
Abstract
The CRISPR–Cas genome editing system is an intrinsic property of a bacteria-based immune system. This employs a guide RNA to detect and cleave the PAM-associated target DNA or RNA in subsequent infections, by the invasion of a similar bacteriophage. The discovery of Cas systems has paved the way to overcome the limitations of existing genome editing tools. In this review, we focus on Cas proteins that are available for gene modifications among which Cas9, Cas12a, and Cas13 have been widely used in the areas of medicine, research, and diagnostics. Since CRISPR has been already proven for its potential research applications, the next milestone for CRISPR will be proving its efficacy and safety. In this connection, we systematically review recent advances in exploring multiple variants of Cas proteins and their modifications for therapeutic applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CRISPR/Cas system has become a significant genome editing tool in the field of molecular biology. CRISPR, an acronym that is explained as a Clustered Regularly Interspaced Short Palindromic Repeats, is inherently equipped by a bacterial defense system to protect itself from an invasive pathogen. CRISPR system works in tandem with an associated protein called Cas proteins (plurality of types like Cas9, Cas12, and Cas13) that are used for targeting and manipulating the deoxyribonucleic acid (DNA) at the specific site in the nucleotide sequence. Genome editing using CRISPR–Cas requires the significant, additional component called guide ribonucleic acid (gRNA) which is assists the CRISPR complex in the target cleavage. Hence, scientists have been persistently bringing out new modifications on the guide RNA for effective genome editing activity. This type of programmable and reprogrammable strategy of gene editing is been explored in various fields such as research, medicine, diagnostics to regulate gene expression, controlling transcription, chromosomal imaging, drug delivery, and even treating the human ophthalmological and hematological diseases fronts in the future. Therefore, in this review, the aspects of different Cas proteins with their associated mechanisms have been briefed out for a better understanding of gene editing.
Historical View
Discovery of CRISPR 80s to Present
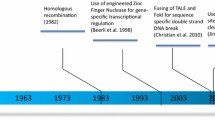
CRISPR–Cas is found in prokaryotes and plays significantly a specific role in the bacterial acquired immune system. Yoshizumi Ishino, a Japanese scientist, discovered the repeated sequences interspersed with spacer sequences in Escherichia coli (1987) while analyzing the gene that is responsible for alkaline phosphatase conversion [1]. Van Embden and his colleagues in the Netherlands threw light into different spacer sequences between DNA repeats in different strains of Mycobacterium tuberculosis (1993). Spacer sequences were used to characterize different strains of M. tuberculosis. This method is known as Spoligotyping, and many such sequences have been discovered in bacteria and archaea [2]. CRISPR sequences were identified in Haloferax (1995) [3] and in Cyanobacterium (1996). CRISPR sequences were discovered in the year 2000 in archaea and bacteria [4]. Jansen and his team termed those repeated nucleotide sequences with spacers found in the genomes of prokaryotes as CRISPR sequences (2002) [5].
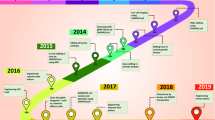
CRISPR-associated proteins, shortly known as “Cas” enzymes (particularly Cas9, Cas12a, and Cas13), use CRISPR sequences as a guide to recognize and cleave target DNA’s complementary strands. CRISPR arrays allow bacteria or archaea to retrospect about viruses. When bacteria encounter the virus, they copy and integrate its DNA copy into their genome as “spacers” between CRISPR short DNA repeats. The nucleotide sequences in the spacers act as a template for the Bacterial RNA to recognize the DNA of an incoming virus and direct the Cas9 enzyme to act upon it; thereby preventing the virus from infecting the bacteria. Doudna and Banfield began researching CRISPRs in 2005. Years later, in 2012, yet another scientist, Emmanuel Charpentier, joined the former for a collaborative study on Cas9 proteins. In 2015, a new Cas protein, cpf1, was discovered to further enhance gene editing [6]. These scientists were subsequently awarded the Nobel Prize in Chemistry on October 7, 2020, in recognition of their contributions to CRISPR–Cas9. These studies have been further being scaled up later on in order to improve the accuracy and efficiency of CRISPR–Cas9 technique (Fig. 1).

Schematic representation of CRISPR discoveries from 1987 to 2020
Existing Genetic Tools
Before the CRISPR–Cas system, several types of endonuclease enzymes that could cut DNA were exposed, besides the already existing Meganucleases, ZFNs (Zinc Finger Nucleases), and TALENs (Transcription activator-like effector nucleases) (Table 1). These enzymes can identify and cut specific nucleotide sequences. The restriction enzymes were used to identify a specific site of DNA, cleave it, and incorporate new genes. Those sequences are found multiple times in a genome. As a result, determining the insertion site ahead of time is extremely difficult. So, scientists sought ways to improve the precision of restriction enzymes and transform them so that they could identify a distinct sequence in the genome. So, the example of enzymes having such one-of-a-kind sequences is Meganucleases. Zinc finger nuclease is another type of endonuclease. These synthetic restriction enzymes are made up of a subunit that can identify the preferred DNA sequence and its restriction sites. Scientists engineer the nuclease proteins by combining zinc finger proteins with the Fok I endonuclease DNA cleavage domain [7]. Transcription activator-like effector nucleases (TALENs) are less difficult to synthesis than of zinc finger nucleases. TALENs are made up of a restriction enzyme subunit that cleaves DNA and a sequence recognition component called a TAL effector. TAL effectors have an ability to cover the long and unique sequences in the genome due to the high number of iterations. Scientists can create TAL effectors for specific DNA sequences more easily than zinc finger endonucleases. It remains, however, a difficult task [8].
General Mechanism of CRISPR
The CRISPR system requires a group of CRISPR-associated genes known as “Cas,” which are usually found next to the CRISPR sequence. Cas genes typically encode proteins that are required for an adaptive immune response [9]. CRISPR–Cas systems are engineered DNA- or RNA-binding proteins that can bind to specific target sequences by designing crRNAs or gRNAs with complementary spacer sequences. Cas proteins can also act as nucleases, making them useful as programmable genome editing tools [10]. There are three major stages in the CRISPR–Cas defense system: adaptation, expression, and interference. In the first stage, the CRISPR locus is adapted by the insertion of new spacer sequences. During expression, the Cas genes are expressed and the CRISPR sequence is transcribed into a long precursor crRNA. Pre-crRNA is converted into mature crRNA through the involvement of Cas proteins (Cas5d) and other accessory factors. The target viral DNA is identified and destroyed by the combined action of crRNA and Cas proteins during the final stage, Interference [11] (Fig. 2).

Mechanism of CRISPR-mediated immunity in bacteria. 1. Adaptation insertion of new spacers into the locus. 2. Expression processing of the CRISPR RNA. 3. Interference detection of the genetic elements by CRISPR RNA and Cas proteins
The CRISPR/Cas9 technique is widely used in science because of its high degree of flexibility and accuracy in identifying, targeting, and cleaving target nucleic acid. This technique is alluring as it allows for very low-cost genetic engineering. It enables the inclusion or elimination of multiple genes at the same time. This technique can quickly manipulate many different genes in an in vitro or in vivo cell lines, a plant that would otherwise take years to complete. Because it is not species-specific, it can be used on organisms that previously have been resistant to genetic engineering. Jennifer Doudna and Emmanuel Charpentier have been honored with the Nobel Prize in Chemistry on October 7, 2020, in recognition of their contributions to CRISPR–Cas9 (Table 2).
Cas9 Protein
Among all the CRISPR effector proteins the well-characterized protein is Cas9, which belongs to the type II CRISPR system which is comprised of subtypes like type II-A, type II-B, and II-C that are sorted on account of Cas9 gene size and the presence of the subtype-specific gene [12]. Cas9 effector protein stemmed from Streptococcus pyogenes, Streptococcus thermophiles, Staphylococcus aureus, Neisseria meningitidis, and Campylobacter jejuni. Cas9 causes double-stranded DNA breaks ensuing in the formation of blunt ends, which may be repaired by a donor template DNA during the non-homologous end joining or homologous recombination (Fig. 3).

