
Abstract
Drug repurposing offers advantages over traditional drug development in terms of cost, speed and improved patient outcomes. The receptor activator of nuclear factor kappa B (RANK) ligand (RANKL) inhibitor denosumab is approved for the prevention of skeletal-related events in patients with advanced malignancies involving bone, including solid tumours and multiple myeloma. Following improved understanding of the role of RANK/RANKL in cancer biology, denosumab has already been repurposed as a treatment for giant cell tumour of bone. Here, we review the role of RANK/RANKL in tumourigenesis, including effects on tumour initiation, progression and metastasis and consider the impact of RANK/RANKL on tumour immunology and immune evasion. Finally, we look briefly at ongoing trials and future opportunities for therapeutic synergy when combining denosumab with anti-cancer agents such as immune checkpoint inhibitors.
Similar content being viewed by others

Drug repurposing as a therapeutic strategy in cancer
Drug discovery and development requires many years of research and financial investment [1]. Repurposing an approved therapeutic agent is generally faster and more cost effective than novel drug development because the largest and most inefficient part of the development process has been bypassed [1]. Repurposing may take 3–12 years, compared with up to 17 years to develop a new drug, and repurposed drugs have the benefit of established safety profiles, which contribute to lower implementation failure rates than seen with novel agents [1]. Drugs with a history of safe use and a favourable toxicology profile may be candidates for repurposing if they have both a plausible mechanism of action and evidence of efficacy in the new indication [1]. Drug repurposing is one of the oldest development pathways for anti-cancer medicines; for example, anthracyclines, which are used extensively in the management of breast cancer and lymphoma, were tested as antibiotics but proved to be too toxic [2]. Thalidomide is another example of a drug successfully repurposed as a cancer treatment. Used to alleviate morning sickness in pregnant women, thalidomide was withdrawn after causing severe teratogenicity, but later demonstrated response rates of 25–35% in relapsed/refractory multiple myeloma and was subsequently approved for treatment of this cancer [3]. This finding prompted development of thalidomide analogues, such as lenalidomide and pomalidomide, which are now typically preferred in clinical practice over the original agent [3].
Recently, there has been renewed interest in repurposing in the oncology field: drugs approved in other indications have been shown to induce cancer cell death, suppress aspects of cancer cell behaviour, or even prevent cancer development. Examples of repurposed drugs that have shown anti-cancer activity in at least one clinical trial include cimetidine, clarithromycin, verapamil and pravastatin [3]. Aspirin is also a potential candidate for repurposing as an anti-cancer agent. There is some evidence, from clinical trials investigating prevention of cardiovascular disease and from observational studies, that it may reduce cancer incidence or death [4]. Similar findings have emerged from interventional studies evaluating the effect of aspirin on mortality and disease recurrence in cancer patients [4]. Potential antineoplastic properties of the oral antidiabetic agent metformin are also being investigated following observations that it reduced cancer incidence by 37% in patients with diabetes [5].
A further avenue open to exploration is repurposing drugs that target the receptor activator of nuclear factor kappa-B (RANK) ligand (RANKL) signalling system [6], which has an established role in regulating bone remodelling [7, 8]. The RANKL inhibitor denosumab was first approved in 2010 and is now indicated for the prevention of skeletal-related events (SREs) in adults with advanced malignancies involving bone [9]. The denosumab label has recently been expanded to include the prevention of SREs in patients with multiple myeloma [9, 10]. Observations that some stromal cells in giant cell tumour of bone (GCTB) express RANKL, possibly leading to the recruitment of RANK-expressing osteoclast-like giant cells, led to increased understanding of the role of RANK–RANKL signalling in the aetiology of GCTB and then to investigations of the therapeutic potential of denosumab in patients with GCTB [11, 12]. Positive clinical trial results led to the approval of denosumab in 2013 for use in adults and skeletally mature adolescents with GCTB that is unresectable or where surgical resection may result in severe morbidity [13]. Accumulating data on the RANK–RANKL pathway raise the possibility that denosumab could be repurposed for use in other types of cancer. Evidence suggests that RANK–RANKL signalling may have osteoclast-independent effects on tumour biology, with RANK/RANKL activity demonstrating a positive correlation with tumour progression and advanced disease [14]. Moreover, trials including denosumab have demonstrated positive effects on survival outcomes in patients with cancer, including increased overall survival (OS) in lung cancer and increased disease-free survival in non-metastatic breast cancer [15, 16]. Recent data also suggest that denosumab and immune checkpoint inhibitors may act synergistically to provide additional benefit [17]. This review explores the RANK–RANKL axis in cancer biology and the potential for repurposing denosumab.
The RANK–RANKL axis: more than bone turnover
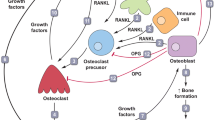
RANKL, its transmembrane signalling receptor RANK, and the decoy receptor osteoprotegerin (OPG) belong to the tumour necrosis factor superfamily [7]. Competition between OPG and RANK to bind RANKL regulates RANK activation and, therefore, its signalling function [7]. Binding of RANKL to RANK initiates various signal-transduction pathways, including production of nuclear factor kappa B [7], the eponymous member of a transcription factor family implicated in cancer development and progression, in inflammation and in crosstalk between these two processes [18, 19]. While the RANK/RANKL/OPG system regulates osteoclast function and bone remodelling in bone homeostasis [7, 8], it also has roles in immunity and development (Fig. 1a–c). Specifically, RANK–RANKL signalling sits at the interface between bone and the immune system, influencing the pathology of diseases such as rheumatoid arthritis [20] and immune functions unrelated to bone including involvement in dendritic cell survival and function, M1 macrophage activation and T-cell differentiation and activation [14, 21, 22]. Additionally, RANK–RANKL signalling functions in epithelial growth and differentiation, including mammary physiology [7, 23], in lymph node development [21], in skeletal muscle [24] and in the central nervous system (CNS) [8]. For example, RANK–RANKL signalling has been implicated in the formation and function of hair follicles, intestinal microfold cells, thymic medullary epithelium and mammary glands [7, 25]. RANK protein is also expressed in bronchial (but not alveolar) epithelial cells [23]. Of note, in mammary gland differentiation and the development of the lobuloalveolar breast tissue architecture required for lactation [26], RANK–RANKL signalling promotes the proliferation and survival of mammary epithelial cells [26] and constitutive signalling may promote the propagation of RANK-expressing stem cells at the expense of normal differentiation [27]. RANKL expression is driven by progesterone-responsive cells in the breast and is required for progesterone-driven epithelial proliferation [28, 29]. In skeletal muscle, the RANK–RANKL axis has been implicated in the regulation of calcium storage, influencing denervated muscle function [24]. Furthermore, in the CNS, the RANK–RANKL axis plays a role in thermoregulation [8]. RANK, RANKL and OPG are, therefore, expressed across a variety of tissue types (Fig. 2a–c). Notably, RANK and OPG have relatively broad expression in normal tissues, while RANKL is largely restricted to osteoblasts and haematopoietic lineages (Fig. 2a), where it is expressed in T lymphocytes and basophils [30,31,32].

