
Abstract
SARS-CoV-2 pandemic has recently made the entire world come to a standstill. The number of cases in the world, especially India, have been increasing exponentially. The need of the hour is to assimilate as much data as possible to fast track the pipeline of bringing in new therapeutic tools against this fatal virus. In this brief communication, we aim to throw light on the various variants of the proteins involved heavily in the pathophysiology of COVID-19, namely Spike protein, ACE2, GRP78, TMPRSS2 and NSP-12. We also portray the molecular docking studies of these proteins with specific drugs that are currently being associated with the same. In our brief study, we come across a few key findings. First of all the combinations of the variants of spike protein and ACE2 binding show overall 25% unfavourable ΔΔG. Second, NSP12 is the most mutation prone among all the NSPs of the SARS-CoV-2 genome and the most common mutations are P323L and A97V. Third, we discovered the variants found in the Indian subpopulation that have greater binding with the currently investigated drugs.
Similar content being viewed by others

Introduction
Since the SARS-CoV-2 that was first reported in Wuhan, China on the 24 December 2019, the world has been at a standstill (Zhu et al. 2020). A mere three months after the first reported case, the WHO declared this disease by the novel virus COVID-19, as a worldwide pandemic. As reported on 16 June, there are as many as 7.9 million people affected worldwide spanning over 215 countries with a jump of over hundred and eighteen thousand cases every day. Even more gnarly is the death count 434,796 and increasing (World Health Organization 2020). Coronavirus disease (COVID-2019) situation reports 2020 (available at https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200616-covid-19-sitrep-148-draft.pdf).
There are quite a few coronaviruses that infect human hosts but with different degrees of infectivity and lethality. Coronaviruses like the NL63, OC43 and HKU1 only cause mild respiratory infections while the Middle Eastern respiratory syndrome (MERS-CoV), severe acute respiratory syndrome (SARS-CoV) and the novel SARS-CoV-2 cause severe respiratory syndromes in the human host. The lethality of SARS-CoV-2 is markedly lower than SARS-CoV and MERS-CoV (0.9–3.3%, 11% and 34%, respectively), but the infectivity of COVID-19 outperforms SARS-CoV and MERS-CoV as the doubling rate of COVID-19 is significantly higher than these two viruses (Sun et al. 2020).
In the present paradigm, the pandemic has disrupted many lives around the globe and brought the world to a standstill. The emphasis in the scientific community is on the various forms of analysis of the virus to aid the therapeutic interventions and vaccine development.
COVID-19 is an enveloped virus having four different structural proteins, N (nucleocapsid), M (membrane), E (envelope) and S (spike) (Yang et al. 2020). The S protein mediates receptor binding and virus entry along with the ACE2 receptor on the host cell surface (Wang et al. 2020a,b). This process is aided by the chaperon proteins GRP78 or HSPA5 and a serine protease TMPRSS2 which primes the spike protein changing its configuration from closed to open (before binding to ACE2) (Hoffmann et al. 2020a, b; Ibrahim et al. 2020).
Thus, here, we have focussed on the S protein and studied the recently sequenced isolates from India to identify the similarities and differences in the protein sequence with respect to Wuhan isolates. We have also analysed the key protein (GRP78, TMPRSS2 and ACE2) allelic variants present in India as assessed their binding affinity in silico through molecular docking studies with current SARS-CoV-2 therapeutic regimens that are employed.
Materials and methods
SARS-CoV-2 sequences are being deposited every single day from all over the world. We analysed all SARS-CoV-2 Indian sequences (197) excluding low coverage (>5% N in the 29.9 kb of each sequence) and with patient status as on 9 June 2020 from the GISAID database (Shu and McCauley 2017). To analyse the sequences, we used the CoV Surfer for all sequences against the Wuhan strain. The results were filtered for duplicates and only high coverage results were retained. The Spike protein variants were extracted from these results and the count of the mutations found were compiled.
Variant analysis
The list of variants for ACE2, GRP78 and TMPRSS2 present in the Indian population were mined from the Index Db (J. Comput. Biol. 2019, pp. 225–234) database along with their allelic frequency. The frequency of the Spike protein variants was calculated by the ratio of the number of patients with occurrence of a particular mutation to the total number of patient sequences obtained from GISAID. These variants were modelled using Swiss Model (Bienert et al. 2017) homology modelling.