Mechanism of action of CRISPR/Cas9 techniques. CRIPSPR/Cas9 gene with cleavage activities creating a double-strand break on the target DNA, which are repaired either by non-homologous end joining (NHEJ) or by homology-directed repair (HDR) with the help of Donor DNA is provided
Mechanisms and Modifications
Cas9 requires crRNA (CRISPR RNA) and tracrRNA (Transactivating crRNA), which complements crRNA’s repeated region [13]. Cas9 binds to a genetically engineered single-guide RNA (sgRNA) made from dual RNA (crRNA:tracrRNA) and recognizes target viral DNA via PAM (Protospacer Adjacent Motif) regions. Cas9 cleaves 3 bp upstream of the PAM sequence after sgRNA base pairs with the target DNA related to the PAM sequence [14]. Cas9 has a bilobed structure with a core cleft that could harbor a crRNA:DNA duplex. A disordered linker that connects the α-helical recognition (REC) and nuclease (NUC) lobes, as well as a highly conserved arginine-rich bridge that forms several interactions with sgRNA. The conserved HNH (Histidine Asparagine Histidine) and RuvC (Resolvase) nuclease domains, as well as a variable C-terminal domain, make up the NUC (Nuclease) lobe which interacts with PAM [15]. The structural conformations of the Cas9 proteins show that guide RNA binding controlling Cas9 activity results in a large conformational reorganization of the protein. As a result of the arrangement of the PAM-interacting residues and the nucleotide sequence of the guide RNA, the protein is capable of DNA binding and PAM recognition [16]. The scrutinized complex binds the guide RNA and scans the DNA; upon recognition of a similar PAM in the non-target strand, it induces the unwinding of the DNA duplex, allowing the guide RNA to probe 10 to 12 nucleotide sequences for complementarily in the target strand’s upstream PAM region [17].
The domain conformational activation of the HNH is subjected to the changes in the linker loops between the HNH and RuvC domains. This allosteric bonding between the nuclease domains results in the cleaving of the HNH domain in the target strand. The RuvC domain conversely cleaves the non-target strand [18]. Among all the Cas9 proteins, spCas9 variant (Streptococcus pyogenes) is the most commonly used genomic tool for gene manipulation, gene disruption, transcriptional activation and repression, and epigenetic modulation. For DNA target recognition, SpCas9 usually recognizes the viral DNA targets and generates a blunt double-strand break (DSB). SpCas9 recognizes a spacer sequence of 20 nucleotides and on the PAM 5-NGG. The PAM 5-NGG recognition limits SpCas9 target sites available in the human genome. Evolutionary approaches have developed variants by altering the PAM specificities for increasing the target site’s availability [19]. The aim of genetically engineered Cas9 variants is to improve target specificity. Several studies have defined how a Cas9 variant is mutated to reduce off-target cleavage activity, resulting in Cas9 and its corresponding gRNA expression from plasmids [20] and their transportation as ribonucleoprotein complexes [21].
Changes in the secondary structures within the short RNA hairpins on the spacer region of gRNAs increase the thermodynamic barrier to crRNA or gRNA strand invasion at off-target sites while generally maintaining on-target action, has increased on-target CRISPR–Cas specificity [22]. The discovery and development of additional Cas9 orthologs that recognize different PAM sequences have increased the number of target sites available. Streptococcus thermophilus Cas9 (stCas9), for example, recognizes the PAM 5′-NNNNGATT and Neisseria meningitidis Cas9 (NmCas9) recognizes 5′-NNNNGATT [23]. These Cas9 orthologs have been repurposed in bacteria and mammalian cells for DNA targeting. Furthermore, PAM sequence 5-NNGRRT (R represents A or G) is recognized by Staphylococcus aureus Cas9 (SaCas9) [23]. Notably, the efficiencies of SaCas9 gene editing are comparable to those of SpCas9, and the smaller size of SaCas9 (1053 amino acids compared to 1368 amino acids of SpCas9) has allowed its use in size-restricted delivery vectors like adeno-associated virus (AAV) [24]. A smaller Cas9 ortholog from Campylobacter jejuni (984 amino acids) was recently reported to recognize the PAM 5-NNNVRYM (V represents A, C, or G; Y represents C or T) and be used for in vivo-targeted genome editing. CasX (980 amino acids), the smallest Cas9 to date, was discovered as a result of additional efforts to identify Cas9 ortholog [25].
Mutations in Cas9 Proteins
Streptococcus pyogenes is the first organism found in the CRISPR/Cas9 system. The binding of gRNA with complementary mismatched target DNA within the bacterial genome leading to off-target activity is one of the major limitations of this system. As a result, shown in Table 3, the modifications of the Cas9 system are made to improve target specification and reduce unexpected cleaning activity [26]. An additional way to reduce off-target cleavage activity increases the proto spacer near the motif length. Nmecas9, a CRISPR–Cas9 system based on Neisseria meningitidis, detects a sequence of 8-nt PAM (5-NNNNGATT) that can reduce off-target cleavage activity [27]. Sacas9 from Staphylococcus aureus detects a 6-nt PAM (5-NNGRRT) sequence of two Cas9 variants from Streptococcus thermophilus (st1cas9 and st3cas9) used to classify two human loci, PRKDC and CARD11, showed lower off-target rates than previously developed SpCas9 [28].
Cas9 Ortholog and Their Function
CRISPR/Cas9 orthologs have also been identified in order to improve the targeted specificities. CRISPR/CpfI Prevotella and Francisella were taken from class II, type endonuclease. CpfI, unlike Cas9, requires a single-directed CRISPR RNA (crRNA) that breaks down and produces 4–5 nucleotides fusion and 5-overhangs [29]. Cas9 orthologs also have the ability to identify RNA as well. The cleavage of ssRNA in SaCas9 and Campylobacter jejuni Cas9 are independent of PAM [10]. SaCas9 suppressed gene expression in E. coli when targeted at RNA [30]. The CRISPR RNA (crRNA) dependent but independent binding of PAM and purification of endogenous RNAs wer observed in C. jejuni Cas9 [31]. Francisella novicida Cas9 (FnCas9) was found to identify bacterial mRNA [32]. It modifies the genetic expression and identifies the RNA genome of the hepatitis C virus in eukaryotic cells [33]. FnCas9 is not PAM-dependent and can be used to identify negative-sense ssRNA viruses from the family Filoviridae, Paramyxoviridae, and Orthomyxoviridae (Table 4). More research is needed to determine the potential effects of RNA targeted at Cas9 on eukaryotic cells [10].
Applications of Cas9
The CRISPR/Cas technology has a very broad application range in the field of research, development, and diagnostics. Primarily, this technique has been used as a genetic tool for target gene (DNA/RNA) modification in the host. Besides, it plays various roles in the field of Genome library screening, Transcriptional regulation (activation/repression), epigenetic modification, live cell imaging, and also therapeutic applications. In human cells, the Gecko (genome scale CRISPR–Cas9 knock out) library that targets 18,080 genes of having 64,751 specific guide RNA sequences via lentiviral delivery allows both negative and positive selection screening. GeCKO library is mainly used for identifying cancer cell viability [34]. The identification of genes influencing the desired phenotype is often accomplished via genetic screening using RNA interference (RNAi) or knockouts. When the resulting ESC (Embryonic stem cells) mutant libraries were screened for resistance to either Clostridium septicum alpha-toxin or 6-thioguanine, 27 known and 4 previously unknown genes were found to be involved in these phenotypes [35]. sgRNAs recruit dCas9 (nuclease dead Cas9) and Cas9 effector fusion proteins to aim for specific loci for modulating the gene expression [36]. The recruitment of dCas9 to the DNA was directed by sgRNA has been shown to interfere with gene transcription (CRISPRi) by preventing transcriptional elongation, polymerase binding, or transcription factor binding, which influence gene expression levels [37]. When targeted the regions upstream of transcriptional start sites, the sgRNA–dCas9 complex, the effector domain, can dictate the cellular response, resulting in gene repression (CRISPRi) (e.g., KRAB, SID domains) or gene activation (CRISPRa) (e.g., VP16, VP64, p65AD domains) [37, 38].
Targeting the HS2 enhancer with dCas9-KRAB fusions was shown to silence globin gene expression with relatively little off-target effects on non-targeted gene expression [39]. Targeting histone epigenetic modifications has been demonstrated by epigenome editing systems. Gene activation with a nuclease-null dCas9 tethered to an acetyltransferase (HAT) enzyme that catalyzes the direct covalent modification of lysine residues on histone tails [40]. The other study used a nuclease-null dCas9 fused to the Krüpel-associated box (KRAB), a naturally occurring transcriptional repression domain involved in recruiting a heterochromatin-forming complex that mediates histone methylation and deacetylation [39]. dCas9 is fused with GFP (Green Fluorescent Protein) that leads to chromatin live cell imaging.