Involvement of the RANK–RANKL axis in bone remodelling, development, immunity and cancer. a In the mammary gland, following the binding of progesterone to its receptor, RANKL is produced. Increased RANKL stimulates mammary gland epithelial cell expansion via paracrine signalling, leading to lactation competence. Image modified from [96] under the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc-sa/3.0/). b Activated T cells and NK cells express RANKL, which stimulates various processes via monocytes, macrophages and dendritic cells including dendritic cell survival and maturation, T-cell activation and NK cell inhibition. c Increased levels of RANKL have been associated with increased tumour growth, stemness, EMT and metastases. Throughout these processes, RANKL acts as a chemoattractant for RANK-expressing cancer. d Increased RANKL produced by bone stromal cells, including cells of the osteoblast lineage, leads to an increased rate of bone remodelling [96]. EMT epithelial–mesenchymal transition, NK natural killer, PR progesterone, RANK receptor activator of nuclear factor kappa B, RANKL receptor activator of nuclear factor kappa B ligand

a RANK, RANKL and OPG tissue expression. Data from the Human Protein Atlas available at www.proteinatlas.org [97]. RANK expression is relatively ubiquitous, while RANKL expression is effectively restricted to haematopoietic cells. OPG expression is highest in mucosal epithelial tissues, consistent with its role in modulating immune responses. b RANKL positive immune cells within a lymph node. Image reproduced from the Human Protein Atlas [97] under the Creative Commons Attribution License (https://creativecommons.org/licenses/by-sa/3.0/); available at https://www.proteinatlas.org/ENSG00000120659-TNFSF11/tissue/lymph+node#img. c RANKL expression in normal breast tissue. Image reproduced from [98] under the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). OPG osteoprotegerin, RANK receptor activator of nuclear factor kappa B, RANKL receptor activator of nuclear factor kappa B ligand, TPM, transcripts per million
The RANK–RANKL axis in cancer biology
Given its varied roles in human physiology, interest has increased in the role of the RANK–RANKL axis in tumourigenesis, specifically in its functions beyond bone metabolism. Cells from various tumour types express RANK, RANKL and OPG (Fig. 3a–c) and RANK–RANKL signalling appears to be involved in tumour initiation, progression and metastasis (Fig. 1d) [6, 7, 25, 33]. With regard to bone metastasis, the effects of RANK/RANKL seem likely to be indirect, mediated by an osteoclast-dependent mechanism, or direct, via interaction of RANKL with RANK-expressing cells [6, 7, 25, 33]. However, there is evidence for involvement of the RANK/RANKL/OPG system in the development and metastasis of melanoma, breast, hepatocellular, lung, prostate and renal cell carcinomas, as well as in primary malignant tumours of the bone (osteosarcoma, multiple myeloma and GCTB) [6, 7, 25, 33]. The relative levels of RANK/RANKL/OPG expression may influence prognosis in numerous cancer types including breast, lung, endometrial, renal cell and gastric cancers, osteosarcoma and multiple myeloma (Fig. 4a, b) [34,35,36,37,38,39,40,41]. In breast cancer, microarray analysis demonstrated that low RANK and high OPG expression were associated with longer OS (p = 0.0078 and p = 0.034, respectively) and disease-free survival (p = 0.059 and p = 0.040, respectively) [34]. Immunohistochemical analysis demonstrated an association between high RANK expression and bone metastasis (p = 0.023) and shorter survival without bone involvement (p = 0.037) [34]. In another study, low RANKL expression has been associated with increased risk for bone metastases (p = 0.018) and shorter disease-free survival (p = 0.018) in breast cancer [40]. In metastatic clear cell renal cell carcinoma, an elevated RANK/OPG ratio has been associated with a shorter median time to metastasis (p = 0.014), progression-free survival (PFS) (p = 0.001) and OS (p = 0.0001) [35]. Moreover, RANK expression in clear cell renal cell carcinoma has been identified as an independent negative prognostic factor for both cancer-specific survival and recurrence-free survival (p < 0.001) [42]. Additionally, high RANK expression has been linked to a shorter PFS (p = 0.01) and OS (p = 0.02) in endometrial cancer [36]. RANK-positive osteosarcomas are also associated with higher mortality than RANK-negative cases (p < 0.05) [41]. A high RANKL expression has been associated with higher stage disease (p = 0.035) and a shorter OS (p = 0.008) in gastric cancer [37]. In lung adenocarcinoma, both high RANK (p = 0.01) and RANKL (p = 0.02) expression have been linked to worse OS [38]. Furthermore, in multiple myeloma, the serum RANKL/OPG ratio was found to correlate with the extent of bone disease (p = 0.023), and a high RANKL/OPG ratio was associated with an increased risk of death (p < 0.001) [39]. Such correlation data should be interpreted with caution as the methods used to evaluate expression levels vary across studies, can include non-validated assays and require expertise to assess the results; therefore, further research is required to confirm these findings.

a Expression of RANK, RANKL and OPG in cells from various solid organ and haematological cancers. The results shown here are based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov/) obtained using the cBioPortal for Cancer Genomic [95]. b RANKL expression in lung adenocarcinoma. c RANKL expression in pancreatic adenocarcinoma. Reproduced from the Human Protein Atlas [97] under the Creative Commons Attribution License (https://creativecommons.org/licenses/by-sa/3.0/); image in b is available at https://www.proteinatlas.org/ENSG00000120659-TNFSF11/pathology/tissue/lung+cancer#img and image in c is available at https://www.proteinatlas.org/ENSG00000120659-TNFSF11/pathology/tissue/pancreatic+cancer#img. ACC adenoid cystic carcinoma, AML acute myeloid leukaemia, ccRCC clear cell renal cell carcinoma, chRCC chromophobe renal cell carcinoma, CS carcinosarcoma, DLBCL diffuse large B-cell lymphoma, GBM glioblastoma multiforme, OPG osteoprotegerin, PCPG pheochromocytoma and paraganglioma, pRCC papillary renal cell carcinoma, RANK receptor activator of nuclear factor kappa B, RANKL receptor activator of nuclear factor kappa B ligand

a Association of RANK, RANKL and OPG expression with survival across different cancer types according to published studies. Each line represents different expression data as labelled; all comparisons are high vs low expression except for the breast cancer results, which are low vs high. b Association of RANK, RANKL and OPG expression (low or high) with 5-year overall survival rates across various solid organ and haematological cancers. OPG expression is significantly (p < 0.001) associated with survival in urothelial, renal and cervical cancers, but extreme gene expression for OPG, RANKL and RANK also show hypothesis-generating associations in other cancers. Log rank p values for Kaplan–Meier plots showing results from analysis of correlation between mRNA expression levels and patient survival from TCGA datasets are plotted. Data extracted from the Human Protein Atlas available at www.proteinatlas.org [97]. aTCGA dataset. bLung-1 dataset. + Indicates a better prognosis with high gene expression. − Indicates a worse prognosis with high gene expression. CI confidence interval, HR hazard ratio, mRNA messenger RNA, NR not reported, OPG osteoprotegerin, RANK receptor activator of nuclear factor kappa B, RANKL receptor activator of nuclear factor kappa B ligand, RCC renal cell carcinoma
Indirect effects of RANK–RANKL signalling on tumour initiation, progression and metastasis: preclinical evidence
RANKL is well known to be involved in a cycle of osteoclast-mediated bone destruction and tumour activity that facilitates bone metastasis [7, 8, 25]. Both tumour cells and tumour-derived factors are involved in the production of RANKL and increase the RANKL/OPG ratio. Increased RANKL in the bone microenvironment upregulates osteoclastogenesis and the activation of mature osteoclasts (bone-resident macrophages), resulting in increased bone resorption. The subsequent release of growth factors and calcium into the bone microenvironment stimulates additional proliferation of tumour cells and release of tumour-derived factors, and thus further increases the RANKL/OPG ratio, thereby promoting continued RANKL-dependent osteoclast-mediated bone destruction [43, 44].