The best model was obtained using the QMEAN4 score and Ramachandran plot. Further, the model quality assessment was conducted by the evaluation of the backbone conformation from the Ramachandran plot by dividing the percentage of amino acid residues of the model in the allowed and disallowed regions. The protein sequences were obtained from UniProt (The UniProt Consortium 2019). The variants were then analysed using iMUTANT (Capriotti et al. 2005), which is a SVM predictor that predicts the protein stability changes upon single-site mutations starting from protein sequence to get the stability of the variants (ΔΔG) at 27°C and 37°C.
Docking analysis
Virtual screening of the list of chosen small molecule inhibitors and variants was carried out using PyRx. AutoDock 4.2 (Morris et al. 2009) was used to analyse the top hits obtained and their respective interactions between the ligands and macromolecules. For comparison, we also conducted the docking analysis with controls and the drugs listed above.
Proteins chosen for analysis
Spike (S) protein and angiotensin converting enzyme 2 (ACE2)
The distinct morphology of the coronavirus is created by the S protein which is a homo-trimer that binds to angiotensin converting enzyme 2 (ACE2) (Kalathiya et al. 2020). The S protein consists of two subunits: S1 and S2, the former consisting of the receptor binding domain and the latter contributing fusion machinery (Bosch et al. 2003). The tight binding of SARS-CoV-2 with ACE2 is one of the likely explanations for the high transmission of the virus (Verdecchia et al. 2020). Needless to say, the analysis of the mutations of the S protein and identification of the conserved regions would go a long way in the creation of an effective drug or vaccine.
Glucose regulating protein 78
The glucose regulating protein 78 or binding immunoglobulin protein (BiP) regulates the unfolded protein response in the endoplasmic reticulum (ER) especially when the cell is under stress (Wang et al. 2009). ER stress activates multiple cellular pathways that exert control at both a transcription as well as translational levels (Hebert-Schuster et al. 2018). The three major pathways are mediated by PERK kinase, IRE1, and ATF6. Pathogens make use of the over expression of GRP78 to gain entry into a cell (Kadowaki et al. 2013).
Several docking studies have been conducted to analyse the mechanism of entry of SARS-CoV-2 using GRP78 binding domain. They report that there are interactions between the substrate binding domain of GRP78 and the receptor binding domain of SARS-CoV-2, especially in regions III (C391-C525) and IV (C480-C488) (Ibrahim et al. 2020). This interaction can now be leveraged to design therapeutics specific to SARS-CoV-2.
Transmembrane serine protease 2 (TMPRSS2)
Plays a central role in viral activation and cell entry in not just SARS-CoV-2 but almost all coronavirus and influenza viruses (Glowacka et al. 2011). This membrane bound molecule has serine proteases that cleave the S protein. Once cleaved, the fusion peptide is exposed for viral entry. TMPRSS2 essentially cleaves the spike protein into two subunits S1 and S2 which allows effective penetration of the virus into the cell (Belouzard et al. 2012). Hence, there has been extensive study on serine protease inhibitors that may be used to prevent cell entry of the virus.
Nonstructural protein-12 (NSP12) or RNA dependent RNA polymerase
NSP12 is a protein encoded in the viral genome (Velthuis et al. 2010). After the infection by SARS-CoV-2, the coronavirus assembles an RNA-synthesis complex with the viral nonstructural proteins (NSP) that takes responsibility of replicating and transcribing the entirety of the viral genome. This NSP12 is the main structure, which is enhanced and supplemented by complexes of nsp7–nsp8, nsp10–nsp14 and nsp10–nsp16 provides high fidelity and processivity in viral replication (Kirchdoerfer et al. 2019).
