
Abstract
Purpose of the Review
Osteoarthritis is widely regarded as a spectrum of conditions that affect all joint tissues, typified by a common entity: cartilage loss. Here, we review recent progress and challenges in chondroprotection and discuss new strategies to prevent cartilage loss in osteoarthritis.
Recent Findings
Advances in clinical, molecular, and cellular characterization are enabling improved stratification of osteoarthritis subtypes. Integration of next-generation sequencing and “omics” approaches with clinically relevant readouts shows promise in delineating both subtypes of disease and meaningful trial end points. Novel delivery strategies are enabling joint-specific delivery.
Summary
Chondroprotection requires a whole joint approach, stratification of patient groups, and use of patient-relevant end points. Drug development should continue to explore new targets, while using modern technologies and recent knowledge to re-visit unsuccessful therapeutics from the past. The overarching goal for chondroprotection is to provide the right treatment(s) for the right patient at the right time.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is a highly prevalent and extremely disabling condition, affecting 30.8 million people in the USA alone [1]. In 1741, J.B. Morgagni first described articular cartilage loss in OA [2]. To this day, radiographic measurement of cartilage loss remains the key diagnostic tool for OA [3]. Consequentially, chondroprotection has been a major focus of disease modifying treatment in OA. Chondroprotective drugs (disease modifying OA drugs, DMOADs) have generally targeted structural changes of disease by aiming to inhibit cartilage-degrading factors or promote cartilage production, with or without effects on symptoms. Of the $81 billion annual healthcare expenditure on OA in the USA, 15% is attributed to prescription drugs [4, 5]. However, an effective means to prevent OA-related damage has proven elusive. Therefore, much of this expenditure centers on drugs that target pain rather than structural changes. Given current limitations in DMOADs, OA progresses and surgical joint replacement is the only viable option. However, joint replacement is by no means a panacea with a mortality rate of 1% and at least 10% of total hip or knee replacements reporting no improvement or worse symptoms at 1 year after surgery [6, 7].
In this review, we consider recent progress and challenges in chondroprotection and how chondroprotection should encompass both direct (within cartilage) and indirect (articular or systemic) strategies. We also discuss how a step change in successful chondroprotection requires reframed thinking in disease stratification, target identification, and drug development, including drug delivery strategies.
Osteoarthritis—Chondroprotection Is Not Chondrocentric
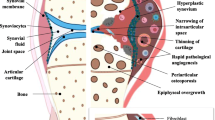
Osteoarthritis (Fig. 1) occurs at synovial joints and was long regarded as a structural disease of wear and tear. The key symptoms of OA are pain and joint instability, with radiographic joint space narrowing due to cartilage loss being a major clinical sign of disease. Alongside OA-related cartilage degradation, osteophytes form at joint margins, and bone remodeling occurs, leading to bone marrow lesions and bone sclerosis [8]. Synovitis (synovial inflammation) and meniscal damage are common and altered levels of inflammatory mediators are detected in OA synovial fluid. Furthermore, OA is now widely accepted as a whole joint disease [9], but the chronicity of tissue involvement in OA remains ambiguous, and cartilage may not be the initial site from which OA propagates. The cross-talk between cartilage and other tissues suggests cartilage loss can occur secondary to other OA-related changes to the joint [10]. We must also remember that synovial joints are, in fact, comprised of diverse tissues that experience different loads; have distinct functional requirements; and possess differing proportions of tissue types. The interactions of these factors likely explain the predilection of OA for certain anatomical sites (commonly knee, hip, spine, hands, and feet).

A representation of an osteoarthritic knee, including the main contributing tissues and their interactions. The exact balance of tissue involvement and interaction is dependent on both joint site and the subtype of OA. Articular cartilage loss typifies OA but the exact balance of tissue involvement and interaction is dependent on joint site, stage and subtype of disease
Chondroprotection offers a preventive strategy that might decelerate structural disease progression and loss of function and, through the manipulation of inflammatory pathways, improve pain. However, the success of such strategies demands a holistic approach that judiciously targets OA pathobiology across all joint tissues.
Current and Recent Targets in Chondroprotection
Over the last 5 years, the quest for successful DMOADs has remained substantial. Recent DMOAD trials (Table 1) encompass chondroprotective factors and drugs targeting inflammatory cytokines, signaling mediators, senescence, and pain. From these studies, it is clear that antibodies against inflammatory mediators like IL-1 and TNF-α have failed in recent OA clinical trials. However, an exploratory analysis of the recent CANTOS trial demonstrated that IL-1 antibodies did reduce the incidence of joint replacements in patients with a history of myocardial infarction [15, 16]. A recent review by Chevalier and Eymard hypothesizes that IL-1 inhibition might not be effective at end-stage OA and, instead, earlier-stage disease or patients with systemic inflammation-related comorbidities may be more amenable to treatment. They also propose that non-intravenous routes of administration should be considered to improve bioavailability [48].
Senescence is a compelling recent target in OA and other diseases of aging. Senescent cells are resistant to apoptosis and secrete inflammatory and catabolic mediators including IL-1, IL-6, and MMP-3. In OA mouse models, senescent cells accumulate in the synovium and cartilage; the small molecule UBX0101 clears senescent cells and ameliorates cartilage damage [49]. Clinical trials are now underway to test UBX0101 for amelioration of painful patellofemoral OA. In addition, repurposing of drugs, like those for osteoporosis, have received particular attention in the OA field. These drugs might serve as potential treatments in subtypes of OA, as osteoporosis and OA have commonalities in bone involvement [50]. Although repurposed Osteoporosis drugs show compelling results in pre-clinical testing in OA, clinical trials in late-stage OA, such as multiple phase III trials investigating calcitonin, have not yet demonstrated obvious benefits [45]. Galcanezumab, a monoclonal antibody targeting calcitonin gene-related peptide, was also unsuccessful in a phase II trial as it did not reduce signs or symptoms of knee OA [46].
Active research into surgical and cell-based strategies continues, but an exhaustive review of the current research landscape is not the purpose of this review. Rather, we seek to present a framework of principles that are applicable to future success of all chondroprotective approaches.
Disease Stratification—Breaking Down a Spectrum of Diseases into Different Subtypes
Advances in clinical, molecular, and genetic characterization of OA suggest it is better regarded as a spectrum, or continuum, of conditions leading to end-stage disease with a common entity: loss of articular cartilage [51]. This is unsurprising given the varied clinical manifestations of OA. However, most current clinical and laboratory-based studies do not consider disease subtypes in their study design.
Disease stratification is best demonstrated in oncology, where the ability to utilize molecular information to predict prognosis and optimal therapeutic strategy has been the first step towards personalized treatment [52]. To apply this to chondroprotection in OA, we must consider the key characteristics by which to stratify patients (Fig. 2).