Genome Editing Technology in Animal Model
The genome editing technology based on CRISPR/Cas9 allows for structural and functional characterization of various diseases in animal models. By means of Cre-loxP Technology, CRISPR/Cas9-based modification of PETN mutations with or without p53 mutations results in knocking out of PETN and P53 genes in mouse liver [41]. The oncogene β-catenin mutation is activated and may be transported to hepatocytes. This could be accomplished by incorporating the Cas9 plasmids [41]. Knocking out of Cas9 in mice model and lung adenocarcinoma models could be generated by inserting a single AAV vector containing loss-of-function mutations in p53, Lkb1, and Kras (G12D) mutations in the lung [42]. Cas9 and sgRNAs are used to target the β-catenin gene. These mutations in oncogenes and tumor suppressor genes develop a new method of using CRISPR/Cas systems in Cancer models. The oncogene β-catenin mutation is activated and may be transported to hepatocytes. This could be accomplished by coinjecting Cas9 plasmids [41]. Knocking of Cas9 in mice model and lung adenocarcinoma models could be generated by inserting a single AAV vector containing loss-of-function mutations in p53, Lkb1, and Kras (G12D) mutations in the lung [42]. Cas9 and sgRNAs are used to target the β-catenin gene. These mutations in oncogenes and tumor suppressor genes develop a new method of using CRISPR/Cas systems in Cancel models. Co-injection of Cas9 mRNA/gRNAs with mutant oligos results in gene knocking mice with precise point mutations in two genes [43]. Cointegration of Cas9mRNA and sgRNA targets the different genes into mice zygotes for generating mutant mouse having biallelic mutations [44]. Microinjection of mRNA encoding Cas9 and specific designed sgRNA into single-celled stage Zebrafish embryo [45]. Many other model organisms, such as Drosophila melanogaster, Caenorhabditis elegans, Axolotl, Xenopus tropicalis, Rattus sp., and Sus barbatus, have used CRISPR–Cas9 technology for efficient genome engineering. The CRISPR/Cas9 system has been demonstrated to be an efficient and reliable method for targeted genome modification.
Therapeutic Applications
CRISPR/Cas9 plays the most exhilarating role in the therapeutic industry for curing many genetic and infectious diseases. Cas9, sgRNA, and a homology-directed repair template were injected into mouse zygotes to correct the dystrophin point mutation in the germline and prevent muscular dystrophy in mutant mice [46]. The researchers used homologous recombination to successfully adjust the cystic fibrosis transmembrane conductor receptor (CFTR) locus in the same diseased human patients cultured intestinal stem cells [47]. CRISPR/Cas9 technology has been used to assess diabetes pathogenesis precisely. To reduce lipid storage in adipocytes, researchers used the CRISPRi technique to silence the expression of Fabp4. An Obese mouse was selected for this study and its body weight was reduced by about 20% as a part of this treatment. It has also reduced type 2 diabetes indicators like insulin resistance, inflammation, high glucose levels, and hepatic steatosis [48]. In Anopheles gambiae, CRISPR/Cas9 enables the inactivation of the FREP1 gene. At the sporozoite and oocyst stages, FREP1 knockout mutants effectively suppressed infection with both rodent and human malaria parasites [49]. The CFTR mutation has been corrected in iPSCs by reprogramming somatic skin fibroblasts from CF patients into embryonic stem cells with the CFTR/Cas9 gRNA vector [50]. CRISPR/Cas9 also plays a major role in preventing HIV by eliminating viral genes from the host cell chromosomes. A sgRNA expression vector targeting the long terminal repeats (LTR) of HIV-1 efficiently cleaves and mutates LTR target sites and suppresses LTR-driven viral gene expression when transfected into HIV-1 provirus-integrated human cells [51]. These findings suggest that the CRISPR–Cas9 technology is the budding technology of the therapeutic industry. Apart from therapeutic applications, CRISPR shows that gene editing technology has the potential to impact a wide range of sectors. Crispr could also be used for pet breeding, allergy-free food production, the production of greener fuels, and pest control which are reviewed in the literature [52,53,54,55].
Cas12 Protein
The sequences extracted from Francisella novicida U112 cultures containing Cas12a-based CRISPR loci revealed that mature crRNAs for Cas12a are 42–44 nucleotides long; with the first 19–20 nucleotides corresponding to the repeat sequence and the remaining 23–25 nucleotides corresponding to the spacer sequence [56]. For type V systems, there are nearly 300,000 putative CRISPR–Cas elements. There is only one predicted RuvC nuclease domain in these Cas nucleases. Rusk (2019) discovered the type V phylogenetic tree to have three major branches: one with Cas12a, Cas12c, Cas12d, and Cas12e; a second branch with Cas12b, Cas12h, and Cas12i; and a third branch with small effectors such as Cas12g, the smallest protein with only 800 amino acids of all the branches [57]. Class 2 type V is further subdivided into the subtypes listed below (V-A, V-B, V-C, and V-U).
However, there is some uncertainty about the protein structure with regard to V-C and V-U. The Cpf1 protein, also known as Cas12a, is encoded by V-A, and its structure paved the way for understanding its mechanism [56]. Cas12a (Cpf1), the prototype of type V effectors, has two RuvC-like nuclease domains but lacks HNH domain. However, a recent structural analysis of the Cas12a–crRNA–target DNA complex elucidates a second nuclease domain with a distinct fold that is functionally equivalent to the Cas9 HNH domain [58].
A T-rich PAM sequence that is (5′-TTN-3′) is required essentially when DNA is cleaved. The positively charged central channel of a nuclease (NUC) lobe may regulate the cleaving of the target strand after catalytic residue mutations in the RuvC domain of Cas12a in the bacterium Acidaminococcus sp. that inhibit the cleaving of the target and non-target strands [59]. Cas12a (cpf1), Cas12b (C2c1), Cas12c (C2c3), Cas12d (CasY), and Cas12e (CasX), the latter of which contains a catalytic domain capable of processing its own mRNA. SpCas9, the PAM for Cas12 proteins, on the other hand, contains thymine (T)-rich sequence and is located at the 5' end of the target sequence. The crRNA–target complex accounted for a distorted PAM duplex with a narrow minor groove that serves as a site for base-specific contacts with amino acid side chains via van der Waals and hydrogen bond interactions [20].
Among the variants of Cas12, Cas12c (C2c3), which has a single large protein (100–200 kDa) with a catalytic RNA-guided cleavage activity of DNA/RNA, aids in the identification of a new Class 2 protein classified as Cas12d. Cas12c and Cas12d share structural similarities. Almost 23 distinct Cas12 varieties from microbes from various habitats have been reported; among these microbes, a CPR (Candidate phyla Radiation) bacterium possesses the CRISPR–Cas genome locus that encodes for a Cas12d protein [60]. Cas12b of type V has been primed to acquire genome editing in human cells. The wild-type BhCas12b cleaves the non-target strand at 37 °C, resulting in a double-strand break. We gain loss-of-function mutation for the BhCas12b by modifying it, which overcomes the limitation of lower editing inefficiency. Despite the fact that Cas12b protein is much smaller than Cas9 and Cas12a, the efficient cas12b nuclease (AacCas12b) derived from Alicyclobacillus acidoterrestris has optimal nuclease or cleavage activity at 48 °C [61].
When infected, mature crRNA molecules guide the subtype-specific interference machinery against invading nucleic acids, effectively silencing foreign genetic material. Cas12a and Cas12b cleave the DNA duplex in both strands shortly after the PAM site is recognized and crRNA binds to the target DNA, leaving staggered ends with 5- and 7-nt [62]. The Cas12 has two varieties namely Cas12a (Cpf1), cas12b (C2c1), and Cas12c (C2c2) of which C2c1 and C2c2 contain RuvC-like endonuclease domains that are correlated to Cpf1. While the third system encompasses an effector with two predicted HEN RNase domains [63]. Another Cas12a, Cpf1, originating from Acidaminococcus sp. and recognized as AsCpf1, contains 1307 amino acids, which are a few amino acids shorter than SpCas9 (1368). The crystal structure of AsCpf1 shows that it has two main lobes: (a) a nuclease lobe (REC) and (b) an alpha helical recognition lobe (NUC) [64]. New Cas12a orthologs with improved crRNA scaffolds boost mammalian genome editing. Just three Cas12a orthologs have been used for mammalian genome editing, and editing performance and targeting coverage also need to be improved [65].
CeCas12a, a novel Cas12a derived from Coprococcus eutactus, is an editable nuclease with higher genome editing efficiencies in human cells than AsCas12a and LbCas12a. CeCas12a has shown to be a more rigorous enzyme for PAM recognition both in vitro and in vivo. These orthologous condenses off-target edits at C-containing PAM at various LbCas1-related sites [66]. Mutations in the NUC domain in Francisella novicida resulted in incomplete inactivation of the target strand cleaving [67]. It was shown that the mutations in the CRISPR genes of cotton crops like AsCpf1 (S542R/K607R and S542R/K548V/N552R) and LbCpf1 (G532R/K595R and G532R/K538V/Y542R) aid to recognize TYCV and TATV PAMs [68]. The study of mutation in the RuvC-I motif of Cas12g1 (D513A) demonstrates the functional variety that emerges along with different types of CRISPR–Cas evolution paths and expands the CRISPR toolbox [69].
