory and inhibitory signals for anti-tumor immune responses and the tumor can sense these signals and react accordingly. Importantly, the definition of immune stimulatory and immunosuppressive signals as (respectively) positive and negative modulators of an anti-tumor immune response may not be the same in solid and hematologic cancers. As an example, PD-1 expression has recently been found to constitute a tumor suppressor gene in T cell lymphoma [116] and blockade of PD-1 has the risk to accelerate T cell leukemia-lymphoma progression [117]. Characterization and interpretation of the immune landscape in tumors is challenging mainly because of the highly dynamic environment. Based on the assumption that T cells are the ultimate mediators of tumor clearance in response to immunotherapy, studying the IFN-γ response pathway and T cell infiltration/activation is showing the relevance of these factors in checkpoint blockade responses. Current efforts to more comprehensively define the immune determinants of response to immunotherapy by single-cell transcriptomic analyses of tumor immune infiltrate will provide new insights into relevant biomarkers and molecular mechanisms underlying checkpoint blockade activity. Important factors to consider in the definition/interpretation of immunotherapy biomarkers are the plasticity of the tumor immune landscape and its heterogeneity across lesions in the same patients [118]. The possibility to assess tumor-intrinsic and immune-microenvironmental factors in different lesions in metastatic patients may provide more accurate indication of the potential efficacy of an immunotherapeutic intervention. As our understanding of pharmacodynamic biomarkers of checkpoint blockade is improving, monitoring their occurrence during treatment will also be of significant aid to guide proper therapeutic decisions. In this regard, the possibility to evaluate activity of immunotherapy by assessing relevant immune changes in peripheral blood during treatment would be invaluable. Systemic factors can be easily and repeatedly monitored, and if they correlate with local anti-tumor activity, they may provide a net measure of the effects of immunotherapy across multiple lesions.
Based on these considerations, a logical approach toward an improved use of checkpoint blockade is to perform coordinate baseline assessments of tumor immune state followed by careful evaluation of pharmacodynamic biomarkers to anticipate clinical outcome and optimize treatments if needed (Fig. 1). Immunotherapy studies are now starting to integrate multiple tumor- and immune-related parameters to clarify the molecular determinants of responses to immunotherapy. As an example of how to organically integrate these factors, the cancer immunogram was proposed to visualize cancer immune states and optimize treatments in a more personalized manner [119]. We should update the parameters to incorporate in such model and monitor its evolution over time during specific treatments to clarify which state(s) more likely renders tumors responsive to immunotherapy (Fig. 1).

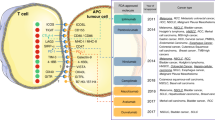
Immunograms of baseline and dynamic biomarkers. Examples of immunograms, including available baseline and dynamic biomarkers, potentially associated with response or lack of response to immune checkpoint blockade (ICB). Systematic assessment of biomarker immunograms before and after treatment in correlation with outcome will help define the tumor-immune state(s) and type of evolution that predict a clinical response
Change history
08 November 2018
The original version of this article unfortunately contained a mistake. The conflict of interest statement was incorrect. The corrected statement is given below.
08 November 2018
The original version of this article unfortunately contained a mistake. The conflict of interest statement was incorrect. The corrected statement is given below.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800.
Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–98.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
•• Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26. This trial together with the one reported in ref 4 demonstrated the clinical benefit of ipilimumab in metastatic melanoma patients and led to the FDA approval of ipilimumab in 2011.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5.
• Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. Pivotal study defining the impact of tumor mutational load on the clinical benefit of CTLA-4 blockade in melanoma patients.
• Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. Pivotal study defining the impact of tumor mutational load on the clinical benefit of PD-1 blockade in NSCLC patients.
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11.
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165(1):35–44.
• Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18(8):1009–21. This study demonstrates the importance of frameshift insertion and deletion mutations in tumor immunogenicity and response to checkpoint blockade.
Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–104.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61.
•• Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415. This study provides the widest characterization of mutational signatures in human cancers.
•• Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. Pivotal study that led to the FDA approval of PD-1 blockade for the treatment of microsatellite-unstable cancers of any origin in 2017.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Holloman WK. Unraveling the mechanism of BRCA2 in homologous recombination. Nat Struct Mol Biol. 2011;18(7):748–54.
Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749–58.
Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–24.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–13.
• McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–9. This study demonstrates the importance of tumor-associated neoantigen clonality in the response to checkpoint blockade of melanoma and NSCLC patients.
Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66(7):3381–5.
Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11(8):2947–53.
Wu C, Zhu Y, Jiang J, Zhao J, Zhang XG, Xu N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 2006;108(1):19–24.
Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(9):3360–5.
•• Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. Pivotal phase 1 trial with PD-1 blockade in melanoma patients providing the first evidence of the association between PD-L1 expression and anti-tumor activity of PD-1 blockade.
Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–46.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–65.
Apolo AB, Infante JR, Balmanoukian A, Patel MR, Wang D, Kelly K, et al. Avelumab, an anti-programmed death-ligand 1 antibody, in patients with refractory metastatic urothelial carcinoma: results from a multicenter, phase Ib study. J Clin Oncol. 2017;35(19):2117–24.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377(20):1919–29.
Hahn NM, Powles T, Massard C, Arkenau H-T, Friedlander TW, Hoimes CJ, et al. Updated efficacy and tolerability of durvalumab in locally advanced or metastatic urothelial carcinoma (UC). J Clin Oncol. 2017;35(15_suppl):4525.
Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–35.
Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18(3):312–22.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, Ouyang J, et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J Clin Oncol. 2018;36(10):942–50.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17.
•• Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–56. This paper summarizes the 3-year overall survival outcomes of advanced melanoma patients enrolled in a phase 3 trial comparing ipilimumab+nivolumab combination with ipilimumab and nivolumab monotherapies. Longer overall survival was found with combination therapy or with nivolumab monotherapy compared to ipilimumab alone.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28.
Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, Kulangara K, et al. PD-L1 Immunohistochemistry assays for lung cancer: results from phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J Thorac Oncol. 2017;12(2):208–22.
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33.
Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105(52):20852–7.
Prestipino A, Emhardt AJ, Aumann K, O’Sullivan D, Gorantla SP, Duquesne S, et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med. 2018;10(429):eaam7729.
Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76(2):227–38.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352(6282):227–31.
Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47(6):1083–99.e6.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–8.
Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268–77.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534(7607):402–6.
•• Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9. Pivotal phase I study of nivolumab in relapsed/refractory Hodgkin lymphoma patients demonstrating activity of PD-1 blockade in this disease associated with particularly high PD-L1 and PD-L2 expression levels on Reed-Stenberg Hodgkin lymphoma cells.
Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol. 2017;35(19):2125–32.
Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–91.
Fish EN, Platanias LC. Interferon receptor signaling in malignancy: a network of cellular pathways defining biological outcomes. Mol Cancer Res. 2014;12(12):1691–703.
Dunn GP, Sheehan KCF, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of <em>JAK1</em> gene expression. Cancer Res. 2005;65(8):3447–53.
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404. e9
Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by <em>JAK1/2</em> mutations. Cancer Discovery. 2017;7(2):188–201.
Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136.
• Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29. This study provides evidence in vivo of clonal selection and outgrowth of tumors with defects in the interferon-receptor signaling pathway and antigen presentation in metastatic melanoma patients developing resistance to PD-1 blockade.
Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery. 2017;7(12):1420–35.
Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–42.
Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547(7664):413–8.
Ozcan M, Janikovits J, von Knebel Doeberitz M, Kloor M. Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology. 2018;7(7):e1445453.
McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–71. e11
Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. 2017;8:15440.
Vey N, Karlin L, Sadot-Lebouvier S, Broussais F, Berton-Rigaud D, Rey J, et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget. 2018;9(25):17675–88.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60.
Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37.
Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016;113(48):E7759–E68.
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95.
Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306.
Pages F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol. 2009;27(35):5944–51.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4.
Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44(3):698–711.
• Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–39. This study provides confirmatory evidence on a large-scale basis of the prognostic value of the Immunoscore in colorectal carcinoma patients.
• Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. This study demonstrates the importance of baseline T-cell density and localization in the tumor bed in the response of metastatic melanoma patients to PD-1/PD-L1 blockade.
Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9(1):204.
Ji R-R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(7):1019–31.