Drugs selected for analysis
Brivudine is an antiviral drug that has been also investigated in cancer studies, as it is an effective inhibitor of HSPs. It is hence being investigated as a way to decrease the levels of GRP78 (or HSPA5) (Heinrich et al. 2011). Camostat mesilate (CM) had been developed in Japan as a protease inhibitor but has been used for various uses ever since. In various studies, it has been proven effective against SARS-CoV, MERS-CoV as well as in SARS-CoV-2. It is known to be a direct inhibitor of the protease TMPRSS2, an essential protein required by SARS-CoV (Uno 2020). Nafamostat mesylate (NM) is also a protease inhibitor that targets and inhibits the lucrative drug target TMPRSS2. Further, in one comparative study between CM and NM, the latter was found to be roughly 15 folds more potent than CM. The study also determined that both drugs did not interfere with cell viability and mortality (Hoffmann et al. 2020a, b).
Originally, perciclovir, a drug for herpesvirus has been found to be very effective as a synthetic nucleoside substitute. In its active form, perciclovir binds to the viral RNA polymerase with very high affinity and impairs viral replication. Anidulafungin is a synthetic antifungal drug. It disrupts fungal cell wall synthesis by inhibiting the enzyme complex 1,3-Δ-D-glucan synthase which is involved in the synthesis of 1,3-Δ-D-glucan, a component of the fungal cell wall. This leads to cell death and inhibition of fungal growth. Suramin is a synthetic sulphated naphthylamine with multiple uses. It was first found to be a treatment for African trypanosomiasis (African sleeping sickness). It acts by inhibiting the enzymes of energy metabolism of the parasite involved in the disease. Reportedly, it can inhibit many other enzymes and proteins like RNA polymerase, reverse transcriptase. It has also been used in the treatment of cancer as well as an antiviral agent against HIV (Dey et al. 2020).
Variants selected for analysis
The variants chosen for analysis are exonic and nonsynonymous variants. For ACE2 (table 1), Var 3 ('p.E668K') showed an overall maximum destabilizing effect and highest allelic frequency followed by Var 1 (p.K26R). Interestingly Var 2 ([p.N546S]) showed an overall stabilizing effect although with lower allelic frequency. In the case of GRP78 of all the variants seen, only Var 2 (p.Val432Ile.) has a mildly stabilizing effect with Var 1 (c.1276A>G, p.Ile426Val.), Var 3 (p.Pro459Ser.) and Var 4 (p.Lys601Met, p.Lys601Thr.) having a destabilizing effect. In the case of TMPRSS2 as well all the exonic, nonsynonymous variants show significant destabilizing effect with Var 3 (p.Thr481Met, p.Thr518Met) being the highest followed by Var 1 (p.Val120Met, p.Val160Met) with the exception of Var 2 (p.Gly8Asp, p.Gly8Val) that has a mild stabilizing effect. Since the number of variants of TMPRSS2 are low, it becomes a lucrative target due to its conserved nature.
Results and discussion
Spike mutations found in India
When the 197 high coverage sequences with patient status were analysed, there were 24 mutations. Of these, the most significant were the D614G, E583D, L54F, Q613 and T572I mutations in the spike protein (Korber et al. 2020) (table 2). Previous studies have shown that the D614G mutation may confer a competitive advantage at furin binding sites (Tang et al. 2020). Mutations are generally seen to occur either coupled with other mutations or stand alone. The L54F mutation is seen to occur coupled with the D614G mutation. This seems to hint at a stability conferred by the coupling of the L54F and D614G mutations. The L54F mutation lies in the N terminal domain of the S protein, T572S in the SD1 domain, and E583 and Q613 in the SD2 domain (Fischer et al. 2020).
Three substantial mutations of NSP12 found in India
When the 197 high coverage sequences with patient status were analysed, there were 14 mutations. Of these, the most significant were the A97V, P323L and V880I mutations in NSP12 (table 3). These mutations have not been studied as extensively as the spike protein and nothing substantial could be found about these mutations. The variants themselves showed rather interesting disparities in their docking studies.
Docking analysis of spike variants and ACE2 variants show that some interactions between spike protein variants and ACE2 variants are more stable than others
The RBD domain of the spike protein is known to bind with the ACE2. Structural variation of the spike protein may cause changes in the binding of the spike protein to ACE2. Table 4 summaries the effect of binding of selected Spike and ACE2 variants. The most significant stable interaction is that of Spike variant 1 (D614G) with ACE2 variant 1 (rs4646116) this is closely followed by Spike variant 1 (D614G) with ACE2 variant 2 (rs756905974). This once again shows the exceptional stability of the D614G mutation.