With respect to bone metastasis, preclinical evidence has shown that high RANK expression in breast cancer cells increases their ability to metastasise to bone [45]. RANKL inhibition promoted tumour apoptosis in a mouse model of bone metastasis from prostate cancer, and in combination with docetaxel, was synergistic in improving survival [46]. Moreover, preclinical data have indicated that the combination of anti-RANKL therapies and AT-406 (an antagonist of inhibitor of apoptosis proteins) may have a beneficial effect on bone metastases [47]. RANKL inhibition also suppressed bone invasion in mouse models of oral squamous cell carcinoma [48]. Furthermore, it has been shown that an elevated RANKL/OPG ratio increases the potential of human non-small cell lung cancer (NSCLC) to metastasise to bone [49]. In mouse models of multiple myeloma, RANKL has been shown to promote the release of myeloma cells from dormancy through osteoclast-mediated remodelling of the endosteal niche, thus promoting disease progression or relapse. In line with these observations, bone resorption has been linked to tumour burden in patients with multiple myeloma [50].
In addition to these well-known bone-related pathways, RANK–RANKL signalling may play a role at several other stages of cancer development, progression and metastasis [7, 25, 33]. RANKL may influence carcinogenesis and metastatic progression through effects on RANK-expressing tumour cells or cells from stem/progenitor compartments that express RANK [7, 25, 33]. Effects on tumour initiation may differ between cancer types and there has been particular interest in the role that RANKL may play in breast cancer given its physiological roles in mammary gland epithelia and the stem cell compartment [7, 25, 33]. Preclinical evidence suggests that RANK/RANKL mediate the pro-tumourigenic role of progesterone in the mammary gland [28, 29, 51]. In a mouse mammary tumour virus model, RANK overexpression promoted tumourigenesis in breast tissue after treatment with carcinogen and progesterone, and RANKL inhibition attenuated tumourigenesis in this and a spontaneous breast tumour model [51]. In another mouse model with breast tissue deleted for RANK, tumourigenesis, tumour growth and stem cell expansion driven by medroxyprogesterone acetate were also attenuated [52]. Furthermore, mammary carcinogenesis driven by BRCA1 mutations relies on autocrine or paracrine RANK–RANKL signalling in mammary progenitor cells [29]. Targeting the RANK–RANKL axis could conceivably, therefore, be beneficial in patients with BRCA1 mutation-positive breast cancer and may provide a rationale for designing clinical trials of cancer prevention [29].
Preclinical data have demonstrated that when expressed in lung epithelial cells or other cells of the tumour microenvironment, RANK may also promote lung cancer development. Signalling via RANK promoted expansion of cancer stem-like cells in a mechanism dependent upon mitochondrial respiration [23]. Female sex hormones are also able to promote lung cancer progression through the RANK–RANKL axis [23]. It has been hypothesised that the influence of sex hormones on RANK/RANKL might in part explain gender-specific differences observed in epidemiological studies of human lung cancer [23, 53]. RANKL expression has been associated with decreased survival in lung adenocarcinoma [23, 38], and RANKL inhibition has reduced tumour progression in mouse models of lung adenocarcinoma [38].
The leucine-rich repeat-containing G-protein-coupled receptor 4 (LGR4) has recently been identified as a RANKL receptor, has been shown to have an antagonistic effect on osteoclast differentiation and to promote the proliferation of tumour cells when overexpressed [6]. Additionally, LGR4 expression has been associated with a poor prognosis and metastasis in breast, prostate and colorectal cancer [54,55,56]; however, LGR4 has been associated with improved survival in lung adenocarcinoma [23]. In multiple myeloma, LGR4 has been implicated in promoting Wnt/β-catenin signalling, which drives proliferation [57]. It could be speculated that patients with tumours expressing high levels of both LGR4 and RANKL might benefit from disruption of RANKL signalling.
Many of the mechanisms underlying the pro-metastatic activities of RANKL, including increased proliferation, migration and invasion of RANK-expressing tumour cells, could be relevant to numerous cancer types [7, 14, 25, 33]. RANK overexpression induced transformation and stemness in untransformed mammary epithelial cells, induced markers of epithelial–mesenchymal transition (EMT), known to be important in acquiring these metastatic capabilities [58] and increased invasiveness, migration and anchorage-independent growth [59]. Additionally, RANK overexpression induced tumourigenesis and promoted metastasis of BRCA1-deficient cells in immunodeficient mice [59]. Furthermore, disruption of RANK–RANKL signalling in mouse models of breast cancer reduced metastasis of RANK-expressing breast cancer cells to lung [7, 60] and of RANK-expressing melanoma cells to bone [61]; conversely, exogenous RANKL promoted lung metastases in mice with RANK-expressing breast tumours [60]. In humans, high RANK/RANKL messenger RNA expression has been significantly associated with metastatic breast adenocarcinomas [59], while RANKL induced markers of EMT in prostate cancer [62] and in head and neck squamous carcinoma cells [63]. RANKL has also been implicated both in the induction of angiogenesis and in increasing vascular permeability via RANK-expressing endothelial cells, which may impact intra/extravasation and metastasis [25, 64]. Vascular endothelial growth factor, which is commonly expressed by tumour cells, enhances endothelial cell responses to RANKL [25].
Potential immunomodulatory role of RANK–RANKL signalling
Immune cells may represent a subset of RANKL-expressing cells that mediate direct effects of RANK–RANKL signalling in cancer [7, 14, 25]. Such cells are commonly found in the tumour microenvironment but their precise roles are varied and not fully understood [7, 14]. The complex role of RANK–RANKL signalling is evident from the fact that it has been implicated in both the generation and regulation of immune responses, suggesting that RANK–RANKL signalling can enhance and suppress immunity and may affect inflammation, T-cell activation and immune evasion [7, 14, 22]. Specifically, RANK–RANKL signalling supports dendritic cell survival and function, M1 macrophage activation and T-cell differentiation and activation [14, 22]. It is also involved in central and peripheral tolerance [7, 14, 25] and in the development of lymph nodes [21]. Additionally, RANKL contributes to regulation of early lymphocyte development [14, 21]. With respect to suppression of immune responses, RANK–RANKL signalling is required for development of medullary thymic epithelial cells, which establish T-cell self-tolerance centrally through expression of the autoimmune regulator (Aire) gene [14, 65]. Peripherally, RANK–RANKL signalling mediates tolerance and immunosuppression in tissues such as pancreas, small intestine (in Peyer’s patches) and skin; defective RANK–RANKL signalling is also implicated in the pathogenesis of autoimmune disease in these tissues [7, 14]. RANK–RANKL signalling is required for the formation and function of regulatory T cells (Tregs) [66, 67]. Specifically, RANKL is involved in the generation and activation of potent Tregs in the pancreas that are involved in preventing diabetes [66]. RANKL also mediates the induction of Tregs in studies of the immune environment in skin [67]. Modelling of the immune response in mice with colitis has shown that RANKL is expressed by Tregs and that blockade of RANK–RANKL signalling can impair Treg function and immune suppression [68].