Applications of Cas12a
Cas12a (Formerly known as Cpf1) is used as an alternative genetic tool of Cas9. Dead Cas12a from Eubacterium eligens (EedCas12a) revealed that it exhibits greater gene repression in the target DNA template strand than deactivated Cas9 from Streptococcus pyogenes (SpdCas9) [67]. It was also demonstrated that Cas12a’s pre-crRNA processing activity makes it a good candidate for multiplex gene regulation [70]. Different literature studies suggest that the multiplex gene regulation of Cas12a was observed in bacteria, plants as well as in mammalian [65, 66]. Cas12a can also be used as a replacement for Cas9 in cell types where Cas9 is toxic, such as some industrial strains of Streptomyces [71]. Targeted mutagenesis in plants can also be accomplished in vivo by co-expression of Cas12a and its related crRNA, as seen in rice. Furthermore, it was demonstrated that mutagenesis was more efficient when pre-crRNAs with full-length direct repeat sequences were used rather than mature crRNAs [72]. Efficient mutagenesis was also observed in soybean and wild tobacco via the delivery of the pre-assembled ribonucleoprotein (RNP) particle. The RNP was constructed using recombinant Cas12a and in vitro transcribed or chemically synthesized crRNAs [67]. Correction of mutations causing Duchene muscular dystrophy (DMD) in patient-derived induced pluripotent stem cells (iPSCs) and mdx mice, a popular model for studying DMD. Dystrophin expression was restored in iPSCs following Cas12a-mediated gene editing, while corrections in the pathophysiological hallmarks of muscular dystrophy were observed in mdx mice [73]. The adenovirus vector was combined with an AsCas12a expression system, resulting in successful mutations in humanized mice with chimeric liver primary human hepatocytes [74]. Cas12a-mediated genome editing was also used to create rat models that resemble human atherosclerosis, and this system may have applications in understanding early-stage atherosclerosis [75].
Biotechnology and Biomedical Engineering
A massive effort is currently underway to redesign all of these tools for biomedical and biotechnological applications. However, recent research has suggested that CRISPR–Cas nucleases could be used in the bioengineering of smart materials such as hydrogels [76]. DNA encapsulates these water-filled polymers. Cas12a was recently used in a study to specifically degrade the DNA scaffold of DNA hydrogels, implying that this smart cutter could be turned into a programmable device to deliver the cargo of DNA encaged hydrogels in a specific location at a specific time. The cleavage properties of Cas12a make it an ideal candidate to promote controlled delivery of the cargo [56]. Cas12a uses a multi-checkpoint mechanism to ensure precise targeting of DNA, which is desirable in a genome editing tool in order to have low off-target effects [56].
Diagnosis
DETECTR, a DNA endonuclease-targeted CRISPR trans reporter, is a diagnostic tool that combines isothermal RPA with Cas12a enzymatic activity [69]. Binding of the crRNA–Cas12a complex to target DNA results in indiscriminate cleavage of ssDNA coupled to a fluorescent reporter in this assay. DETECTR was used to differentiate between HPV16 and HPV18 in crude DNA extracts from cultured human cells and clinical samples [69]. DETECTR was used to test crude extracts from 25 human anal swab samples, with the results compared to an approved PCR-based assay. Following DNA extraction, DETECTR was correctly identified. HPV16 and HPV18 were detected in clinical specimens within 1 h, with results agreeing with PCR results in 25/25 and 23/25 samples, respectively. Cas12a has a wide range of applications. Currently, many studies are going on this genomic tool to find its more uses in research, diagnostics, and the health sector field.
Cas13 Protein
RNA editing has several advantages over traditional DNA editing technology. CRISPR/Cas13 is an example of a major RNA editing technology. Cas13 enzymes, unlike Cas9 and Cpf1, do not require a PAM sequence but do require a protospacer flanking site (PFS) to target RNA. However, LWACas13s do not require a PFS site. Cas13 enzymes lack the RuvC and HNH domains required for DNA cleavage, preventing them from directly editing the genome. A CRISPR/Cas13-based RNA editing system would most likely be reversible, avoiding genomic off-targets introduced by non-homologous end joining (NHEJ).
Cas13 proteins such as Cas13a, Cas13b, and Cas13c are found in CRISPR/Cas systems of class 2 subtype VI that use a single effector protein [77]. The Cas13 protein and CRISPR RNA (guide RNA, 60–65nt) recognize a 28–30nt sequence on the target RNA through a spacer sequence which can make up the active endoRNase. Cas13a fundamental studies have shown two catalytic activities [78]. Cas13a first catalyzes the processing of pre-crRNAs to supply mature crRNAs, the cleavage of the target RNA is intervened by a separate catalytic activity. When the guide RNA base pairs with the target RNA, a catalytic site is formed between two HEPN domains of Cas13. Cas13 has several properties that make it an excellent candidate for an RNA-modulating tool. To begin, Cas13 is frequently programmed to target potentially any RNA by changing the crRNA spacer sequence. Second, unlike Cas9 and Cpf1, which need an exact PAM (protospacer adjacent motif) to be present in the DNA, Cas13 (mainly Cas13b) does not need a specific sequence element in the target site called PFS. Third, among the CRISPR/Cas systems, the effector complex is one of the most basic. Cas13-crRNA may be a two-component system that is easier to control and deliver than trimeric or multimeric systems. Fourth, because Cas13 is responsible for both crRNA biogenesis and silencing, multiple crRNAs can be loaded simultaneously [79]. In knockdown experiments, they discovered that PspCas13b is highly specific to on-target sequences. According to preliminary transcriptomic and gene-level analyses, PspCas13b may be more specific in knockdown experiments than short-hairpin RNAs (shRNAs). The efficiency of silencing was also impressive, with 60–90% suppression [80]. However, the central seed tolerates single mismatches to some extent, caution may be required when designing guide RNAs to avoid off-target effects. It is unclear how some Cas13 achieve such high selectivity, and whether the specificity can be maintained in all cellular and molecular contexts (Table 5).
RNA Editing Technology of Cas13
CRISPR/Cas13 is a RNA-targeting genome editing system. When ssRNA get recognized, Cas13a (C2c2) gets activated [77]. Cas13a is derived from Leptotrichia wadei and Cas13b is derived from Prevotella sp. P5–125 [81]. Cas13 subtype Cas13a cleaves nitrogenous base uracil anywhere after binding to the target RNA. This process is known as collateral cleavage. This cleavage process spreads to neighboring untargeted RNAs. Cas13a has been found to cut specific target mRNAs spontaneously in both bacteria and eukaryotic cells [77, 81]. dCas13a, a Cas13a variant, retains the ability to bind targeted RNA and was used for RNA imaging in live cells [81]. dCas13a, like RCas9, has been targeted to mRNA to visualize the formation of stress granules [82]. The subtypes of Cas13 endonuclease have distinctive functions in various animals or plant model systems. It is extremely difficult to design crRNA due to the large number of Orthologs and subtypes of cas13. LwaCas13a, PspCas13, and RfxCas13d have been described in the table for targeting RNAs of various genes in mammalian cells. The comparison between Cas13a, cas13b, and Cas13d shows that RfxCas143d has the most important knockdown in HEK293T cells [83]. LwaCas13a has been used as a model system for plant or agriculture-based applications. It has a significant knockdown in rice protoplasts (Table 6).
Applications of Cas13
Research and Development
Cas13 has multiple applications in the field of research and development. Cas13 requires a Cas13 variant for RNA knockdown and to reduce off-target activity. CRISPR sequence of Cas13 requires mainly PFS sequence to identify and cleave the target RNA. The smallest Cas13 variant, also known as Cas13d (CasRx), is delivered by Adeno-associated virus vector. Cas13d does not require a PFS sequence to identify the target DNA. This variant of cas13d has high specificity and it reduces off-target activities of Cas, Cas13a plays a unique role in RNA imaging. The catalytically inactive Cas13 (dCas13a) is bounded with GFP (Green Fluorescent Protein), ZFN (Zinc finger nucleases), and KRAB (Kruppel-associated box domain) [78]. The fusion leads to the recognition of endogenous, untagged, and unmodified mRNA [84]. Cas13a (Formerly known as C2c2) protects plants from viruses over time. Cas13a codon optimization is required for proper protein functioning, which can reduce off-target activity while increasing multiplex target activities of RNA-targeting Cas protein [85]. Apoptosis (programmed cell death) is activated inside the cell by non-specific RNA degradation of Cas13a in response to the infection by a cellular pathogen [72]. Cas13b, catalytically inactive Cas13, is fused with eukaryotic RNA-modifying enzyme N6-methyladenosine (m6A) bounded with YTHDF1 Protein to trigger the assembly of translational mechanism. Cas13 variant can be fused with YTHDF2, which can induce RNA degradation. In this way, Cas13 regulates gene expression in eukaryotic cells [86].