Overview of OA stratification categories and their interactions. Clinical signs and symptoms, risk factors, and molecular signatures interact and can define OA subtypes. The combinatorial effects of different stratification measures will define distinct subtypes and stages of OA. For example, risk factors such as trauma will drive changes in molecular and cellular signatures and altered clinical signs and symptoms. As OA progresses stratification categories will continue to feedback on each other, therefore individual stratification measures (or combinations thereof) may be specific to a particular stage of disease
Clinical Phenotypes
A first step to classify OA can be through the affected joint(s). Clinically adopted imaging, pain, and function scores can also incorporate affected joint surface(s) and stage of disease. There are several studies where these scores reveal patterns in chondroprotective response. Individuals with severe cartilage loss have a reduced likelihood of improved pain following intra-articular steroid injection [53, 54]. Meanwhile, strontium ranelate inhibition of cartilage loss was more effective in patients with meniscal extrusion and bone marrow lesions than in those with meniscal extrusion alone [55]. Osteophytes and synovitis may also predict the rate of structural progression [8, 56], which varies between individuals, influencing both prognosis and chondroprotective strategy [57]. Potential predictors of structural progression include pain [58], inertia [57], bone marrow lesions [59], joint loading [60], baseline Kellgren–Lawrence (KL) grade, obesity, and alignment [61]. However, few studies have the power and quality to reliably deconvolute clinical predictors of cartilage loss or other clinical outcomes [62].
Risk Factors
Risk factors for OA, including age, sex, and trauma, may hint at the underlying mechanism and determine the broadly applicable stratification steps. Age is associated with inflammaging and senescence and predicts improvement in pain following corticosteroid injection for knee OA [54]. Mechanisms underlying pain differ in males and females, and, consequentially, the mechanisms underlying cartilage damage may also be modulated by sex. Younger males are less likely to require joint replacement [63], and morphological differences between sexes might contribute to OA development and severity [64, 65]. Although few studies have investigated the effects of OA risk factors on pharmacologic driven chondroprotection, there is evidence that OA progression is reduced in post-menopausal women undergoing hormone replacement therapy [66, 67]. Site- and hormone-based interactions are also seen in the presentation of hand OA at the time of menopause, in contrast to other forms of OA that present post-menopause [68, 69]. Comorbidities, including obesity, diabetes, and metabolic syndrome, are additional risk factors for OA with clinically related readouts such as high serum cholesterol, which is associated with generalized OA [70]. Lifestyle, diet, and alteration of the microbiome may also affect OA onset and progression [71].
Joint loading and trauma significantly impact OA development. Lower step rate is associated with a greater risk of increased cartilage damage in knee OA patients [72]. Following traumatic ACL injury, initial cartilage damage is more common in the lateral and medial compartments of the knee than in the medial alone [73]. Sport-related knee injuries lead to a significantly greater likelihood of joint replacement [74], yet younger patients have differing underlying biology and immune status, higher physical demands, and greater need to avoid surgical joint replacement. These factors greatly impact the requirements of therapeutic chondroprotection while identifying a relatively defined group on which to test DMOADs [75].
The breadth of OA risk factors and clinical phenotypes demonstrates the wealth of potential disease subtypes. Risk factors alone, although identifiable prior to clinical presentation for OA, are not adequate to reliably predict OA occurrence or prognosis. A key advantage of stratifying OA patients based on observable clinical factors (pain, function, and imaging scores) is data accessibility: there is often no invasive testing needed and readouts are readily available in the clinic. When considering timing of DMOAD intervention and identification of disease mechanisms, patients often do not present to clinic until they have “end-stage” disease where pain or function is severely affected. Indeed, the structural features of OA do not consistently correlate with each other or with symptoms. Patients can suffer severe pain, but show limited radiographic evidence of OA, or have late-stage radiographic OA, but limited pain. However, cross-talk between cartilage damage and pain can occur, with pain-targeting strategies (e.g., corticosteroids) leading directly or indirectly to cartilage loss [76,77,78].
Neither the clinical phenotype nor the combination of risk factors directly reveals mechanism of disease, preventing meaningful identification, development, and evaluation of chondroprotective strategies [79]. In fact, a number of molecular mechanisms (endotypes) may contribute to each risk factor-induced effect and clinical phenotype. While a first step in stratification for chondroprotection may utilize clinical scoring and risk factors, it is unlikely that these will be meaningful in isolation. Future studies should explore the interrelationships among structure, function, and pain and integrate molecular endotyping to generate robust stratification systems.
Molecular Signatures
Tissue-specific mechanisms will underlie OA, some unique to an individual tissue and some common across multiple tissues and joints. Recent studies have defined OA subtypes to be bone-, cartilage-, and inflammation- or synovium-driven [80]. It is logical to expand these principles to examine the contributions of the fat pad and synovial fluid, as well as meniscus, muscle, tendon, and ligament [9]. The contribution of each joint tissue to cartilage damage will vary between patients, ultimately combining to define one of the clinical subtypes on the OA spectrum.
Genotypes
OA has a complex polygenic genetic architecture, with multiple alleles acting in concert to both increase and decrease risk of disease and distinguish disease subtypes. This is demonstrated through the identification of novel and non-overlapping genetic risk loci for hip and knee OA in a meta-analysis of 17,151 hip OA patients, 23,877 knee OA patients, and > 560,000 controls [81]. A further genome-wide analysis of UK Biobank data identified 15 novel signals for hip OA, 7 for knee OA, and 6 shared between hip and knee OA [82••]. A number of loci were within genes involved in cartilage development or homeostasis (COL11A1, COL11A2, FGFR3, GDF5, TGFB1, IL11). The genetic susceptibility loci showed site-specific association and shared correlation with phenotypes including obesity, BMI, bone mineral density, and age at first live birth. Furthermore, the OA-associated loci functionally affected genes in their vicinity, altering gene expression in degraded compared to intact cartilage.
Cells and Transcriptomics
Improved granularity in OA stratification is possible by defining the cellular and molecular basis of OA using cytometry and next-generation sequencing approaches. These add to traditional histochemistry-based approaches and offer the possibility of a new disease taxonomy. Bulk genome-wide expression analysis of chondrocytes in articular cartilage revealed two major patient subgroups, differing in complement activation, innate immune responses, and Wnt and TGFβ signaling response [83•]. Single-cell sequencing has also identified cell subsets associated with both cartilage zone and level of damage [84••]. Although many approaches are cartilage-based, plasma miRNA signatures can distinguish levels of OA cartilage damage as either significant or minimal, also indicating epigenetic mediators as disease modulators [85]. CD14+CD16+ macrophages, enriched in the synovial fluid of knee OA patients at the time of intervention, correlate with stiffness and function scores [86•]. In the synovium, RNA-Seq of synovial macrophages has also revealed two distinct subtypes of OA – an inflammatory subtype in which macrophages have a proliferative (Ki-67 positive) signature and a ‘classical’ subtype in which macrophages possess a tissue remodelling signature [87].