Docking analysis of chosen drugs and variants of the proteins selected for the study
The selected variants of GRP78 were screened against Brivudine using PyRx. The results were used to identify the variant that showed the best binding to Brivudine. This was followed by 100 runs on Autodock to achieve the clustering histogram for statistically significant binding energy and interacting residues (table 5).
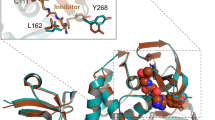
The control showed a binding affinity of around –6.5 to –6 kcal/mol. There was a difference in binding affinity with structural variation of GRP78. Of the three variants, Var 1 showed the best binding affinity with Brividuine with a binding affinity of –6.5 kcal/mol. Var 2 and Var 3 showed decreased affinity with Brivudine. The bond lengths varied from 2.1Å to 3.4Å for both control as well as Var 1. The detailed interactions are shown in (figure 1a (control), 1b (Var 1)). Var 1 showed higher binding affinity with no significant change in interactions. Both Camostat and Nafamostat mesylate are potent seriene inhibitors in the case of TMPRSS2. Nafamostat meslyate showed much better binding when compared to Camostat to the control as is seen in the clustering histograms (table 5). When screened against all the variants, Nafamostat outperformed Camostat with the best binding being shown by Var 1. The detailed interactions are shown in (figure 1c (control), 1d (Var 1)).

Interactions between the chosen drugs and variants. (a) GRP control with brivudine, (b) GRP Var 1 with Brivudine. (c) TMPRSS2 control with NM, (d) TMPRSS2 Var 1 with NM, (e) NSP 12 control with Anidulafungin, (f) NSP12 Var 1 with Anidulafungin. The interactions are shown with yellow dotted lines. More interactions correspond to greater binding.
Anidulafungin was the most effective drug in the context of NSP12 binding energy with especially a high binding energy at –13.43 kcal/mol. This binding energy was seen primarily in the first variant of the three usually found in Indian populations with a frequency of around 0.3, which is the highest observed. Since anidulafungin is usually administered as an antifungal, it is interesting to note that it is effective in binding to NSP12. The control however has relatively poorer binding of around –4.02 kcal/mol. Further, we see lesser bonds in the docked model of the control than that of the variant 1 (figure 1e (control), 1f (Var 1)).
Conclusion
With the SARS-CoV-2 outbreak on the rise, particularly in the Indian subcontinent, it becomes more imperative that we understand the various factors that could contribute towards finding a potential treatment strategy for the pandemic. In this brief study, we focussed on the variants that are present in the Indian population and their effect in in silico binding to experimental SARS-CoV-2 therapeutic options. Of all the possible combinations for the variants of spike protein and ACE2 binding, 25% show overall unfavourable ΔΔG. We see that NSP12 is the most mutation prone among all the NSPs of the SARS-CoV-2 genome and the most common mutations are P323L and A97V. With respect to the Spike protein, the most frequent mutation seen is the D614G followed by T572I. We also see that particular Indian variants (Var 1 of TMPRSS2 and Nafamostat Mesylate and Var 1 of NSP12 and Anidulafungin) show higher binding affinity to small molecule inhibitors as compared to their control counterparts. There is more research required into repurposing of small molecule inhibitors and their efficacy in the context of the Indian population. This could hopefully pave way for more robust and personalized treatment strategies.
References
Belouzard S., Millet J. K., Licitra B. N. and Whittaker G. R. 2012 Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4, 1011–1033.
Bienert S., Waterhouse A., de Beer T. A. P., Tauriello G., Studer G., Bordoli L. and Schwede T. 2017 The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res. 45, D313–D319.
Bosch B. J., van der Zee R., de Haan C. A. and Rottier P. J. 2003 The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77, 8801–8811.
Capriotti E., Fariselli P. and Casadio R. 2005 I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310.
Dey S. K., Rahman M. M., Siddiqi U. R. and Howlader A. 2020 Analyzing the epidemiological outbreak of COVID-19: a visual exploratory data analysis approach. J. Med. Virol. 92, 632–638.