RNA Editing Technology
Another important application of inactivated Cas13 is RNA editing technology, REPAIRv1 (RNA Editing for Programmable A to I Replacement) [81]. Here, dCas13 was fused with human adenosine deaminase acting on RNA (ADAR) enzymes called ADAR1 and ADAR2. The programmable replacement of adenosine with inosine has been recognized as guanine during complementary base pairing. The replacement of A to I causes temporary alteration of translation of a selected gene. The specificity of this system is improved by REPAIRv2, which introduces a mutation within the ADAR2 enzyme [81].
Diagnosis
An emerging application of Cas13 in the field of diagnosis is SHERLOCK (Specific High-Sensitivity Enzymatic Reporter unLOCKing) Technology. This can be used for in vitro detection of DNA and RNA in a sample [87]. In this method sample, the nucleic acid is amplified to dsDNA by recombinase polymerase. Here, RNA sample undergoes reverse transcription and produces dsDNA, which serves as a template of T7 polymerase that produces ssRNA transcripts. Cas13 scans those ssRNA transcripts. The activated Cas13 cleaves the reporter upon recognition and releases the signal. SHERLOCK Technology is a highly sensitive and effective diagnostic technique used for the detection of Zika and dengue viruses [87]. The improved version of SHERLOCK was SHERLOCKv2. This was a multiplex detection method [88]. Multiple variants of Cas13 prefer different motifs of RNA of nt sequences AU, AC, UC, or GA. Different reporters can be used to detect various RNA in the sample. SHERLOCKv2 uses a Csm6 protein that is allosterically activated by Cas13 via cyclic oligoadenylates upon its own activation to improve signal strength. The combination of Csm6 and Cas13 collateral RNA cleavage results in a stronger signal from cleaved reporters [88].
Role of CRISPR/Cas System in COVID-19
A CRISPR/Cas12-based diagnostic method for the detection of the COVID-19 virus has been developed called SARS_Cov2 DETECTR. The total detection time is around 40 min and it has 95% accuracy. This assay has been started with RNA extraction, then reverse transcription and isothermal amplification by using RT_LAMP. Cas12 and guide RNA target E (Envelope) and N (Nucleoprotein) gene of SARS-Cov2. This assay also includes a laminar flow strip for visual readouts, with a single band close to where the sample was applied indicating a negative test and two higher bands or a single higher band indicating cleavage of the fluorescent probe and thus positive for SARS-Cov2 [89]. By the recently discovered RNA-guided RNA-targeting endonuclease, Cas13 can be used as a potential therapeutic tool against COVID-19.PAC_MAN (Prophylactic Antiviral CRISPR in human cells).
PAC-MAN (Prophylactic Antiviral CRISPR in huMAN cells) was created using the Ruminococcus flavefaciens-derived VI-D CRISPR–Cas13d variant, which was chosen for its small size, which allows for easier packaging in viral vehicles, high specificity, and strong catalytic activity in human cells. Given the evidence indicating relatively high mutation and recombination rates of SARS-CoV-2, this technique was developed to simultaneously target multiple regions for RNA degradation, paving the way for a much-needed pan coronavirus targeting strategy [90].
Future Prospects
CRISPR–Cas technology is transforming the new face of research. As compared with the other genome editing tools, it is very cheap, feasible, and more efficient in manipulating the genes. Many of the future challenges in the field of research, medicine, or agriculture are mainly depend on this technology from simple organisms to complex organisms also from targeting one gene to multiple genes. Although this technology has a lot of promise for medicinal development, efficacy and long-term safety are still major problems in clinical trials. Off-target editing, massive DNA deletions, and other rearrangements are all serious safety concerns that have yet to be effectively addressed in preclinical research. Recent studies have shown that CRISPR/Cas9-mediated in vivo genome editing of disease genes can improve disease phenotype and pathology in animal models without causing serious side effects [91]. Meanwhile, researchers have been contributing towards the significant progress in the CRISPR–Cas system to increase the target specificity effectively.
Technological and ethical concerns are still present, which makes an impact on not having the next level of discoveries. But it has the potential to eradicate genetic disorders, making non-pathogens, bringing back extinct species, treating invasive diseases, etc. Continuing the research will have clear, precise information about the CRISPR–Cas system, and more advancement could occur in the near future. Human genome editing for reproductive purposes, according to bioethicists and experts, should not be tried at this time, but work to make gene therapy safe and successful should continue. The National Academy of Sciences, National Academy of Medicine, Chinese Academy of Sciences, and the Royal Society of the United Kingdom call together an international summit to discuss recent scientific advances in human gene editing as well as the range of ethical and governance issues that these advances raise. Most of the biotech startup has already started working on this system and have a hope to improvise also to develop the cutting-edge research and innovations. The possibilities of keen understanding will create excitement among sciences to develop novel strategies in the genome editing approach.
Data Availability
Not applicable.
References
Ishino, Y., Shinagawa, H., Makino, K., Amemura, M., & Nakata, A. (1987). Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology, 169(12), 5429–5433. https://doi.org/10.1128/jb.169.12.5429-5433.1987
Groenen, P. M. A., Bunschoten, A. E., van Soolingen, D., & van Errtbden, J. D. A. (1993). Nature of DNA polymorphism in the direct repeat cluster of Mycobacterium tuberculosis; application for strain differentiation by a novel typing method. Molecular Microbiology, 10(5), 1057–1065. https://doi.org/10.1111/j.1365-2958.1993.tb00976.x
Mojica, F. J. M., Ferrer, C., Juez, G., & Rodríguez-Valera, F. (1995). Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Molecular Microbiology, 17(1), 85–93. https://doi.org/10.1111/j.1365-2958.1995.mmi_17010085.x
Mojica, F. J. M., Diez-Villasenor, C., Soria, E., & Juez, G. (2000). Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Molecular Microbiology, 36(1), 244–246. https://doi.org/10.1046/j.1365-2958.2000.01838.x
Jansen, R., van Embden, J. D. A., Gaastra, W., & Schouls, L. M. (2002). Identification of genes that are associated with DNA repeats in prokaryotes. Molecular Microbiology, 43(6), 1565–1575. https://doi.org/10.1046/j.1365-2958.2002.02839.x
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell, 163(3), 759–771. https://doi.org/10.1016/j.cell.2015.09.038
Kim, Y. G., Cha, J., & Chandrasegaran, S. (1996). Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences, 93(3), 1156–1160. https://doi.org/10.1073/pnas.93.3.1156
Bibikova, M., Carroll, D., Segal, D. J., Trautman, J. K., Smith, J., Kim, Y.-G., & Chandrasegaran, S. (2001). Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Molecular and Cellular Biology, 21(1), 289–297. https://doi.org/10.1128/MCB.21.1.289-297.2001
Porteus, M. H., & Baltimore, D. (2003). Chimeric nucleases stimulate gene targeting in human cells. Science, 300(5620), 763–763. https://doi.org/10.1126/science.1078395
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science, 315(5819), 1709–1712. https://doi.org/10.1126/science.1138140
Brouns, S. J. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J. H., Snijders, A. P. L., et al. (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science, 321(5891), 960–964. https://doi.org/10.1126/science.1159689
Pickar-Oliver, A., & Gersbach, C. A. (2019). The next generation of CRISPR–Cas technologies and applications. Nature Reviews Molecular Cell Biology, 20(8), 490–507. https://doi.org/10.1038/s41580-019-0131-5