Expression of RANK and RANKL is largely segregated among immune cells [7, 25, 69]. RANKL is primarily expressed in T cells and is upregulated upon T-cell stimulation, enhancing T-cell proliferation and function. In contrast, RANK is expressed in monocytes, macrophages and dendritic cells and is induced in T cells and natural killer (NK) cells by specific cytokines [7, 14, 25, 69]. With respect to the development of immune responses, RANK–RANKL signalling in dendritic cells enhances their survival and function, including their ability to prime and activate T cells, and to enhance T-cell memory [70, 71], by inducing expression of activating cytokines [7, 14, 25, 72]. Evidence suggests that RANK and the co-stimulatory molecule CD40 may act co-operatively in dendritic cell-mediated activation of T-cell responses [14, 71]. Disruption of RANK–RANKL signalling can antagonise dendritic cell function and T-cell–mediated immune responses [73]. RANK–RANKL signalling in macrophages also enhances their survival and function, including antigen presentation [14, 74]. However, the high levels of antigens and immunosuppressive factors typically found in tumour microenvironments can result in T-cell exhaustion, associated with the upregulation of markers, such as immune checkpoint proteins; therefore, although dendritic cells may initially mediate T-cell differentiation and activation, extensive inflammation can induce immune suppression [75].
The importance of RANK–RANKL signalling in tumour immunology, and in stimulating or suppressing anti-tumour immune responses in cancer, is not yet fully understood [7, 14, 25]. Preclinical studies suggest that RANKL can promote the negative selection of tumour-specific T cells by inducing the expression of self-antigens that are shared with tumours in the thymus, thereby promoting central tolerance to tumour antigens and tumour immune evasion [76]. RANKL inhibition can reversibly decrease self-antigen expression in the thymus and can spare from deletion T cells that target melanoma, thus enhancing the anti-tumour immune response and improving survival [76]. Furthermore, synergy has been observed between the suppression of central tolerance in Aire-deficient mice and peripheral immune checkpoint inhibition using anti-cytotoxic T-lymphocyte–associated antigen-4 (CTLA-4) antibodies with respect to slowing tumour growth and improving survival through increasing T cells that target melanoma [77]. Co-treatment with anti-RANKL and anti-CTLA-4 antibodies could not reproduce the synergistic effect of Aire-deficiency and anti-CTLA-4 antibodies on survival, although synergistic improvements in survival with co-treatment could be achieved if mice were also vaccinated with tumour cells producing granulocyte–macrophage colony-stimulating factor [77].
In addition to establishing central tolerance to tumours [7, 14, 76], RANK–RANKL signalling plays an important role within tumours where it may contribute to the development of a tolerogenic immune microenvironment [25, 60, 78, 79]. RANK is highly expressed on tumour-associated macrophages (TAMs), and RANKL is expressed by tumour-infiltrating T cells [7, 25, 69]. Tregs inhibit effector T-cell activity and can, therefore, play a role in tumour immune evasion [80], and as discussed, RANKL plays a crucial role in the expansion of Tregs in a variety of situations. Evidence suggests that RANK–RANKL signalling in M2 TAMs produces chemokines that recruit immunosuppressive Tregs to the tumour microenvironment [7, 25, 79]. Tregs have been shown to produce RANKL, which functions as a chemoattractant for RANK-expressing monocytes [81], suggesting that Tregs may perpetuate tumour progression [7, 14, 25, 60]. Distant metastasis may also be influenced by RANKL expression in tumour-infiltrating T lymphocytes [7, 14, 25]. Specifically, Tregs producing RANKL have been implicated in the development of lung metastases in a RANK-expressing breast cancer mouse model [60]. Although modulation of the tumour immune microenvironment could facilitate tumour progression, RANKL inhibition in mouse models of lung adenocarcinoma that responded to anti-RANKL therapy showed only modest changes in the tumour microenvironment, including non-specific depletion of tumour T cells [38]. In addition to their suggested role in modulating the tumour immune microenvironment, M2 TAMs have also been implicated in angiogenesis and metastasis [25]. NK cells play an important role in tumour immunosurveillance [82]; RANKL can stimulate production of NK cell inhibitory factors by acute myeloid leukaemia cells, which also upregulate RANK expression by NK cells, promoting RANK–RANKL signalling that further contributes to NK cell inhibition [33].
Therapeutic potential of targeting the RANK–RANKL axis in cancer
Denosumab mimics the activity of endogenous OPG in binding to RANKL, although it does not inhibit the pro-apoptotic protein TRAIL [7, 14, 25]. Various trials have recently completed or are in progress, particularly in breast and lung carcinoma, that are providing insight into the repurposing of denosumab to an anti-cancer therapy (Table 1). Anti-RANKL therapy may have the potential to help patients with primary bone tumours other than GCTB, or who have associated lesions, such as aneurysmal bone cysts [83]. Moreover, denosumab is hypothesised to have a number of anti-tumourigenic effects including increasing bone metastasis-free survival (BMFS), reducing proliferation and stemness in early tumourigenesis and augmenting checkpoint immunotherapy (Fig. 5a, b).