Fischer S. C., Corujo-Simon E., Lilao-Garzon J., Stelzer E. H. K. and Muñoz-Descalzo S. 2020 The transition from local to global patterns governs the differentiation of mouse blastocysts. PLoS One 15, e0233030.
Glowacka I., Bertram S., Müller M. A., Allen P., Soilleux E., Pfefferle S. et al. 2011 Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 85, 4122–4134.
Hebert-Schuster M., Rotta B. E., Kirkpatrick B., Guibourdenche J. and Cohen M. 2018 The interplay between glucose-regulated protein 78 (GRP78) and steroids in the reproductive system. Int. J. Mol. Sci. 19, 1842.
Heinrich J., Tuukkanen A., Schroeder M., Fahrig T. and Fahrig R. 2011 RP101 (brivudine) binds to heat shock protein HSP27 (HSPB1) and enhances survival in animals and pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 137, 1349–1361.
Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S. et al. 2020a SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.
Hoffmann M., Schroeder S., Kleine-Weber H., Müller M. A., Drosten C. and Pöhlmann S. 2020b Nafamostat Mesylate blocks activation of SARS-CoV-2: New treatment option for COVID-19. Antimicrob. Agents Chemother. 64, 6.
Ibrahim I. M., Abdelmalek D. H., Elshahat M. E. and Elfiky A. A. 2020 COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 80, 554–562.
Kadowaki H. and Nishitoh H. 2013 Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (basel). 4, 306–333.
Kalathiya U., Padariya M., Mayordomo M., Lisowska M., Nicholson J., Singh A. et al. 2020 Highly conserved homotrimer cavity formed by the SARS-CoV-2 spike glycoprotein: A novel binding site. J. Clin. Med. 9, 1473.
Kirchdoerfer R. N. and Ward A. B. 2019 Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 10, 1–9.
Korber B., Fischer W. M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Foley B. et al. 2020 Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. Sheffield COVID-19 Genomics Group. bioRxiv. 069054.
Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S. and Olson A. J. 2009 Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 16, 2785–2791.
Shu Y. and McCauley J. 2017 GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 22, 30494.
Sun J., He W. T., Wang L., Lai A., Xiang J., Zhai X. et al. 2020 COVID-19: epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol. Med. 26, 483–495.
Tang L., Schulkins A., Chen C., Deshayes K. and Kenney J. S. 2020 The SARS-CoV-2 spike protein D614G mutation shows increasing dominance and may confer a structural advantage to the Furin cleavage domain. Preprints 2020050407.
Te Velthuis A. J., Arnold J. J., Cameron C. E., van den Worm S. H. and Snijder E. J. 2010 The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 38, 203–214.
The UniProt Consortium 2019 UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515.
Uno Y. 2020 Camostat mesilate therapy for COVID-19. Intern. Emerg. Med. 15, 1577–1578.
Verdecchia P., Cavallini C., Spanevello A. and Angeli F. 2020 The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 76, 14–20.
Wang M., Wey S., Zhang Y., Ye R. and Lee A. S. 2009 Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 11, 2307–2316.
Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G. et al. 2020a Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904.
Wang W., Xu Y., Gao R., Lu R., Han K., Wu G. and Tan W. 2020b Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 323, 1843–1844.
World Health Organization 2020 Coronavirus diseases (COVID-19) situation report-148 (https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200616-covid-19-sitrep-148-draft.pdf).
Yang P. and Wang X. 2020 COVID-19: a new challenge for human beings. Cell. Mol. Immunol. 17, 555–557.
Zhu N., Zhang D., Wang W., Li X., Yang B., Song J. et al. 2020 China novel coronavirus investigating and research team. A novel coronavirus from patients with Pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733.
Acknowledgements
The authors thank Dr Rakesh Tuli, Ex-Director of NBRI, Lucknow and Executive Director of NABI, Mohali for his scientific discussions and encouragement. The facilities provided by BITS Pilani are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: Upendra Nongthomba
Rights and permissions
About this article

Cite this article
Sridhar, S., Saha, G., Lata, S. et al. Molecular docking studies of Indian variants of pathophysiological proteins of SARS-CoV-2 with selected drug candidates. J Genet 100, 64 (2021). https://doi.org/10.1007/s12041-021-01313-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12041-021-01313-2