Kitano S, Postow MA, Ziegler CG, Kuk D, Panageas KS, Cortez C, et al. Computational algorithm-driven evaluation of monocytic myeloid-derived suppressor cell frequency for prediction of clinical outcomes. Cancer Immunology Research. 2014;2(8):812–21.
Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15(1):69–77.
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–62.
De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539(7629):443–7.
Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:50–8.
Kroemer G, Zitvogel L. The breakthrough of the microbiota. Nat Rev Immunol. 2018;18:87.
Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33(4):570–80.
• Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84. Landmark study (together with Ref 85) showing the impact of gut microbiome on the anti-tumor activity of checkpoint blockade in mice.
• Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–9. Landmark study (together with Ref 84) showing the impact of gut microbiome on the anti-tumor activity of checkpoint blockade in mice.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7.
Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368–79.
Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia. 2017;19(10):848–55.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103.
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–8.
Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016;7:10391.
Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. 2013;6:1.
Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210(9):1685–93.
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–710.
Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112(19):6140–5.
Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One. 2014;9(2):e87705.
Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. 2018;33(4):649–63. e4
Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015;43(Database issue):D423–31.
Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–7.
Delyon J, Mateus C, Lefeuvre D, Lanoy E, Zitvogel L, Chaput N, et al. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol. 2013;24(6):1697–703.
Ku GY, Yuan J, Page DB, Schroeder SE, Panageas KS, Carvajal RD, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer. 2010;116(7):1767–75.
Martens A, Wistuba-Hamprecht K, Yuan J, Postow MA, Wong P, Capone M, et al. Increases in absolute lymphocytes and circulating CD4+ and CD8+ T cells are associated with positive clinical outcome of melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22(19):4848–58.
Wang W, Yu D, Sarnaik AA, Yu B, Hall M, Morelli D, et al. Biomarkers on melanoma patient T Cells associated with ipilimumab treatment. J Transl Med. 2012;10(1):146.
Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res. 2010;16(10):2861–71.
Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, et al. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunology Research. 2013;1(4):229–34.
Cassidy MR, Wolchok RE, Zheng J, Panageas KS, Wolchok JD, Coit D, et al. Neutrophil to lymphocyte ratio is associated with outcome during ipilimumab treatment. EBioMedicine. 2017;18:56–61.
Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545(7652):60–5.
Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, et al. Non-conventional inhibitory CD4(+)Foxp3(−)PD-1(hi) T cells as a biomarker of immune checkpoint blockade activity. Cancer Cell. 2018;33(6):1017–32. e7
Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med. 2014;6(238):238ra70.
Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. 2014;20(9):2424–32.
Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23.
Hosoi A, Takeda K, Nagaoka K, Iino T, Matsushita H, Ueha S, et al. Increased diversity with reduced “diversity evenness” of tumor infiltrating T-cells for the successful cancer immunotherapy. Sci Rep. 2018;8(1):1058.
Oh DY, Cham J, Zhang L, Fong G, Kwek SS, Klinger M, et al. Immune toxicities elicted by CTLA-4 blockade in cancer patients are associated with early diversification of the T-cell repertoire. Cancer Res. 2017;77(6):1322–30.
Chen P-L, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discovery. 2016;6:827–37.
Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934–49. e15
Wartewig T, Kurgyis Z, Keppler S, Pechloff K, Hameister E, Ollinger R, et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552(7683):121–5.
Ratner L, Waldmann TA, Janakiram M, Brammer JE. Rapid progression of adult T-cell leukemia–lymphoma after PD-1 inhibitor therapy. N Engl J Med. 2018;378(20):1947–8.
Jimenez-Sanchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170(5):927–38. e20
Blank CU, Haanen JB, Ribas A, Schumacher TN. Cancer immunology. The “cancer immunogram”. Science. 2016;352(6286):658–60.
Acknowledgements
We would like to thank the Swim Across America, Ludwig Institute for Cancer Research, the Parker Institute for Cancer Immunotherapy, and the NIH/NCI Cancer Center Support Grant (P30 CA008748) for their support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on CART and Immunotherapy
Rights and permissions
About this article

Cite this article
Zappasodi, R., Wolchok, J.D. & Merghoub, T. Strategies for Predicting Response to Checkpoint Inhibitors. Curr Hematol Malig Rep 13, 383–395 (2018). https://doi.org/10.1007/s11899-018-0471-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-018-0471-9