Targeting of the RANK–RANKL pathway may improve patient outcomes by a reducing self-renewal of cancer stem cells and b augmenting immunotherapy, tipping the balance in favour of the immune system. c Concomitant immune checkpoint inhibitor and denosumab therapy is associated with a higher response rate than has been reported for immune checkpoint inhibitors alone [17, 99, 100]a. Red colouring indicates studies of patients with melanoma; blue colouring indicates studies of patients with NSCLC; square = nivolumab; triangle = pembrolizumab; circle = atezolizumab, hexagon = avelumab; diamond = ICI + denosumab. aAs the trials differ in study design and methodology, such cross-trial comparisons should be viewed as hypothesis-generating only. ICI immune checkpoint inhibitor, NSCLC non-small lung cell carcinoma, RANK receptor activator of nuclear factor kappa B, RANKL receptor activator of nuclear factor kappa B ligand
In a phase 3 clinical trial of 1432 patients with castration-resistant prostate cancer at high risk of bone metastasis, denosumab significantly increased BMFS, but did not improve OS, compared with placebo [84]. A single-arm phase 2a study (D-BEYOND), conducted to determine whether a short course of neoadjuvant denosumab (two 120 mg doses a week apart) can decrease tumour proliferation rates (based on Ki67 immunohistochemistry) in newly diagnosed, early-stage breast cancer in pre-menopausal women, found that a short course of denosumab did not reduce proliferation rate, but did induce a significant increase in tumour-infiltrating lymphocytes. These data support the hypothesis that denosumab may potentiate immunotherapy efficacy [85]. ABCSG-18, a phase 3 study using adjuvant denosumab (60 mg every 6 months) in patients with oestrogen and/or progesterone-positive non-metastatic breast cancer receiving aromatase inhibitors has demonstrated a 50% reduction in fractures alongside a clinically meaningful impact on disease-free survival (secondary endpoint) after a median follow-up of 72 months (hazard ratio = 0.823, 95% confidence interval 0.69–0.98, p = 0.026) [16]. However, the primary analysis of the phase 3 clinical trial D-CARE in non-metastatic high-risk early-stage breast cancer patients reported that the interventional scheme of denosumab (120 mg monthly for 6 months, then every 3 months for 5 years) failed to demonstrate an improvement over placebo with respect to the trial’s unconventional composite primary endpoint of BMFS [86]. This was defined as the time from randomisation to the first observation of bone metastasis or death and, at the primary analysis, death accounted for 42% of the BMFS endpoint [86], confounding the ability to observe an effect of denosumab in preventing the development of bone metastasis. Moreover, in contrast to ABCSG-18, there was a lack of disease-free survival benefit in D-CARE [16, 86]. This may however be attributed to the heterogeneous high-risk early-stage breast cancer population and investigational dosing regimen used in D-CARE [86].
In a retrospective analysis of a phase 3 clinical trial of denosumab versus zoledronic acid in the treatment of bone metastases from solid tumours, denosumab was associated with increased OS in 811 patients with bone metastases from any lung cancer [15]. This outcome aligns with evidence from mouse models indicating that RANKL inhibition impairs the progression of lung adenocarcinomas [38]. However, recent prospective data from a phase 2 trial and the phase 3 SPLENDOUR trial showed that the addition of denosumab to standard first-line therapy in patients with metastatic NSCLC (with or without bone metastases) did not improve OS or PFS [87, 88]. Consistent with the above retrospective analysis [15], a slight improvement in OS was observed in the subgroup of patients with bone metastases receiving denosumab in the phase 2 study, but this effect was not significant [87, 88].
Given the multifactorial roles of the RANK–RANKL axis in the immune system, there has been interest in the potential of RANKL inhibitors to improve response to immune checkpoint inhibitors in the treatment of cancer [6, 89]. Inhibitors of immune checkpoint proteins such as CTLA-4 and anti-programmed death-1 (PD-1) or its ligand have revolutionised treatment for some patients [89]. These agents have been approved in several tumour types, including melanoma, head and neck squamous carcinoma, NSCLC, renal cell and urothelial carcinoma, gastric cancer and Hodgkin’s lymphoma [89]. However, their effectiveness is limited by variable responses to treatment in individual patients; there is a need to improve their activity overall and to develop biomarkers that can predict which patients are likely to derive benefit from current strategies [89]. Preclinical data and emerging data from real-world practice suggest that denosumab and immune checkpoint inhibitors may act synergistically [69, 90]. For example, in one case report, a patient with melanoma presented 13 months after initial diagnosis with an axillary nodal mass and widespread bone metastases and demonstrated an unexpected dramatic partial response following concomitant treatment with denosumab and single-agent ipilimumab (anti-CTLA-4) [90].
Preclinical studies suggest a synergistic effect of anti-RANKL in combination with anti-CTLA-4 and PD-1 antibodies, which have demonstrated tumour growth suppression in mouse models of melanoma, prostate and colon cancer [91]. In mouse models of melanoma, the addition of RANKL inhibition to anti-CTLA-4 antibodies did not deplete Tregs, but increased tumour-infiltrating CD4 + and CD8 + T cells compared with anti-PD-1 and anti-CTLA-4 therapy [69, 91]. T-cell effector function (measured by cytokine production) was also enhanced in tumours treated with both agents compared with tumours treated with either single agent [69, 91]. Interestingly, the synergistic effect of anti-RANKL and anti-PD-1 antibodies on lung metastasis was dependent upon NK cells, whereas the synergistic effect on subcutaneous tumour growth was dependent upon T cells [90, 91]. Building on these data, real-world evidence using Flatiron Health’s electronic health record database from ~ 255 cancer clinics across the US, including patients with advanced melanoma or NSCLC, found that a longer mean duration of concomitant immune checkpoint inhibitor and denosumab therapy was associated with improved overall response rate (p = 0.0172 and p < 0.0001 for melanoma and NSCLC, respectively) [17]. The response rate associated with the combination appears to be higher than reported historical response rates to immune checkpoint inhibitors alone (Fig. 5c). Additional survival analyses supported the association of concomitant therapy and improved survival outcomes, primarily in NSCLC (p < 0.0001) [17].
Prospective studies combining denosumab and checkpoint inhibitor immunotherapy are in progress. A phase 2 Australia and New Zealand Urogenital and Prostate Cancer Trials Group investigator-sponsored trial (KEYPAD) of denosumab and the PD-1 inhibitor pembrolizumab in patients with vascular endothelial growth factor receptor tyrosine kinase inhibitor-refractory clear cell renal cell carcinoma (NCT03280667), and an Australia and New Zealand Melanoma Trials Group phase 1b/2 trial (CHARLI) of ipilimumab–nivolumab–denosumab and nivolumab–denosumab in patients with unresectable stage III/IV melanoma (NCT03161756) are currently recruiting (Table 1). Of note, denosumab has an established safety profile in clinical practice, including when used in combination with novel anti-cancer agents, with no new detected safety signals to date [9, 92].
Discussion and conclusions
Expanding knowledge of biological mechanisms as well as bioinformatic and combinatorial screening approaches are being used to identify existing agents that may be repurposed to benefit people with cancer. Improved knowledge relating to the role played by the RANK–RANKL axis in the cancer setting has highlighted that agents targeting this pathway may be able to modulate different stages of tumour progression from initiation to metastasis. In this respect, trials are investigating denosumab in different cancers with promising results. Such trials also offer the opportunity to identify potentially meaningful clinical synergy with checkpoint inhibitor immunotherapies through characterisation of the immune effects of denosumab on intratumoural cell populations.
A focus for further work will be to establish if there are groups of patients likely to derive benefit from denosumab as an anti-RANKL agent in cancer in combination with anti-cancer therapy. For example, the RANKL system is strongly influenced by sex hormones [8, 29], and RANKL activity is increased in post-menopausal women lacking oestrogen owing to decreased OPG expression [6, 29]. Endogenous levels of OPG are lower in BRCA1 mutation carriers, who are at greater risk of breast cancer than the general population, compared with levels in women without the mutation [93]. A trial will assess the effect of denosumab on normal breast tissue in these individuals as well as BRCA2 mutation carriers and high-risk, non-BRCA carriers [94]. Moreover, RANK/RANKL/OPG and LGR4 expression data for primary tumours, such as renal, breast and lung, may assist in identifying patients most likely to benefit from denosumab therapy [95].