Rath, D., Amlinger, L., Rath, A., & Lundgren, M. (2015). The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie, 117, 119–128. https://doi.org/10.1016/j.biochi.2015.03.025
Chylinski, K., Le Rhun, A., & Charpentier, E. (2013). The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biology, 10(5), 726–737. https://doi.org/10.4161/rna.24321
Deltcheva, E., Chylinski, K., Sharma, C. M., Gonzales, K., Chao, Y., Pirzada, Z. A., et al. (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature, 471(7340), 602–607. https://doi.org/10.1038/nature09886
Garneau, J. E., Dupuis, M. -È., Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature, 468(7320), 67–71. https://doi.org/10.1038/nature09523
Anders, C., Niewoehner, O., Duerst, A., & Jinek, M. (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature, 513(7519), 569–573. https://doi.org/10.1038/nature13579
Jiang, F., Zhou, K., Ma, L., Gressel, S., & Doudna, J. A. (2015). A Cas9-guide RNA complex preorganized for target DNA recognition. Science, 348(6242), 1477–1481. https://doi.org/10.1126/science.aab1452
Dagdas, Y. S., Chen, J. S., Sternberg, S. H., Doudna, J. A., & Yildiz, A. (2017). A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Science Advances, 3(8), 0027. https://doi.org/10.1126/sciadv.aao0027
Jiang, F., Taylor, D. W., Chen, J. S., Kornfeld, J. E., Zhou, K., Thompson, A. J., et al. (2016). Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science, 351(6275), 867–871. https://doi.org/10.1126/science.aad8282
Sternberg, S. H., LaFrance, B., Kaplan, M., & Doudna, J. A. (2015). Conformational control of DNA target cleavage by CRISPR–Cas9. Nature, 527(7576), 110–113. https://doi.org/10.1038/nature15544
Hu, J. H., Miller, S. M., Geurts, M. H., Tang, W., Chen, L., Sun, N., et al. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature, 556(7699), 57–63. https://doi.org/10.1038/nature26155
Kleinstiver, B. P., Prew, M. S., Tsai, S. Q., Topkar, V. V., Nguyen, N. T., Zheng, Z., et al. (2015). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature, 523(7561), 481–485. https://doi.org/10.1038/nature14592
Chen, J. S., & Doudna, J. A. (2017). The chemistry of Cas9 and its CRISPR colleagues. Nature Reviews Chemistry, 1(10), 0078. https://doi.org/10.1038/s41570-017-0078
Kleinstiver, B. P., Tsai, S. Q., Prew, M. S., Nguyen, N. T., Welch, M. M., Lopez, J. M., et al. (2016). Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nature Biotechnology, 34(8), 869–874. https://doi.org/10.1038/nbt.3620
Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D. A., Yan, W. X., & Zhang, F. (2016). Rationally engineered Cas9 nucleases with improved specificity. Science, 351(6268), 84–88. https://doi.org/10.1126/science.aad5227
Vakulskas, C. A., Dever, D. P., Rettig, G. R., Turk, R., Jacobi, A. M., Collingwood, M. A., et al. (2018). A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nature Medicine, 24(8), 1216–1224. https://doi.org/10.1038/s41591-018-0137-0
Kocak, D. D., Josephs, E. A., Bhandarkar, V., Adkar, S. S., Kwon, J. B., & Gersbach, C. A. (2019). Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nature Biotechnology, 37(6), 657–666. https://doi.org/10.1038/s41587-019-0095-1
Esvelt, K. M., Mali, P., Braff, J. L., Moosburner, M., Yaung, S. J., & Church, G. M. (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature Methods, 10(11), 1116–1121. https://doi.org/10.1038/nmeth.2681
Ran, F. A., Cong, L., Yan, W. X., Scott, D. A., Gootenberg, J. S., Kriz, A. J., et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature, 520(7546), 186–191. https://doi.org/10.1038/nature14299
Burstein, D., Harrington, L. B., Strutt, S. C., Probst, A. J., Anantharaman, K., Thomas, B. C., et al. (2017). New CRISPR–Cas systems from uncultivated microbes. Nature, 542(7640), 237–241. https://doi.org/10.1038/nature21059
Kim, H., Kim, S.-T., Ryu, J., Kang, B.-C., Kim, J.-S., & Kim, S.-G. (2017). CRISPR/Cpf1-mediated DNA-free plant genome editing. Nature Communications, 8(1), 14406. https://doi.org/10.1038/ncomms14406
Yamada, M., Watanabe, Y., Gootenberg, J. S., Hirano, H., Ran, F. A., Nakane, T., et al. (2017). Crystal structure of the minimal Cas9 from Campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Molecular Cell, 65(6), 1109–1121. https://doi.org/10.1016/j.molcel.2017.02.007
Le Rhun, A., Escalera-Maurer, A., Bratovič, M., & Charpentier, E. (2019). CRISPR-Cas in Streptococcus pyogenes. RNA Biology, 16(4), 380–389. https://doi.org/10.1080/15476286.2019.1582974
Lee, C. M., Cradick, T. J., & Bao, G. (2016). The Neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells. Molecular Therapy, 24(3), 645–654. https://doi.org/10.1038/mt.2016.8
Cebrian-Serrano, A., & Davies, B. (2017). CRISPR-Cas orthologues and variants: Optimizing the repertoire, specificity and delivery of genome engineering tools. Mammalian Genome, 28(7–8), 247–261. https://doi.org/10.1007/s00335-017-9697-4
Müller, M., Lee, C. M., Gasiunas, G., Davis, T. H., Cradick, T. J., & Siksnys, V. (2016). Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing of the human genome. Molecular Therapy, 24(3), 636–644. https://doi.org/10.1038/mt.2015.218
Jaganathan, D., Ramasamy, K., Sellamuthu, G., Jayabalan, S., & Venkataraman, G. (2018). CRISPR for crop improvement: An update review. Frontiers in Plant Science. https://doi.org/10.3389/fpls.2018.00985
Lino, C. A., Harper, J. C., Carney, J. P., & Timlin, J. A. (2018). Delivering CRISPR: A review of the challenges and approaches. Drug Delivery, 25(1), 1234–1257. https://doi.org/10.1080/10717544.2018.1474964
Dugar, G., Leenay, R. T., Eisenbart, S. K., Bischler, T., Aul, B. U., Beisel, C. L., & Sharma, C. M. (2018). CRISPR RNA-dependent binding and cleavage of endogenous RNAs by the Campylobacter jejuni Cas9. Molecular Cell, 69(5), 893–905. https://doi.org/10.1016/j.molcel.2018.01.032
Price, A. A., Sampson, T. R., Ratner, H. K., Grakoui, A., & Weiss, D. S. (2015). Cas9-mediated targeting of viral RNA in eukaryotic cells. Proceedings of the National Academy of Sciences, 112(19), 6164–6169. https://doi.org/10.1073/pnas.1422340112
Poltronieri, P., Sun, B., & Mallardo, M. (2015). RNA viruses: RNA roles in pathogenesis, coreplication and viral load. Current Genomics, 16(5), 327–335. https://doi.org/10.2174/1389202916666150707160613
Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343(6166), 84–87. https://doi.org/10.1126/science.1247005
Koike-Yusa, H., Li, Y., Tan, E.-P., Velasco-Herrera, M. D. C., & Yusa, K. (2014). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nature Biotechnology, 32(3), 267–273. https://doi.org/10.1038/nbt.2800
Pham, H., Kearns, N. A., & Maehr, R. (2016). Transcriptional regulation with CRISPR/Cas9 effectors in mammalian cells. Methods in Molecular Biology. https://doi.org/10.1007/978-1-4939-3067-8_3
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., & Lim, W. A. (2021). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell, 184(3), 844. https://doi.org/10.1016/j.cell.2021.01.019
Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell, 154(2), 442–451. https://doi.org/10.1016/j.cell.2013.06.044
Thakore, P. I., D’Ippolito, A. M., Song, L., Safi, A., Shivakumar, N. K., Kabadi, A. M., et al. (2015). Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nature Methods, 12(12), 1143–1149. https://doi.org/10.1038/nmeth.3630
Hilton, I. B., D’Ippolito, A. M., Vockley, C. M., Thakore, P. I., Crawford, G. E., Reddy, T. E., & Gersbach, C. A. (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nature Biotechnology, 33(5), 510–517. https://doi.org/10.1038/nbt.3199
Xue, W., Chen, S., Yin, H., Tammela, T., Papagiannakopoulos, T., Joshi, N. S., et al. (2014). CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature, 514(7522), 380–384. https://doi.org/10.1038/nature13589
Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell, 159(2), 440–455. https://doi.org/10.1016/j.cell.2014.09.014