Protein, Lipid and Metabolomic Biomarkers
There has been significant focus on using biomarkers to stratify OA and correlate with clinical signs and symptoms. Investigated biomarkers include ECM components and breakdown products, cytokines, adipokines, metabolites, lipids, and enzymes in blood, serum, urine, and synovial fluid, as well as tissues of the OA joint. Differing levels of knee cartilage loss are positively correlated with serum cartilage oligomeric matrix protein and fibulin-3 levels, whereas IL-6 is associated with increased synovitis and pain [88]. The presence of IL-17 in synovial fluid may also discriminate an OA subtype with faster cartilage loss, reduced osteophytes, and increased adipokines. Increased pain in hip OA patients is associated with increased IL-6, visfatin, and leptin, while in knee OA patients, increased leptin, reduced adiponectin and reduced adiponectin–leptin ratio are associated with pain [89,90,91]. Given the association of OA with metabolic disease, it is feasible that lipid and metabolomic markers may define specific OA subtypes and impact disease progression as they do in cancer [92,93,94].
To help crystallize critical OA stratification panels, multidimensional computational strategies should help us to deconvolute and integrate the myriad of interrelationships between transcriptomic and biomarker signatures, as well as clinical signs, symptoms, and risk factors. The same approach should be applied to determining meaningful end points to assess interventions. The combinatorial information from a range of genetic, epigenetic, transcriptomic, protein, lipid, or metabolomic biomarkers is important. Different combinations may mediate different effects, and single stratifiers may not convey any meaningful effect or association. In turn, molecular or cellular endotypes may contribute to one clinical phenotype, and some clinical phenotypes may share the same endotypic contributors. An integrated approach will therefore provide a more robust framework for successful chondroprotection and clinical translation.
Integrating Stratification with Chondroprotective Strategies
To better deliver OA chondroprotection, phenotypes and endotypes need to be utilized intelligently—both in disease stratification and in the identification of meaningful end points. This will enable identification of pathobiological mechanisms, molecular targets, responsive groups, and markers of therapeutic efficacy. This is demonstrated in rheumatoid arthritis, where distinct transcriptomic signatures correlate with drug responses and define the clinical subtypes of disease [95]. Stratification and meaningful end points must be incorporated into all stages of the translational pipeline—in discovery, pre-clinical, and clinical studies of chondroprotection (Fig. 3).

Integration of OA stratification categories with the translational cycle. The key stratification categories should be applied to studies of disease development and to pre-clinical and clinical drug development pathways. To refine critical stratification measures and identify relevant end points for pre-clinical and clinical studies, nextgeneration sequencing, “omics”, and cytometry approaches should be integrated with risk factors, clinical signs and symptoms. Tissue-based end points and stratification measures should be derived using well-phenotyped healthy and diseased tissues from the joint. Where appropriate, embedding tissue collection and analysis within enrolment and outcome stages of clinical trials would inform future studies across the translational cycle
Pre-clinical Studies
Cellular or molecular signatures may not have functional effects on cartilage damage or OA pathobiology and may instead simply postcode a disease state or subtype. Therefore, successful delivery of chondroprotection relies on well-conducted functional studies and on the extension of cross-sectional and longitudinal studies of OA endotypes—all informed by clinical signs and risk factors. Tissues across the whole joint plus urine, blood, and synovial fluid should be incorporated. Cellular and molecular signatures of tissues obtained from diseased joints, healthy joints and joints following t herapeutic intervention are also essential. These signatures will enable identification of underlying mechanisms of disease and produce meaningful readouts to assess whether an intervention results in a “healthy” or “effectively treated” signature. The use of the spectrum of relevant tissues and fluids allows more robust endotype identification, while identifying less invasive surrogates for tissue-based signatures. Stratification panels will undergo continuous evolution, changing not only with disease subset but also with the stage of cartilage damage and other clinical readouts and risk-factors.
Studies of disease mechanisms, drug target identification, and drug testing rely on in vitro and in vivo methods, supplemented with in silico and mathematical modeling, to identify key drivers, stratification sets, and end points. Functional assays and modeling of identified biomarkers, cell types, and molecular signatures will enable validation of potential targets for chondroprotective agents and testing of therapeutic efficacy. Models and end points used in vitro and in vivo need to account for differing endotypes and changing molecular signatures associated with OA progression or cessation. As with human disease, these models should not be chondrocentric. Firstly, chondroprotection alone is not adequate for patient-beneficial changes in pain and function. Secondly, OA is a whole joint disease, with multiple studies showing significant cross-talk among bone, synovium, muscle, fat pad, meniscus, ligament, and cartilage. Finally, we should consider comorbidities, risk factors, and the mechanical environment that will all affect cellular, molecular, and functional readouts in vitro and in vivo.
For in vitro studies, well-phenotyped human tissue is the optimal source of cells and tissues. Molecular and cellular interrogation, co-cultures, and microfluidic and organ-on-a-chip platforms alone and in combination with bioreactor-driven loading will enable study of multidirectional interactions between cartilage and other critical tissues and mediators. Since the joint itself presents challenges for sampling, the development of minimally invasive biopsy methods will be necessary. The soft tissues of the joint are more amenable to biopsy, and, given their likely involvement in secreting factors that propagate OA pain and cartilage loss, research efforts may be better focused here than on interrogation of cartilage itself. However, differing cell subsets and molecular signatures in cartilage may still contribute to OA, driving diversity in disease mechanism, changes in pain and function, or response to therapeutics. Given the importance of pain and function to patients, it would be fruitful to focus on in vitro signatures that relate to these clinical readouts in order to improve the identification of potential target molecules and therapeutics.
In vivo validation is embedded within the translational pathway, and a variety of in vivo models of OA are available [96]. To establish mechanisms, targets, or drug efficacies, the multiple subtypes and risk factors of OA must be considered in the selection of an appropriate in vivo model. For example, the commonly used destabilization of medial meniscus (DMM) mouse model uses injury to induce OA in the knee of young male animals, yet only 12% of knee OA patients have post-traumatic disease and many of these patients will be female. Cartilage damage in the DMM is sex dependent, and, given the distinct pain pathways in males compared to females, stratification by sex is preferable in all in vivo models [97]. In addition, the age of mice at DMM and their diet affects disease progression and molecular response [98, 99]. This stresses that age, sex, joint site, diet, obesity, and loading regime pre- and post-disease or therapeutic intervention should be considered carefully, as should the genetic background of animals and the incorporation of comorbidity models [100]. End points in animal studies should encompass traditional cartilage readouts (e.g., OARSI scores) alongside effects on molecular signatures of other joint tissues and pain and function measures that relate to human disease.