Overall, given the increased understanding of the pleiotropic roles of RANK/RANKL/OPG signalling in tumours and immune cells, the repurposing of denosumab, with its potential to enhance the efficacy of immuno-oncology drugs, could be a cost-effective component of such a combination approach for the treatment of people with cancer.
References
McCabe B, Liberante F, Mills KI. Repurposing medicinal compounds for blood cancer treatment. Ann Hematol. 2015;94:1267–76.
Bachur NR. Anthracycline antibiotics, in Cancer management in man. In: Woolley PV, editor. Cancer growth and progression. Dordrecht: Springer; 1989.
Bertolini F, Sukhatme VP, Bouche G. Drug repurposing in oncology–patient and health systems opportunities. Nat Rev Clin Oncol. 2015;12:732–42.
Langley RE, Burdett S, Tierney JF, Cafferty F, Parmar MK, Venning G. Aspirin and cancer: has aspirin been overlooked as an adjuvant therapy? Br J Cancer. 2011;105:1107–13.
Hart T, Dider S, Han W, Xu H, Zhao Z, Xie L. Toward repurposing metformin as a precision anti-cancer therapy using structural systems pharmacology. Sci Rep. 2016;6:20441.
de Groot AF, Appelman-Dijkstra NM, van der Burg SH, Kroep JR. The anti-tumor effect of RANKL inhibition in malignant solid tumors—a systematic review. Cancer Treat Rev. 2018;62:18–28.
Gonzalez-Suarez E, Sanz-Moreno A. RANK as a therapeutic target in cancer. FEBS J. 2016;283:2018–33.
Hanada R, Hanada T, Sigl V, Schramek D, Penninger JM. RANKL/RANK-beyond bones. J Mol Med (Berl). 2011;89:647–56.
Amgen. XGEVA (denosumab) Food and Drug Administration prescribing information. 2018; https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125320s182lbl.pdf. Accessed 8 Jan 2019.
Amgen. European Commission approves expanded indication for Amgen’s XGEVA® (denosumab) for the prevention of skeletal-related events in patients with multiple myeloma. 2018; https://www.amgen.com/media/news-releases/2018/04/european-commission-approves-expanded-indication-for-amgens-xgeva-denosumab-for-the-prevention-of-skeletalrelated-events-in-patients-with-multiple-myeloma/. Accessed 8 Jan 2019.
Thomas D, Henshaw R, Skubitz K, Chawla S, Staddon A, Blay JY, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11:275–80.
Wu PF, Tang JY, Li KH. RANK pathway in giant cell tumor of bone: pathogenesis and therapeutic aspects. Tumour Biol. 2015;36:495–501.
Amgen. FDA approves Amgen’s XGEVA® (denosumab) for the treatment of giant cell tumor of bone. 2013; https://www.amgen.com/media/news-releases/2013/06/fda-approves-amgens-xgeva-denosumab-for-the-treatment-of-giant-cell-tumor-of-bone/. Accessed 8 Jan 2019.
Cheng ML, Fong L. Effects of RANKL-targeted therapy in immunity and cancer. Front Oncol. 2014;3:329.
Scagliotti GV, Hirsh V, Siena S, Henry DH, Woll PJ, Manegold C, et al. Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: subgroup analysis from a randomized phase 3 study. J Thorac Oncol. 2012;7:1823–9.
Gnant M, Pfeiler G, Steger GG, Egle D, Greil R, Fitzal F, et al. Adjuvant denosumab in early breast cancer: Disease-free survival analysis of 3,425 postmenopausal patients in the ABCSG-18 trial. J Clin Oncol. 2018;36:500.
Liede A, Hernandez RK, Wade SW, Bo R, Nussbaum NC, Ahern E, et al. An observational study of concomitant immunotherapies and denosumab in patients with advanced melanoma or lung cancer. Oncoimmunology. 2018;7:e1480301.
Xia Y, Shen S, Verma IM. NF-kappaB, an active player in human cancers. Cancer Immunol Res. 2014;2:823–30.
Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–7.
Danks L, Takayanagi H. Immunology and bone. J Biochem. 2013;154:29–39.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23.
Huang R, Wang X, Zhou Y, Xiao Y. RANKL-induced M1 macrophages are involved in bone formation. Bone Res. 2017;5:17019.
Rao S, Sigl V, Wimmer RA, Novatchkova M, Jais A, Wagner G, et al. RANK rewires energy homeostasis in lung cancer cells and drives primary lung cancer. Genes Dev. 2017;31:2099–112.
Dufresne SS, Dumont NA, Boulanger-Piette A, Fajardo VA, Gamu D, Kake-Guena SA, et al. Muscle RANK is a key regulator of Ca2 + storage, SERCA activity, and function of fast-twitch skeletal muscles. Am J Physiol Cell Physiol. 2016;310:C663–72.
Renema N, Navet B, Heymann MF, Lezot F, Heymann D. RANK-RANKL signalling in cancer. Biosci Rep. 2016;36:e00366.
Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50.
Pellegrini P, Cordero A, Gallego MI, Dougall WC, Munoz P, Pujana MA, et al. Constitutive activation of RANK disrupts mammary cell fate leading to tumorigenesis. Stem Cells. 2013;31:1954–65.
Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, Delaloye JF, et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med. 2013;5:182.
Rao S, Cronin SJF, Sigl V, Penninger JM. RANKL and RANK: from mammalian physiology to cancer treatment. Trends Cell Biol. 2018;28:213–23.
Page G, Miossec P. RANK and RANKL expression as markers of dendritic cell-T cell interactions in paired samples of rheumatoid synovium and lymph nodes. Arthritis Rheum. 2005;52:2307–12.
Poli C, Martin JC, Braudeau C, Beriou G, Hemont C, Charrier C, et al. Receptor activating NF-kappaB ligand (RANKL) is a constitutive intracellular protein in resting human basophils and is strongly induced on their surface by interleukin 3. Immunobiology. 2015;220:692–700.
Walsh NC, Alexander KA, Manning CA, Karmakar S, Wang JF, Weyand CM, et al. Activated human T cells express alternative mRNA transcripts encoding a secreted form of RANKL. Genes Immun. 2013;14:336–45.
Sisay M, Mengistu G, Edessa D. The RANK/RANKL/OPG system in tumorigenesis and metastasis of cancer stem cell: potential targets for anticancer therapy. Onco Targets Ther. 2017;10:3801–10.
Santini D, Schiavon G, Vincenzi B, Gaeta L, Pantano F, Russo A, et al. Receptor activator of NF-kB (RANK) expression in primary tumors associates with bone metastasis occurrence in breast cancer patients. PLoS One. 2011;6:e19234.
Beuselinck B, Jean-Baptiste J, Couchy G, Job S, De Reynies A, Wolter P, et al. RANK/OPG ratio of expression in primary clear-cell renal cell carcinoma is associated with bone metastasis and prognosis in patients treated with anti-VEGFR-TKIs. Br J Cancer. 2015;113:1313–22.
Wang J, Liu Y, Wang L, Sun X, Wang Y. Clinical prognostic significance and pro-metastatic activity of RANK/RANKL via the AKT pathway in endometrial cancer. Oncotarget. 2016;7:5564–75.