Li, C., Brant, E., Budak, H., & Zhang, B. (2021). CRISPR/Cas: A nobel prize award-winning precise genome editing technology for gene therapy and crop improvement. Journal of Zhejiang University Science B, 22(4), 253–284. https://doi.org/10.1631/jzus.B2100009
Bashar, K. K., & Hanif, M. A. (2021). Crop gene editing against biotic stresses via CRISPR/Cas9 tools: A review. Archives of Phytopathology and Plant Protection, 54(15–16), 1159–1181. https://doi.org/10.1080/03235408.2021.1895476
Flotte, T. R. (2021). CRISPR keeps things fresh: Next-generation tools for gene editing. Human Gene Therapy, 32(5–6), 235–236. https://doi.org/10.1089/hum.2021.29152.trf
Abdeen, A. A., Cosgrove, B. D., Gersbach, C. A., & Saha, K. (2021). Integrating biomaterials and genome editing approaches to advance biomedical science. Annual Review of Biomedical Engineering, 23(1), 493–516. https://doi.org/10.1146/annurev-bioeng-122019-121602
Yang, H., Wang, H., Shivalila, C. S., Cheng, A. W., Shi, L., & Jaenisch, R. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell, 154(6), 1370–1379. https://doi.org/10.1016/j.cell.2013.08.022
Wang, H., Yang, H., Shivalila, C. S., Dawlaty, M. M., Cheng, A. W., Zhang, F., & Jaenisch, R. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell, 153(4), 910–918. https://doi.org/10.1016/j.cell.2013.04.025
Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L., Tsai, S. Q., Sander, J. D., et al. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology, 31(3), 227–229. https://doi.org/10.1038/nbt.2501
Schwank, G., Koo, B.-K., Sasselli, V., Dekkers, J. F., Heo, I., Demircan, T., et al. (2013). Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell, 13(6), 653–658. https://doi.org/10.1016/j.stem.2013.11.002
Long, C., McAnally, J. R., Shelton, J. M., Mireault, A. A., Bassel-Duby, R., & Olson, E. N. (2014). Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science, 345(6201), 1184–1188. https://doi.org/10.1126/science.1254445
Chung, J. Y., Ain, Q. U., Song, Y., Yong, S.-B., & Kim, Y.-H. (2019). Targeted delivery of CRISPR interference system against Fabp4 to white adipocytes ameliorates obesity, inflammation, hepatic steatosis, and insulin resistance. Genome Research, 29(9), 1442–1452. https://doi.org/10.1101/gr.246900.118
Dong, Y., Simões, M. L., Marois, E., & Dimopoulos, G. (2018). CRISPR/Cas9 -mediated gene knockout of Anopheles gambiae FREP1 suppresses malaria parasite infection. PLoS Pathogens, 14(3), e1006898. https://doi.org/10.1371/journal.ppat.1006898
Crane, A. M., Kramer, P., Bui, J. H., Chung, W. J., Li, X. S., Gonzalez-Garay, M. L., et al. (2015). Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports, 4(4), 569–577. https://doi.org/10.1016/j.stemcr.2015.02.005
Ebina, H., Misawa, N., Kanemura, Y., & Koyanagi, Y. (2013). Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Scientific Reports, 3(1), 2510. https://doi.org/10.1038/srep02510
Paul, B., & Montoya, G. (2020). CRISPR-Cas12a: Functional overview and applications. Biomedical Journal, 43(1), 8–17. https://doi.org/10.1016/j.bj.2019.10.005
Rusk, N. (2019). Spotlight on Cas12. Nature Methods, 16(3), 215–215. https://doi.org/10.1038/s41592-019-0347-5
Yan, F., Wang, W., & Zhang, J. (2019). CRISPR-Cas12 and Cas13: The lesser known siblings of CRISPR-Cas9. Cell Biology and Toxicology, 35(6), 489–492. https://doi.org/10.1007/s10565-019-09489-1
Gao, W., Long, L., Tian, X., Xu, F., Liu, J., Singh, P. K., et al. (2017). Genome editing in cotton with the CRISPR/Cas9 system. Frontiers in Plant Science. https://doi.org/10.3389/fpls.2017.01364
Yan, W. X., Hunnewell, P., Alfonse, L. E., Carte, J. M., Keston-Smith, E., Sothiselvam, S., et al. (2019). Functionally diverse type V CRISPR-Cas systems. Science, 363(6422), 88–91. https://doi.org/10.1126/science.aav7271
Harrington, L. B., Ma, E., Chen, J. S., Witte, I. P., Gertz, D., Paez-Espino, D., et al. (2020). A scoutRNA is required for some type V CRISPR-Cas systems. Molecular Cell, 79(3), 416–424. https://doi.org/10.1016/j.molcel.2020.06.022
Strecker, J., Jones, S., Koopal, B., Schmid-Burgk, J., Zetsche, B., Gao, L., et al. (2019). Engineering of CRISPR-Cas12b for human genome editing. Nature Communications, 10(1), 212. https://doi.org/10.1038/s41467-018-08224-4
Loureiro, A., & da Silva, G. (2019). CRISPR-Cas: Converting a bacterial defence mechanism into a state-of-the-art genetic manipulation tool. Antibiotics, 8(1), 18. https://doi.org/10.3390/antibiotics8010018
Shmakov, S., Abudayyeh, O. O., Makarova, K. S., Wolf, Y. I., Gootenberg, J. S., Semenova, E., et al. (2015). Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Molecular Cell, 60(3), 385–397. https://doi.org/10.1016/j.molcel.2015.10.008
Dong, D., Ren, K., Qiu, X., Zheng, J., Guo, M., Guan, X., et al. (2016). The crystal structure of Cpf1 in complex with CRISPR RNA. Nature, 532(7600), 522–526. https://doi.org/10.1038/nature17944
Teng, F., Li, J., Cui, T., Xu, K., Guo, L., Gao, Q., et al. (2019). Enhanced mammalian genome editing by new Cas12a orthologs with optimized crRNA scaffolds. Genome Biology, 20(1), 15. https://doi.org/10.1186/s13059-019-1620-8
Chen, P., Zhou, J., Wan, Y., Liu, H., Li, Y., Liu, Z., et al. (2020). A Cas12a ortholog with stringent PAM recognition followed by low off-target editing rates for genome editing. Genome Biology, 21(1), 78. https://doi.org/10.1186/s13059-020-01989-2
Kim, H. K., Song, M., Lee, J., Menon, A. V., Jung, S., Kang, Y.-M., et al. (2017). In vivo high-throughput profiling of CRISPR–Cpf1 activity. Nature Methods, 14(2), 153–159. https://doi.org/10.1038/nmeth.4104
Zetsche, B., Heidenreich, M., Mohanraju, P., Fedorova, I., Kneppers, J., DeGennaro, E. M., et al. (2017). Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nature Biotechnology, 35(1), 31–34. https://doi.org/10.1038/nbt.3737
Li, S.-Y., Cheng, Q.-X., Wang, J.-M., Li, X.-Y., Zhang, Z.-L., Gao, S., et al. (2018). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discovery, 4(1), 20. https://doi.org/10.1038/s41421-018-0028-z
Xu, X.-R.S., Gantz, V. M., Siomava, N., & Bier, E. (2017). CRISPR/Cas9 and active genetics-based trans-species replacement of the endogenous Drosophila kni-L2 CRM reveals unexpected complexity. eLife. https://doi.org/10.7554/eLife.30281
Makarova, K. S., Zhang, F., & Koonin, E. V. (2017). SnapShot: Class 2 CRISPR-Cas systems. Cell, 168(1–2), 328-328.e1. https://doi.org/10.1016/j.cell.2016.12.038
Tsukamoto, T., Sakai, E., Iizuka, S., Taracena-Gándara, M., Sakurai, F., & Mizuguchi, H. (2018). Generation of the adenovirus vector-mediated CRISPR/Cpf1 system and the application for primary human hepatocytes prepared from humanized mice with chimeric liver. Biological and Pharmaceutical Bulletin, 41(7), 1089–1095. https://doi.org/10.1248/bpb.b18-00222
Lee, J. K., Jeong, E., Lee, J., Jung, M., Shin, E., Kim, Y., et al. (2018). Directed evolution of CRISPR-Cas9 to increase its specificity. Nature Communications, 9(1), 3048. https://doi.org/10.1038/s41467-018-05477-x
Han, D., Li, J., & Tan, W. (2019). CRISPR propels a smart hydrogel. Science, 365(6455), 754–755. https://doi.org/10.1126/science.aay4198
Abudayyeh, O. O., Gootenberg, J. S., Konermann, S., Joung, J., Slaymaker, I. M., Cox, D. B. T., et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. https://doi.org/10.1126/science.aaf5573
Knott, G. J., East-Seletsky, A., Cofsky, J. C., Holton, J. M., Charles, E., O’Connell, M. R., & Doudna, J. A. (2017). Guide-bound structures of an RNA-targeting A-cleaving CRISPR–Cas13a enzyme. Nature Structural & Molecular Biology, 24(10), 825–833. https://doi.org/10.1038/nsmb.3466
Liu, L., Li, X., Wang, J., Wang, M., Chen, P., Yin, M., et al. (2017). Two distant catalytic sites are responsible for C2c2 RNase activities. Cell, 168(1–2), 121–134. https://doi.org/10.1016/j.cell.2016.12.031
DeNardo, L., & Luo, L. (2017). Genetic strategies to access activated neurons. Current Opinion in Neurobiology, 45, 121–129. https://doi.org/10.1016/j.conb.2017.05.014
Taxman, D. J., Moore, C. B., Guthrie, E. H., & Huang, M.T.-H. (2010). Short hairpin RNA (shRNA): Design, delivery, and assessment of gene knockdown. In Methods in Molecular Biology. https://doi.org/10.1007/978-1-60761-657-3_10