Future work should include next-generation transcriptomic, “omic,” and cytometry platforms, in combination with multidimensional co-culture techniques, to develop high-throughput screening of chondroprotection agents. However, a clearly defined molecular signature of “healthy” or “effectively repaired” tissues will be crucial to such strategies. To this end, it is critical that future clinical trials incorporate tissue end points into their design.
Clinical Trials
Many promising drugs have completed pre-clinical and early-phase clinical trials, but most have failed in phase III trials. It is plausible that this is due to the heterogeneity in endotypes of the patient populations within trials or to the design of targeting of patients with too advanced disease. A priori patient selection into trials requires validated inclusion criteria, including clinical markers, risk factors, and endotypes of disease to identify individuals most likely to respond to treatment [101]. Identifying molecular endotypes that predispose to OA or mark early OA is a first step in selecting patient groups within the effective therapeutic window for pharmacologic intervention—most likely before joint damage, function, and pain are too severe. Furthermore, stratification by molecular endotype for “basket” trials (one pharmacologic, multiple OA joint sites with a shared endotype) or “umbrella” trials (multiple pharmacologics targeting a molecular endotype at one joint site) may hold greater promise than traditional phase trials. Given that 31 million adults are affected by OA in the USA alone [1], successful targeting of a small subset of patients would be life changing for millions of people. For both surgical and pharmacologic interventions, reversal of all disease symptoms is unlikely. However, enabling optimal response to treatment is within reach. Clinical observations of imaging, pain, and function provide commonly used end points for clinical trials, often in combination with activity levels and patient reported outcome measures (PROMS). Combining these with meaningful molecular readouts at an accessible and relevant tissue level is also essential at trial end points where efficacy is inferred, especially given the interaction of all these factors. Without careful selection of end points, the ability to separate responders from non-responders is limited and effective (direct or indirect) chondroprotection may be missed. Furthermore, tissue readouts following clinical trials provide essential information to feed back into the discovery pipeline [102]. Finally, epidemiology studies of any clinically adopted therapeutics would enable large-scale evaluation of population-level efficacy.
Future Look
The OA field continues to evolve, with the advent of new technologies contributing to improved strategies for therapeutic delivery and the identification of novel, unexplored targets. A reframed translational pathway, encompassing integration of stratification with discovery, pre-clinical, and clinical studies, should be applied to these advances.
Administration, Delivery, and Release of Drugs
A particular challenge in OA is effective drug administration to ensure delivery and release at the site of action. OA is mainly localized to the particular joint affected by the disease, and many drugs, such as the MMP inhibitors, have failed due to systemic side effects. Intra-articular injections have received increased interest, as their benefits in targeted delivery compared to oral and intravenous formulations may outweigh their disadvantages. In order to decrease the frequency, and associated patient-burden, of these injections, the half-life of the drugs, as well as slow-release methods, will need to be considered.
Recent advances in delivery and release vehicles of drugs make intra-articular injections more attractive. Hydrogels, micro- and nanoparticles, and liposomes are all potential carriers for drugs, but microparticles are the most likely to provide an adequate retention time for appropriate drug release over a therapeutically useful period [103]. However, as even intra-articular approaches can affect multiple tissues, tissue-specific targeting may be necessary. A novel approach used electrostatic interactions to get drugs into the cartilage and closer to chondrocytes [104]. These cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment, as shown in a rat model of OA [105••]. The delivery of drugs specifically to the cartilage, where necessary, would be a major breakthrough in the field.
In addition to the use of these strategies for drugs that have already undergone development, nanomicelles can be employed to carry mRNA, as shown using the cartilage-anabolic transcription factor RUNX1, which significantly suppressed disease progression after injection into mouse OA knee joints [106]. Virus-like particles have also been coupled to recombinant nerve growth factor to create a vaccine. Pre-clinical studies showed that both prophylactic and therapeutic vaccination resulted in the attenuation of chronic pain behavior in surgically induced murine OA [107].
Emerging Targets
Emerging targets for chondroprotection include mediators of oxidative stress, and of cholesterol and lipid metabolism, as well as nuclear receptors.
Oxidative stress occurs during disease and aging and is detrimental to many cells. Oxidative stress is evident in OA cartilage, as OA chondrocytes have increased reactive oxygen species-induced DNA damage and lipid peroxidation products. Overexpression of the antioxidant heme oxygenase-1 (HO1) was shown to protect against cartilage damage in post-traumatic and aging models of OA [108]. Oxidative stress targeting could encompass both small molecules and bioactives. S-methylisothiorea, an inhibitor of inducible nitric oxide synthase, produced anti-nociceptive and anti-arthritic effects in a rat OA model by inhibiting cartilage damage and suppressing nitric oxide in synovial fluid [109]. In addition, the bioactive sulfurophane induces HO1 and protects against DMM-induced cartilage damage [110].
Given that obesity-associated comorbidities are widely accepted risk factors for OA, it is unsurprising that cholesterol and its metabolic regulators are potential targets. High cholesterol diets alter chondrocyte cholesterol metabolism and uptake and induce more severe OA following DMM. Knockdown of CH25H–CYP7B1–RORα axis members—involved in cholesterol uptake, metabolism, and transcriptional response—decrease cartilage damage [111]. RORα, a nuclear receptor, is already a target in other diseases. Nuclear receptors are transcription factors activated by lipophilic ligands including vitamin D, glucocorticoids, retinoic acid, free fatty acids, and eicosanoids—all of which show altered expression and disease modulation in OA models and are amenable to therapeutic targeting [70]. Lipid mediators are not only pro-inflammatory like eicosanoids but also drive inflammation resolution. Resolvin D1 inhibits synovial macrophage-driven cartilage damage [112], and the resolvin precursor 17-HDHA is associated with pain but not cartilage loss [113]. These emerging targets converge on the chronic inflammatory pathways that underlie OA. Although chondroprotection is a key goal, the molecular targets should incorporate OA natural history, targeting tissues that drive cartilage loss, pain, and dysfunction in each relevant disease subtype.