Zhang X, Song Y, Song N, Zhang L, Wang Y, Li D, et al. Rankl expression predicts poor prognosis in gastric cancer patients: results from a retrospective and single-center analysis. Braz J Med Biol Res. 2018;51:e6265.
Faget J, Contat C, Zangger N, Peters S, Meylan E. RANKL signaling sustains primary tumor growth in genetically engineered mouse models of lung adenocarcinoma. J Thorac Oncol. 2017;13:387–98.
Terpos E, Szydlo R, Apperley JF, Hatjiharissi E, Politou M, Meletis J, et al. Soluble receptor activator of nuclear factor kappaB ligand-osteoprotegerin ratio predicts survival in multiple myeloma: proposal for a novel prognostic index. Blood. 2003;102:1064–9.
Timotheadou E, Kalogeras KT, Koliou GA, Wirtz RM, Zagouri F, Koutras A, et al. Evaluation of the prognostic value of RANK, OPG, and RANKL mRNA expression in early breast cancer patients treated with anthracycline-based adjuvant chemotherapy. Transl Oncol. 2017;10:589–98.
Trieb K, Windhager R. Receptor activator of nuclear factor kappaB expression is a prognostic factor in human osteosarcoma. Oncol Lett. 2015;10:1813–5.
Steven A, Leisz S, Fussek S, Nowroozizadeh B, Huang J, Branstetter D, et al. Receptor activator of NF-kappaB (RANK)-mediated induction of metastatic spread and association with poor prognosis in renal cell carcinoma. Urol Oncol. 2018;36:502.e15–24.
Dougall WC. Molecular pathways: osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin Cancer Res. 2012;18:326–35.
Boudot C, Henaut L, Thiem U, Geraci S, Galante M, Saldanha P, et al. Overexpression of a functional calcium-sensing receptor dramatically increases osteolytic potential of MDA-MB-231 cells in a mouse model of bone metastasis through epiregulin-mediated osteoprotegerin downregulation. Oncotarget. 2017;8:56460–72.
Blake ML, Tometsko M, Miller R, Jones JC, Dougall WC. RANK expression on breast cancer cells promotes skeletal metastasis. Clin Exp Metastasis. 2014;31:233–45.
Miller RE, Roudier M, Jones J, Armstrong A, Canon J, Dougall WC. RANK ligand inhibition plus docetaxel improves survival and reduces tumor burden in a murine model of prostate cancer bone metastasis. Mol Cancer Ther. 2008;7:2160–9.
Casimiro S, Alho I, Bettencourt M, Pires R, Lipton A, Costa L. RANKL enhances the effect of an antagonist of inhibitor of apoptosis proteins (cIAPs) in RANK-positive breast cancer cells. J Bone Oncol. 2013;2:116–22.
Shin M, Matsuo K, Tada T, Fukushima H, Furuta H, Ozeki S, et al. The inhibition of RANKL/RANK signaling by osteoprotegerin suppresses bone invasion by oral squamous cell carcinoma cells. Carcinogenesis. 2011;32:1634–40.
Peng X, Guo W, Ren T, Lou Z, Lu X, Zhang S, et al. Differential expression of the RANKL/RANK/OPG system is associated with bone metastasis in human non-small cell lung cancer. PLoS One. 2013;8:e58361.
Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun. 2015;6:8983.
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010;468:103–7.
Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010;468:98–102.
Rodriguez-Lara V, Hernandez-Martinez JM, Arrieta O. Influence of estrogen in non-small cell lung cancer and its clinical implications. J Thorac Dis. 2018;10:482–97.
Wu J, Xie N, Xie K, Zeng J, Cheng L, Lei Y, et al. GPR48, a poor prognostic factor, promotes tumor metastasis and activates beta-catenin/TCF signaling in colorectal cancer. Carcinogenesis. 2013;34:2861–9.
Yue Z, Yuan Z, Zeng L, Wang Y, Lai L, Li J, et al. LGR4 modulates breast cancer initiation, metastasis, and cancer stem cells. FASEB J. 2018;32:2422–37.
Luo W, Tan P, Rodriguez M, He L, Tan K, Zeng L, et al. Leucine-rich repeat-containing G protein-coupled receptor 4 (Lgr4) is necessary for prostate cancer metastasis via epithelial-mesenchymal transition. J Biol Chem. 2017;292:15525–37.
Ren Z, van Andel H, de Lau W, Hartholt RB, Maurice MM, Clevers H, et al. Syndecan-1 promotes Wnt/beta-catenin signaling in multiple myeloma by presenting Wnts and R-spondins. Blood. 2018;131:982–94.
Fabregat I, Malfettone A, Soukupova J. New insights into the crossroads between EMT and stemness in the context of cancer. J Clin Med. 2016;5:E37.
Palafox M, Ferrer I, Pellegrini P, Vila S, Hernandez-Ortega S, Urruticoechea A, et al. RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res. 2012;72:2879–88.
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470:548–53.
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440:692–6.
Odero-Marah VA, Wang R, Chu G, Zayzafoon M, Xu J, Shi C, et al. Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008;18:858–70.
Yamada T, Tsuda M, Takahashi T, Totsuka Y, Shindoh M, Ohba Y. RANKL expression specifically observed in vivo promotes epithelial mesenchymal transition and tumor progression. Am J Pathol. 2011;178:2845–56.
Min JK, Cho YL, Choi JH, Kim Y, Kim JH, Yu YS, et al. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: impaired permeability and angiogenesis in eNOS-deficient mice. Blood. 2007;109:1495–502.
Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–37.
Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4(+)CD25(+) Treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity. 2002;16:183–91.
Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, et al. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med. 2006;12:1372–9.
Totsuka T, Kanai T, Nemoto Y, Tomita T, Okamoto R, Tsuchiya K, et al. RANK-RANKL signaling pathway is critically involved in the function of CD4 + CD25 + regulatory T cells in chronic colitis. J Immunol. 2009;182:6079–87.
Ahern E, Harjunpaa H, Barkauskas D, Allen S, Takeda K, Yagita H, et al. Co-administration of RANKL and CTLA4 antibodies enhances lymphocyte-mediated antitumor immunity in mice. Clin Cancer Res. 2017;23:5789–801.
Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. 1997;186:2075–80.
Wiethe C, Dittmar K, Doan T, Lindenmaier W, Tindle R. Enhanced effector and memory CTL responses generated by incorporation of receptor activator of NF-kappa B (RANK)/RANK ligand costimulatory molecules into dendritic cell immunogens expressing a human tumor-specific antigen. J Immunol. 2003;171:4121–30.
Josien R, Wong BR, Li HL, Steinman RM, Choi Y. TRANCE, a TNF family member, is differentially expressed on T cell subsets and induces cytokine production in dendritic cells. J Immunol. 1999;162:2562–8.
Yun TJ, Tallquist MD, Aicher A, Rafferty KL, Marshall AJ, Moon JJ, et al. Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J Immunol. 2001;166:1482–91.
Seshasayee D, Wang H, Lee WP, Gribling P, Ross J, Van Bruggen N, et al. A novel in vivo role for osteoprotegerin ligand in activation of monocyte effector function and inflammatory response. J Biol Chem. 2004;279:30202–9.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30.