Abudayyeh, O. O., Gootenberg, J. S., Essletzbichler, P., Han, S., Joung, J., Belanto, J. J., et al. (2017). RNA targeting with CRISPR–Cas13. Nature, 550(7675), 280–284. https://doi.org/10.1038/nature24049
Deng, H.-X., Zhai, H., Shi, Y., Liu, G., Lowry, J., Liu, B., et al. (2021). Efficacy and long-term safety of CRISPR/Cas9 genome editing in the SOD1-linked mouse models of ALS. Communications Biology, 4(1), 396. https://doi.org/10.1038/s42003-021-01942-4
Nuñez, J. K., Kranzusch, P. J., Noeske, J., Wright, A. V., Davies, C. W., & Doudna, J. A. (2014). Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nature Structural & Molecular Biology, 21(6), 528–534. https://doi.org/10.1038/nsmb.2820
Dolan, A. E., Hou, Z., Xiao, Y., Gramelspacher, M. J., Heo, J., Howden, S. E., et al. (2019). Introducing a spectrum of long-range genomic deletions in human embryonic stem cells using type I CRISPR-Cas. Molecular Cell, 74(5), 936–950. https://doi.org/10.1016/j.molcel.2019.03.014
Lee, H., Dhingra, Y., & Sashital, D. G. (2019). The Cas4-Cas1-Cas2 complex mediates precise prespacer processing during CRISPR adaptation. eLife. https://doi.org/10.7554/eLife.44248
Brendel, J., Stoll, B., Lange, S. J., Sharma, K., Lenz, C., Stachler, A.-E., et al. (2014). A Complex of cas proteins 5, 6, and 7 is required for the biogenesis and stability of clustered regularly interspaced short palindromic repeats (CRISPR)-derived RNAs (crRNAs) in Haloferax volcanii. Journal of Biological Chemistry, 289(10), 7164–7177. https://doi.org/10.1074/jbc.M113.508184
Hrle, A., Maier, L.-K., Sharma, K., Ebert, J., Basquin, C., Urlaub, H., et al. (2014). Structural analyses of the CRISPR protein Csc2 reveal the RNA-binding interface of the type I-D Cas7 family. RNA Biology, 11(8), 1072–1082. https://doi.org/10.4161/rna.29893
Kalwani, P., Rath, D., & Ballal, A. (2020). Novel molecular aspects of the CRISPR backbone protein ‘Cas7’ from cyanobacteria. Biochemical Journal, 477(5), 971–983. https://doi.org/10.1042/BCJ20200026
Nishimasu, H., Ran, F. A., Hsu, P. D., Konermann, S., Shehata, S. I., Dohmae, N., et al. (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell, 156(5), 935–949. https://doi.org/10.1016/j.cell.2014.02.001
Batool, A., Malik, F., & Andrabi, K. I. (2021). Expansion of the CRISPR/Cas genome-sculpting toolbox: Innovations, applications and challenges. Molecular Diagnosis & Therapy, 25(1), 41–57. https://doi.org/10.1007/s40291-020-00500-8
Shmakov, S. A., Makarova, K. S., Wolf, Y. I., Severinov, K. V., & Koonin, E. V. (2018). Systematic prediction of genes functionally linked to CRISPR-Cas systems by gene neighborhood analysis. Proceedings of the National Academy of Sciences, 115(23), E5307–E5316. https://doi.org/10.1073/pnas.1803440115
Schunder, E., Rydzewski, K., Grunow, R., & Heuner, K. (2013). First indication for a functional CRISPR/Cas system in Francisella tularensis. International Journal of Medical Microbiology, 303(2), 51–60. https://doi.org/10.1016/j.ijmm.2012.11.004
Smargon, A. A., Cox, D. B. T., Pyzocha, N. K., Zheng, K., Slaymaker, I. M., Gootenberg, J. S., et al. (2017). Cas13b is a type VI-B CRISPR-associated RNA-guided rnase differentially regulated by accessory proteins Csx27 and Csx28. Molecular Cell, 65(4), 618-630.e7. https://doi.org/10.1016/j.molcel.2016.12.023
Harrington, L. B., Burstein, D., Chen, J. S., Paez-Espino, D., Ma, E., Witte, I. P., et al. (2018). Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science, 362(6416), 839–842. https://doi.org/10.1126/science.aav4294
Heler, R., Wright, A. V., Vucelja, M., Bikard, D., Doudna, J. A., & Marraffini, L. A. (2017). Mutations in Cas9 enhance the rate of acquisition of viral spacer sequences during the CRISPR-Cas immune response. Molecular Cell, 65(1), 168–175. https://doi.org/10.1016/j.molcel.2016.11.031
Ikeda, A., Fujii, W., Sugiura, K., & Naito, K. (2019). High-fidelity endonuclease variant HypaCas9 facilitates accurate allele-specific gene modification in mouse zygotes. Communications Biology, 2(1), 371. https://doi.org/10.1038/s42003-019-0627-8
Kleinstiver, B. P., Pattanayak, V., Prew, M. S., Tsai, S. Q., Nguyen, N. T., Zheng, Z., & Joung, J. K. (2016). High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 529(7587), 490–495. https://doi.org/10.1038/nature16526
Okafor, I. C., Singh, D., Wang, Y., Jung, M., Wang, H., Mallon, J., et al. (2019). Single molecule analysis of effects of non-canonical guide RNAs and specificity-enhancing mutations on Cas9-induced DNA unwinding. Nucleic Acids Research. https://doi.org/10.1093/nar/gkz1058
Casini, A., Olivieri, M., Petris, G., Montagna, C., Reginato, G., Maule, G., et al. (2018). A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nature Biotechnology, 36(3), 265–271. https://doi.org/10.1038/nbt.4066
Chatterjee, P., Jakimo, N., Lee, J., Amrani, N., Rodríguez, T., Koseki, S. R. T., et al. (2020). An engineered ScCas9 with broad PAM range and high specificity and activity. Nature Biotechnology, 38(10), 1154–1158. https://doi.org/10.1038/s41587-020-0517-0
Hou, Z., Zhang, Y., Propson, N. E., Howden, S. E., Chu, L.-F., Sontheimer, E. J., & Thomson, J. A. (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proceedings of the National Academy of Sciences, 110(39), 15644–15649. https://doi.org/10.1073/pnas.1313587110
Tan, Y., Chu, A. H. Y., Bao, S., Hoang, D. A., Kebede, F. T., Xiong, W., et al. (2019). Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proceedings of the National Academy of Sciences, 116(42), 20969–20976. https://doi.org/10.1073/pnas.1906843116
Kim, E., Koo, T., Park, S. W., Kim, D., Kim, K., Cho, H.-Y., et al. (2017). In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nature Communications, 8(1), 14500. https://doi.org/10.1038/ncomms14500
Liu, J.-J., Orlova, N., Oakes, B. L., Ma, E., Spinner, H. B., Baney, K. L. M., et al. (2019). CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature, 566(7743), 218–223. https://doi.org/10.1038/s41586-019-0908-x
Acharya, S., Mishra, A., Paul, D., Ansari, A. H., Azhar, M., Kumar, M., et al. (2019). Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proceedings of the National Academy of Sciences, 116(42), 20959–20968. https://doi.org/10.1073/pnas.1818461116
Nelles, D. A., Fang, M. Y., O’Connell, M. R., Xu, J. L., Markmiller, S. J., Doudna, J. A., & Yeo, G. W. (2016). Programmable RNA tracking in live cells with CRISPR/Cas9. Cell, 165(2), 488–496. https://doi.org/10.1016/j.cell.2016.02.054
Konermann, S., Lotfy, P., Brideau, N. J., Oki, J., Shokhirev, M. N., & Hsu, P. D. (2018). Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell, 173(3), 665–676. https://doi.org/10.1016/j.cell.2018.02.033
Cox, D. B. T., Gootenberg, J. S., Abudayyeh, O. O., Franklin, B., Kellner, M. J., Joung, J., & Zhang, F. (2017). RNA editing with CRISPR-Cas13. Science, 358(6366), 1019–1027. https://doi.org/10.1126/science.aaq0180
Huynh, N., Depner, N., Larson, R., & King-Jones, K. (2020). A versatile toolkit for CRISPR-Cas13-based RNA manipulation in Drosophila. Genome Biology, 21(1), 279. https://doi.org/10.1186/s13059-020-02193-y
Terns, M. P. (2018). CRISPR-based technologies: Impact of RNA-targeting systems. Molecular Cell, 72(3), 404–412. https://doi.org/10.1016/j.molcel.2018.09.018
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
BN, KMS, and SA contributed in literature search and writing the article; SA, KMS, and AT contributed towards drafting of the manuscript, tables, and figures, while MP, KM, and AS analyzed the data, performed the editing, proofreading, and approved the manuscript for communication.
Corresponding author
Ethics declarations
Conflict of interest
All authors have declared no conflict of interest.
Ethical Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bharathkumar, N., Sunil, A., Meera, P. et al. CRISPR/Cas-Based Modifications for Therapeutic Applications: A Review. Mol Biotechnol 64, 355–372 (2022). https://doi.org/10.1007/s12033-021-00422-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-021-00422-8