Conclusion
Significant progress has been made in understanding OA and in developing technologies to interrogate and treat this complex disease. It is now clear that chondroprotection should target all joint tissues not just cartilage. The field should continue to adopt collaborative strategies that combine cutting-edge cytometric, omics, and computational technologies. Future approaches should target the taxonomy of OA, embedding disease stratification as well as meaningful tissue, patient, and clinical end points within all stages of the translational pathway. Improved definitions of OA subtypes, meaningful end points, and targets will enable us to re-purpose or re-investigate existing therapeutics, as well as to accelerate the development of novel agents. The overarching goal for chondroprotection must be to provide the right treatment(s) for the right patient at the right time.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Cisternas MG, Murphy L, Sacks JJ, Solomon DH, Pasta DJ, Helmick CG. Alternative methods for defining osteoarthritis and the impact on estimating prevalence in a US population-based survey. Arthritis Care Res. 2016;68:574–80.
Benedek TG. A history of the understanding of cartilage. Osteoarthritis Cartilage. 2006;14:203–9.
Bekkers JEJ, Creemers LB, Dhert WJA, Saris DBF. Diagnostic modalities for diseased articular cartilage-from defect to degeneration: a review. Cartilage. 2010;1:157–64.
Biener AI, Decker SL, Rohde F. Source of increased health care spending in the United States. J Am Med Assoc. 2019;321:1147.
Zhao X, Shah D, Gandhi K, Wei W, Dwibedi N, Webster L, et al. Clinical, humanistic, and economic burden of osteoarthritis (OA) among noninstitutionalized adults in the United States (US). Value Health. 2018;21:S4.
Liddle AD, Pandit H, Judge A, Murray DW. Patient-reported outcomes after total and unicompartmental knee arthroplasty: a study of 14 076 matched patients from the national joint registry for England and Wales. Bone Joint J. 2015;97-B:793–801.
Judge A, Cooper C, Williams S, Dreinhoefer K, Dieppe P. Patient-reported outcomes one year after primary hip replacement in a European collaborative cohort. Arthritis Care Res. 2010;62:480–8.
Mathiessen A, Slatkowsky-Christensen B, Kvien TK, Haugen IK, Berner Hammer H. Ultrasound-detected osteophytes predict the development of radiographic and clinical features of hand osteoarthritis in the same finger joints 5 years later. RMD Open. 2017;3:e000505.
Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–707.
Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19:18.
Wang J. Efficacy and safety of adalimumab by intra-articular injection for moderate to severe knee osteoarthritis: an open-label randomized controlled trial. J Int Med Res. 2018;46:326–34.
Aitken D, Laslett LL, Pan F, Haugen IK, Otahal P, Bellamy N, et al. A randomised double-blind placebo-controlled crossover trial of HUMira (adalimumab) for erosive hand OsteoaRthritis – the HUMOR trial. Osteoarthritis Cartilage. 2018;26:880–7.
Lindsley HB, Schue J, Tawfik O, Bolce R, Smith DD, Hinson G, et al. FRI0304 treatment of knee osteoarthritis with intra-articular infliximab improves total WOMAC score. High baseline levels of synovial cellularity predict improvement. Ann Rheum Dis. 2013;71:417.
Huerta JS, Gálvez-Romero J, Lόpez-Rodriguez W, Lόpez A. SAT0524 utility of tocilizumab in clinical manifestations of erosive osteoarthrosis of hands regional hospital Issste Puebla, MΈxico. Ann Rheum Dis. 2017;76:974.
Ridker PM, Everett BM, Thuren T, MacFadyen J, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31.
Schieker M, Mindeholm L, Praestgaard J, Scotti C, Solomon D, Thuren T, DreyerK, Roubenoff R, Ridker P. Interleukin-1β inhibition with canakinumab associates with reduced rates of total hip and knee replacement (THR/TKR) and osteoarthritis (OA) symptoms: exploratory results from the canakinumab anti-inflammatory thrombosis outcomes study (CANTOS) [abstract]. Arthritis Rheumatol. 2018;70 (suppl 10).
Fleischmann RM, Bliddal H, Blanco FJ, Schnitzer TJ, Peterfy C, Chen S, et al. A phase 2 trial of lutikizumab, an anti-interleukin 1α/β dual variable domain immunoglobulin, in knee osteoarthritis patients with synovitis. Arthritis Rheum. 2019. https://doi.org/10.1002/art.40840.
Deshmukh V, Hu H, Barroga C, Bossard C, KC S, Dellamary L, et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthritis Cartilage. 2018;26:18–27.
Deshmukh V, Seo T, Swearingen C, Yazici Y. FRI0552 Sm04690, a wnt pathway inhibitor: anti-inflammatory and cartilage protective effects in preclinical oa models. Ann Rheum Dis. 2018;77:801–2.
Onuora S. Osteoarthritis: Wnt inhibitor shows potential as a DMOAD. Nat Rev Rheumatol. 2017;13:634.
Yazici Y, McAlindon TE, Gibofsky A, et al. Results from a 52-week randomized, double-blind, placebo-controlled, phase 2 study of a novel, intra-articular wnt pathway inhibitor (SM04690) for the treatment of knee osteoarthritis. Osteoarthritis Cartilage. 2018;26:S293–4.
Grothe K, Flechsenhar K, Paehler T, Ritzeler O, Beninga J, Saas J, et al. IκB kinase inhibition as a potential treatment of osteoarthritis – results of a clinical proof-of-concept study. Osteoarthritis Cartilage. 2017;25:46–52.
Brandt KD, Mazzuca SA, Katz BP, Lane KA, Buckwalter KA, Yocum DE, et al. Effects of doxycycline on progression of osteoarthritis: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2005;52:2015–25.
Snijders GF, Van Den Ende CHM, Van Riel PLCM, Van Den Hoogen FHJ, Den Broeder AA. The effects of doxycycline on reducing symptoms in knee osteoarthritis: results from a triple-blinded randomised controlled trial. Ann Rheum Dis. 2011;70:1191–6.
Siebuhr A, Bay-Jensen A-C, Thudium CT, Karsdal MA, Serruys B, Werkmann D, et al. The anti-ADAMTS-5 nanobody®, M6495, protects against cartilage breakdown in cartilage and synovial joint tissue explant models. Osteoarthritis Cartilage. 2018;26:S187.
Lindström E, Rizoska B, Henderson I, Terelius Y, Jerling M, Edenius C, et al. Nonclinical and clinical pharmacological characterization of the potent and selective cathepsin K inhibitor MIV-711. J Transl Med. 2018;16:125.
Conaghan PG, Bowes MA, Kingsbury SR, Brett A, Guillard G, Tunblad K, et al. Six months’ treatment with MIV-711, a novel cathepsin K inhibitor induces osteoarthritis structure modification: results from a randomized double-blind placebo-controlled phase IIA trial. Osteoarthritis Cartilage. 2018;26:S25–6.