Khan IS, Mouchess ML, Zhu ML, Conley B, Fasano KJ, Hou Y, et al. Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J Exp Med. 2014;211:761–8.
Bakhru P, Zhu ML, Wang HH, Hong LK, Khan I, Mouchess M, et al. Combination central tolerance and peripheral checkpoint blockade unleashes antimelanoma immunity. JCI Insight. 2017;2:e93265.
Demoulin SA, Somja J, Duray A, Guenin S, Roncarati P, Delvenne PO, et al. Cervical (pre)neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells via RANKL secretion. Oncoimmunology. 2015;4:e1008334.
Fujimura T, Kambayashi Y, Furudate S, Asano M, Kakizaki A, Aiba S. Receptor activator of NF-kappaB ligand promotes the production of CCL17 from RANK + M2 macrophages. J Invest Dermatol. 2015;135:2884–7.
Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–18.
Breuil V, Schmid-Antomarchi H, Schmid-Alliana A, Rezzonico R, Euller-Ziegler L, Rossi B. The receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) is a new chemotactic factor for human monocytes. FASEB J. 2003;17:1751–3.
Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27:5932–43.
Savvidou OD, Bolia IK, Chloros GD, Papanastasiou J, Koutsouradis P, Papagelopoulos PJ. Denosumab: current use in the treatment of primary bone tumors. Orthopedics. 2017;40:204–10.
Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. 2012;379:39–46.
Nguyen B, Maetens N, Salgado R, Venet D, Vuylsteke P, Polastro L, et al. D-BEYOND: A window of opportunity trial evaluating denosumab, a RANK-ligand (RANKL) inhibitor and its biological effects in young pre-menopausal women diagnosed with early breast cancer [abstract CT101/22]. AACR Annual Meeting 14–18 April 2018. http://www.abstractsonline.com/pp8/#!/4562/presentation/11232. Accessed 8 Jan 2019.
Coleman RE, Finkelstein D, Barrios CH, Martin M, Iwata H, Glaspy JA, et al. Adjuvant denosumab in early breast cancer: First results from the international multicenter randomized phase III placebo controlled D-CARE study. J Clin Oncol. 2018;36(15):501.
De Boer R, Pedrazzoli P, Biesma B, Natale R, Lee CK, Zhu L, et al. First-line treatment of metastatic non-small cell lung cancer (NSCLC): a randomized, double-blind, phase 2 trial of denosumab in combination with chemotherapy [abstract 402]. Eur J Cancer. 2018;103:e23–147.
Peters S, Danson SJ, Hasan B, Reinmuth N, Majem M, Tournoy KG, et al. A randomised phase III trial evaluating the addition of denosumab to standard first-line treatment in advanced NSCLC: the ETOP and EORTC SPLENDOUR trial [abstract 1385PD]. Ann Oncol. 2018;29(mdy292):008.
Thallinger C, Fureder T, Preusser M, Heller G, Mullauer L, Holler C, et al. Review of cancer treatment with immune checkpoint inhibitors: current concepts, expectations, limitations and pitfalls. Wien Klin Wochenschr. 2017;130:83–91.
Smyth MJ, Yagita H, McArthur GA. Combination anti-CTLA-4 and anti-RANKL in metastatic melanoma. J Clin Oncol. 2016;34:e104–6.
Ahern E, Harjunpää H, O’Donnell JS, Allen S, Dougall WC, Teng MWL, et al. RANKL blockade improves efficacy of PD1-PD-L1 blockade or dual PD1-PD-L1 and CTLA4 blockade in mouse models of cancer. Oncoimmunology. 2018;7:e1431988.
Liede A, Wade S, Lethen J, Hernandez RK, Warner D, Abernethy AP, et al. An observational study of concomitant use of emerging therapies and denosumab or zoledronic acid in prostate cancer. Clin Ther. 2018;40:536–49.
Kotsopoulos J, Singer C, Narod SA. Can we prevent BRCA1-associated breast cancer by RANKL inhibition? Breast Cancer Res Treat. 2017;161:11–6.
Australian New Zealand Clinical Trials Registry. 2018; https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12614000694617. Accessed 8 Jan 2019.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2016;6:11.
von Moos R, Haynes I. Where do bone-targeted agents RANK in breast cancer treatment? J Clin Med. 2013;2:89–102.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Azim HA Jr, Peccatori FA, Brohee S, Branstetter D, Loi S, Viale G, et al. RANK-ligand (RANKL) expression in young breast cancer patients and during pregnancy. Breast Cancer Res. 2015;17:24.
Gulley JL, Rajan A, Spigel DR, Iannotti N, Chandler J, Wong DJL, et al. Avelumab for patients with previously treated metastatic or recurrent non-small-cell lung cancer (JAVELIN Solid Tumor): dose-expansion cohort of a multicentre, open-label, phase 1b trial. Lancet Oncol. 2017;18:599–610.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–65.
Acknowledgements
Medical writing support including development of a draft outline and first full draft in consultation with the authors was provided by Jack Dean, MBChB at Oxford PharmaGenesis (Oxford, UK); development of subsequent drafts in collaboration with the authors, assembling tables and figures, copyediting and fact checking was provided by Louise Niven, DPhil at Aspire Scientific (Bollington, UK).
Funding
Medical writing support was funded by Amgen (Europe) GmbH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SP has received educational grants, provided consultation, attended advisory boards and/or provided lectures for the following organisations: AbbVie, Amgen, AstraZeneca, Biocartis, Boehringer Ingelheim, Bristol-Myers Squibb, Clovis, Daiichi Sankyo, Debiopharm, Eli Lilly, F. Hoffmann-La Roche, Foundation Medicine, Illumina, Janssen, Merck Serono, Merck Sharp and Dohme, Merrimack, Novartis, Pfizer, Pharma Mar, Regeneron, Sanofi, Seattle Genetics and Takeda. PC has attended advisory boards and provided lectures for Amgen. IM-R has received grants, travel accommodation, or held an advisory role with Amgen, Bioncotech, Bristol-Myers Squibb, Merck Serono, Merck Sharp and Dohme, Novartis, Pierre Fabre and Roche. DN is an Amgen employee and holds Amgen stock. CG has provided consultation, attended advisory boards, received travel support and/or provided lectures for the following organisations (all fees direct to institution): AbbVie, AstraZeneca, Bristol-Myers Squibb, Ipsen, Merck KGaA, Merck Sharp and Dohme, Pfizer and Sanofi. CG has directed research grant support (all fees direct to institution) from Amgen, Bristol-Myers Squibb and Merck Sharp and Dohme. CG is a consultant for Novotech CRO Pty and is the coordinating principal investigator of the ANZUP-sponsored KEYPAD study.
Ethical approval
The manuscript does not report clinical or patient data acquired by any of the authors.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed content
The manuscript does not report clinical or patient data acquired by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Peters, S., Clézardin, P., Márquez-Rodas, I. et al. The RANK–RANKL axis: an opportunity for drug repurposing in cancer?. Clin Transl Oncol 21, 977–991 (2019). https://doi.org/10.1007/s12094-018-02023-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-018-02023-5