Gigout A, Guehring H, Froemel D, Meurer A, Ladel C, Reker D, et al. Sprifermin (rhFGF18) enables proliferation of chondrocytes producing a hyaline cartilage matrix. Osteoarthritis Cartilage. 2017;25:1858–67.
Reker D, Kjelgaard-Petersen CF, Siebuhr AS, Michaelis M, Gigout A, Karsdal MA, et al. Sprifermin (rhFGF18) modulates extracellular matrix turnover in cartilage explants ex vivo. J Transl Med. 2017;15:250.
Dahlberg LE, Aydemir A, Muurahainen N, Gühring H, Fredberg Edebo H, Krarup-Jensen N, et al. A first-in-human, double-blind, randomised, placebo-controlled, dose ascending study of intra-articular rhFGF18 (sprifermin) in patients with advanced knee osteoarthritis. Clin Exp Rheumatol. 2016;34:445–50.
Clegg DO, Reda DJ, Harris CL, Klein MA, O’Dell JR, Hooper MM, et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med. 2006;354:795–808.
Hochberg MC, Martel-Pelletier J, Monfort J, Möller I, Castillo JR, Arden N, et al. Combined chondroitin sulfate and glucosamine for painful knee osteoarthritis: a multicentre, randomised, double-blind, non-inferiority trial versus celecoxib. Ann Rheum Dis. 2016;75:37–44.
Zhu X, Sang L, Wu D, Rong J, Jiang L. Effectiveness and safety of glucosamine and chondroitin for the treatment of osteoarthritis: a meta-analysis of randomized controlled trials. J Orthop Surg Res. 2018;13:170.
Wu B, Li Y-M, Liu Y-C. Efficacy of intra-articular hyaluronic acid injections in hip osteoarthritis: a meta-analysis of randomized controlled trials. Oncotarget. 2017;8:86865–76.
Richette P, Chevalier X, Ea HK, Eymard F, Henrotin Y, Ornetti P, et al. Hyaluronan for knee osteoarthritis: an updated meta-analysis of trials with low risk of bias. RMD Open. 2015;1:e000071.
McGuire D, Lane N, Segal N, Metyas S, Barthel H, Miller M, et al. TPX-100 leads to marked, sustained improvements in subjects with knee osteoarthritis: pre-clinical rationale and results of a controlled clinical trial. Osteoarthritis Cartilage. 2018;26:S243.
Walicke PA, Hefti F, Bales R, Lu S-P, Ruckle JL, Brown MT, et al. First-in-human randomized clinical trials of the safety and efficacy of tanezumab for treatment of chronic knee osteoarthritis pain or acute bunionectomy pain. Pain reports. 2018;3:e653.
Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010;363:1521–31.
Sanga P, Katz N, Polverejan E, Wang S, Kelly KM, Haeussler J, et al. Long-term safety and efficacy of fulranumab in patients with moderate-to-severe osteoarthritis pain: a phase II randomized, double-blind, placebo-controlled extension study. Arthritis Rheum. 2017;69:763–73.
Maloney J, Kivitz A, Schnitzer TJ, Dakin P, Di Martino S, Gao H, et al. Fasinumab in the treatment of hip and knee osteoarthritic pain: efficacy and safety in a 36-week randomized, double-blind placebo-controlled clinical trial. Osteoarthritis Cartilage. 2017;25:S56–7.
Stevens R, Petersen D, Ervin J, Nezzer J, Nieves Y, Campbell J, et al. OP0167 efficacy and safety of CNTX-4975 in subjects with moderate to severe osteoarthritis knee pain: 24-week, randomized, double-blind, placebo-controlled, dose-ranging study. Ann Rheum Dis. 2017;76:121.
Rayegani SM, Raeissadat SA, Enayati E, Rahimi Dehgolan S. AB0974 the efficacy and safety of intra-articular botulinum toxin injection in knee osteoarthritis. Ann Rheum Dis. 2019;77:1610.
Hsieh LF, Wu CW, Chou CC, Yang SW, Wu SH, Lin YJ, et al. Effects of botulinum toxin landmark-guided intra-articular injection in subjects with knee osteoarthritis. PM R. 2016;8:1127–35.
Ledin H, Good L, Aspenberg P. Denosumab reduces early migration in total knee replacement: a randomized controlled trial involving 50 patients. Acta Orthop. 2017;88:255–8.
Karsdal MA, Byrjalsen I, Alexandersen P, Bihlet A, Andersen JR, Riis BJ, et al. Treatment of symptomatic knee osteoarthritis with oral salmon calcitonin: results from two phase 3 trials. Osteoarthritis Cartilage. 2015;23:532–43.
Jin Y, Smith C, Monteith D, Brown R, Camporeale A, McNearney TA, et al. CGRP blockade by galcanezumab was not associated with reductions in signs and symptoms of knee osteoarthritis in a randomized clinical trial. Osteoarthritis Cartilage. 2018;26:1609–18.
Hunter DJ, Pike MC, Jonas BL, Kissin E, Krop J, McAlindon T. Phase 1 safety and tolerability study of BMP-7 in symptomatic knee osteoarthritis. BMC Musculoskelet Disord. 2010;11:232.
Chevalier X, Eymard F. Anti-IL-1 for the treatment of OA: dead or alive? Nat Rev Rheumatol. 2019;15:191–2.
Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23:775–81.
Geusens PP, Van Den Bergh JP. Osteoporosis and osteoarthritis: shared mechanisms and epidemiology. Curr Opin Rheumatol. 2016;28:97–103.
Waarsing JH, Bierma-Zeinstra SMA, Weinans H. Distinct subtypes of knee osteoarthritis: data from the osteoarthritis initiative. Rheumatol. 2015;54:1650–8.
Russnes HG, Lingjærde OC, Børresen-Dale AL, Caldas C. Breast cancer molecular stratification: from intrinsic subtypes to integrative clusters. Am J Pathol. 2017;187:2152–62.
Maricar N, Parkes MJ, Callaghan MJ, Hutchinson CE, Gait AD, Hodgson R, et al. Structural predictors of response to intra-articular steroid injection in symptomatic knee osteoarthritis. Arthritis Res Ther. 2017;19:88.
Fatimah N, Salim B, Raja E u H, Nasim A. Predictors of response to intra-articular steroid injections in patients with osteoarthritis of the knee joint. Clin Rheumatol. 2016;35:2541–7.
Roubille C, Martel-Pelletier J, Raynauld JP, Abram F, Dorais M, Delorme P, et al. Meniscal extrusion promotes knee osteoarthritis structural progression: protective effect of strontium ranelate treatment in a phase III clinical trial. Arthritis Res Ther. 2015;17:82.
Haugen IK, Slatkowsky-Christensen B, Bøyesen P, Sesseng S, Van Der Heijde D, Kvien TK. MRI findings predict radiographic progression and development of erosions in hand osteoarthritis. Ann Rheum Dis. 2016;75:117–23.
Felson D, Niu J, Sack B, Aliabadi P, McCullough C, Nevitt MC. Progression of osteoarthritis as a state of inertia. Ann Rheum Dis. 2013;72:924–9.
Wang Y, Teichtahl AJ, Abram F, Hussain SM, Pelletier JP, Cicuttini FM, et al. Knee pain as a predictor of structural progression over 4 years: data from the osteoarthritis initiative, a prospective cohort study. Arthritis Res Ther. 2018;20:250.
Roemer FW, Kwoh CK, Fujii T, Hannon MJ, Boudreau RM, Hunter DJ, et al. From early radiographic knee osteoarthritis to joint arthroplasty: determinants of structural progression and symptoms. Arthritis Care Res. 2018;70:1778–86.
Henriksen M, Creaby MW, Lund H, Juhl C, Christensen R. Is there a causal link between knee loading and knee osteoarthritis progression? A systematic review and meta-analysis of cohort studies and randomised trials. BMJ Open. 2014;4:e005368.
Collins JE, Katz JN, Losina E. Identifying rapid structural disease progression in knee osteoarthritis [abstract]. Arthritis Rheum. 2017;7:9–11.
Deveza LA, Melo L, Yamato TP, Mills K, Ravi V, Hunter DJ. Knee osteoarthritis phenotypes and their relevance for outcomes: a systematic review. Osteoarthritis Cartilage. 2017;25:1926–41.
Collins JE, Deshpande BR, Katz JN, Losina E. Race- and sex-specific incidence rates and predictors of total knee arthroplasty: seven-year data from the osteoarthritis initiative. Arthritis Care Res. 2016;68:965–73.
Logerstedt DS, Zeni J, Snyder-Mackler L. Sex differences in patients with different stages of knee osteoarthritis. Arch Phys Med Rehabil. 2014;95:2376–81.
Fang WH, Huang GS, Chang HF, Chen CY, Kang CY, Wang CC, et al. Gender differences between WOMAC index scores, health-related quality of life and physical performance in an elderly Taiwanese population with knee osteoarthritis. BMJ Open. 2015;5:e008542.
Roman-Blas JA, Castañeda S, Largo R, Herrero-Beaumont G. Osteoarthritis associated with estrogen deficiency. Arthritis Res Ther. 2009;11:241.
Jung JH, Bang CH, Song GG, Kim C, Kim J-H, Choi SJ. Knee osteoarthritis and menopausal hormone therapy in postmenopausal women. Menopause. 2018;26:598–602.
Prieto-Alhambra D, Judge A, Javaid MK, Cooper C, Diez-Perez A, Arden NK. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: influences of age, gender and osteoarthritis affecting other joints. Ann Rheum Dis. 2014;73:1659–64.
Watt FE. Hand osteoarthritis, menopause and menopausal hormone therapy. Maturitas. 2016;83:13–8.
Sun MMG, Beier F, Ratneswaran A. Nuclear receptors as potential drug targets in osteoarthritis. Curr Opin Pharmacol. 2018;40:81–6.
Schott EM, Farnsworth CW, Grier A, Lillis JA, Soniwala S, Dadourian GH, et al. Targeting the gut microbiome to treat the osteoarthritis of obesity. JCI Insight. 2018;3:e95997.
Hart HF, Gross KD, Crossley KM, Barton CJ, Felson DT, Guermazi A, et al. Is step rate associated with worsening of patellofemoral and tibiofemoral joint osteoarthritis in women and men? The multicenter osteoarthritis study. Arthritis Care Res. 2019. https://doi.org/10.1002/acr.23864.
Swärd P, Kostogiannis I, Neuman P, von Porat A, Boegård T, Roos H. Differences in the radiological characteristics between post-traumatic and non-traumatic knee osteoarthritis. Scand J Med Sci Sports. 2010;20:731–9.
Ackerman IN, Bohensky MA, Kemp JL, de Steiger R. Likelihood of knee replacement surgery up to 15 years after sports injury: a population-level data linkage study. J Sci Med Sport. 2018;22:629–34.
Gelber AC, Hochberg MC, Mead LA, Wang NY, Wigley FM, Klag MJ. Joint injury in young adults and risk for subsequent knee and hip osteoarthritis. Ann Intern Med. 2000;133:321–8.
Neogi T. The epidemiology and impact of pain in osteoarthritis. Osteoarthritis Cartilage. 2013;21:1145–53.
McAlindon TE, LaValley MP, Harvey WF, Price LL, Driban JB, Zhang M, et al. Effect of intra-articular triamcinolone vs saline on knee cartilage volume and pain in patients with knee osteoarthritis a randomized clinical trial. JAMA. 2017;317:1967–75.
Lapane KL, Yang S, Driban JB, Liu SH, Dubé CE, McAlindon TE, et al. Effects of prescription nonsteroidal antiinflammatory drugs on symptoms and disease progression among patients with knee osteoarthritis. Arthritis Rheum. 2015;67:724–32.
Kraus VB, Blanco FJ, Englund M, Karsdal MA, Lohmander LS. Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthritis Cartilage. 2015;23:1233–41.
Karsdal MA, Michaelis M, Ladel C, Siebuhr AS, Bihlet AR, Andersen JR, et al. Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: lessons learned from failures and opportunities for the future. Osteoarthritis Cartilage. 2016;24:2013–21.
Styrkarsdottir U, Lund SH, Thorleifsson G, Zink F, Stefansson OA, Sigurdsson JK, et al. Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat Genet. 2018;50:1681–7.
•• Zengini E, Hatzikotoulas K, Tachmazidou I, et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet. 2018;50:549–58 This work shows how genetic data can be linked with clinical and cellular readouts.
• Soul J, Dunn SL, Anand S, Serracino-Inglott F, Schwartz JM, Boot-Handford RP, et al. Stratification of knee osteoarthritis: two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann Rheum Dis. 2018;77:423 This work shows the utility of bulk RNAseq in identifying OA patient subgroups.
•• Ji Q, Zheng Y, Zhang G, et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann Rheum Dis. 2019;78:100–10 The first use of single-cell RNAseq to track progression in OA.
Ali SA, Shestopaloff K, Gandhi R, Kapoor M. Next generation sequencing discovers known and novel circulating microRNAs in knee osteoarthritis. Osteoarthritis Cartilage. 2018;26:S182.
• Gómez-Aristizábal A, Gandhi R, Mahomed NN, Marshall KW, Viswanathan S. Synovial fluid monocyte/macrophage subsets and their correlation to patient-reported outcomes in osteoarthritic patients: a cohort study. Arthritis Res Ther. 2019;21:26 The identification of immune cell subsets within synovial fluid illustrates that cells in synovial fluid, as well as cytokines, may drive OA subtypes.
Wood MJ, Leckenby A, Reynolds G, Spiering R, Pratt AG, Rankin KS, et al. Macrophage proliferation distinguishes 2 subgroups of knee osteoarthritis patients. JCI Insight. 2019;4:e125325.
Mobasheri A, Bay-Jensen AC, van Spil WE, Larkin J, Levesque MC. Osteoarthritis year in review 2016: biomarkers (biochemical markers). Osteoarthritis Cartilage. 2017;25:199–208.
Snelling SJB, Bas S, Puskas GJ, Dakin SG, Suva D, Finckh A, et al. Presence of IL-17 in synovial fluid identifies a potential inflammatory osteoarthritic phenotype. PLoS One. 2017;12:e0175109.
Bas S, Finckh A, Puskas GJ, Suva D, Hoffmeyer P, Gabay C, et al. Adipokines correlate with pain in lower limb osteoarthritis: different associations in hip and knee. Int Orthop. 2014;38:2577–83.
Hosseininia S, Önnerfjord P, Dahlberg LE. Targeted proteomics of hip articular cartilage in OA and fracture patients. J Orthop Res. 2019;37:131–5.
Yang L, Cui X, Zhang N, Li M, Bai Y, Han X, et al. Comprehensive lipid profiling of plasma in patients with benign breast tumor and breast cancer reveals novel biomarkers. Anal Bioanal Chem. 2015;407:5065–77.
Shen S, Yang L, Li L, Bai Y, Cai C, Liu H. A plasma lipidomics strategy reveals perturbed lipid metabolic pathways and potential lipid biomarkers of human colorectal cancer. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1068–1069:41–8.
Zhai G. Alteration of metabolic pathways in osteoarthritis. Metabolites. 2019;9:11.
Sumitomo S, Nagafuchi Y, Tsuchida Y, Tsuchiya H, Ota M, Ishigaki K, et al. Transcriptome analysis of peripheral blood from patients with rheumatoid arthritis: a systematic review. Inflamm Regen. 2018;38:21.
Malfait A-M, Little CB. On the predictive utility of animal models of osteoarthritis. Arthritis Res Ther. 2015;17:225.
Ma HL, Blanchet TJ, Peluso D, Hopkins B, Morris EA, Glasson SS. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage. 2007;15:695–700.
Loeser RF, Olex AL, McNulty MA, Carlson CS, Callahan MF, Ferguson CM, et al. Microarray analysis reveals age-related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum. 2012;64:705–17.
Hülser M-L, Schreiyäck C, Luo Y, Bozec A, Schett G, Neum ann E, Müller-Ladner U. AB0046 Metabolism and osteoarthritis are linked by adipokines. Ann Rheum Dis. 2017;76:1063.
Kuyinu EL, Narayanan G, Nair LS, Laurencin CT. Animal models of osteoarthritis: classification, update, and measurement of outcomes. J Orthop Surg Res. 2016;11:19.
Li H, Wang D, Yuan Y, Min J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res Ther. 2017;19:248.
Hunter DJ, Nevitt M, Losina E, Kraus V. Biomarkers for osteoarthritis: current position and steps towards further validation. Best Pract Res Clin Rheumatol. 2014;28:61–71.
Maudens P, Jordan O, Allémann E. Recent advances in intra-articular drug delivery systems for osteoarthritis therapy. Drug Discov Today. 2018;23:1761–75.
Evans CH. Catering to chondrocytes. Sci Transl Med. 2018;10:eaav7043.
•• Geiger BC, Wang S, Padera RF, Grodzinsky AJ, Hammond PT. Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci Transl Med. 2018;10:eaat8800 This work illustrates a novel and promising means of tissue-specific drug delivery in chondroprotection.
Aini H, Itaka K, Fujisawa A, Uchida H, Uchida S, Fukushima S, et al. Messenger RNA delivery of a cartilage-anabolic transcription factor as a disease-modifying strategy for osteoarthritis treatment. Sci Rep. 2016;6:18743.
von Loga IS, El-Turabi A, Jostins L, Miotla-Zarebska J, Mackay-Alderson J, Zeltins A, et al. Active immunisation targeting nerve growth factor attenuates chronic pain behaviour in murine osteoarthritis. Ann Rheum Dis. 2019;78:672–5.
Takada T, Miyaki S, Ishitobi H, Hirai Y, Nakasa T, Igarashi K, et al. Bach1 deficiency reduces severity of osteoarthritis through upregulation of heme oxygenase-1. Arthritis Res Ther. 2015;17:285.
More AS, Kumari RR, Gupta G, Lingaraju MC, Balaganur V, Pathak NN, et al. Effect of iNOS inhibitor S-methylisothiourea in monosodium iodoacetate-induced osteoathritic pain: implication for osteoarthritis therapy. Pharmacol Biochem Behav. 2013;103:764–72.
Davidson RK, Jupp O, De Ferrars R, et al. Sulforaphane represses matrix-degrading proteases and protects cartilage from destruction in vitro and in vivo. Arthritis Rheum. 2013;65:3130–40.
Choi WS, Lee G, Song WH, Koh JT, Yang J, Kwak JS, et al. The CH25H–CYP7B1–RORα axis of cholesterol metabolism regulates osteoarthritis. Nature. 2019;566:254–8.
Sun AR, Wu X, Liu B, Chen Y, Armitage CW, Kollipara A, et al. Pro-resolving lipid mediator ameliorates obesity induced osteoarthritis by regulating synovial macrophage polarisation. Sci Rep. 2019;9:426.
Valdes AM, Ravipati S, Menni C, Abhishek A, Metrustry S, Harris J, et al. Association of the resolvin precursor 17-HDHA, but not D-or E-series resolvins, with heat pain sensitivity and osteoarthritis pain in humans. Sci Rep. 2017;7:10748.
Acknowledgments
The authors would like to thank Dr. Mathew Baldwin from The Botnar Research Centre for his helpful discussions and proofreading. We would also like to thank Yvonne van de Grint for her invaluable help with the figures.
Funding
Jolet Y. Mimpen and Sarah J.B. Snelling are supported by the NIHR Oxford Biomedical Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Osteoarthritis
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mimpen, J.Y., Snelling, S.J.B. Chondroprotective Factors in Osteoarthritis: a Joint Affair. Curr Rheumatol Rep 21, 41 (2019). https://doi.org/10.1007/s11926-019-0840-y
Published:
DOI: https://doi.org/10.1007/s11926-019-0840-y