
Abstract
Background
In the Netherlands, a screening programme was set up in 1994 in order to identify all patients with familial hypercholesterolaemia (FH). After 15 years of screening, we evaluated the geographical distribution, possible founder effects and clinical phenotype of the 12 most prevalent FH gene mutations.
Methods
Patients who carried one of the 12 most prevalent mutations, index cases and those identified between 1994 and 2009 through the screening programme and whose postal code was known were included in the study. Low-density lipoprotein cholesterol (LDL-C) levels at the time of screening were retrieved. The prevalence of identified FH patients in each postal code area was calculated and visualised in different maps.
Results
A total of 10,889 patients were included in the study. Mean untreated LDL-C levels ranged from 4.4 to 6.4 mmol/l. For almost all mutations, a region of high prevalence could be observed. In total, 51 homozygous patients were identified in the Netherlands, of which 13 true homozygous for one of the 12 most prevalent mutations. The majority of them were living in high-prevalence areas for that specific mutation.
Conclusions
Phenotypes with regard to LDL-C levels varied between the 12 most prevalent FH mutations. For most of these mutations, a founder effect was observed. Our observations can have implications with regard to the efficiency of molecular screening and physician’s perception of FH and to the understanding of the prevalence and distribution of homozygous patients in the Netherlands.
Similar content being viewed by others


Introduction
Familial hypercholesterolaemia (FH) is an autosomal co-dominant monogenic disorder of lipoprotein metabolism. It is characterised by severely elevated levels of low-density lipoprotein cholesterol (LDL-C) from birth onwards, clinically leading to premature atherosclerosis and cardiovascular disease (CVD). The estimated prevalence of heterozygous FH is 1 per 400 in the Dutch population [1, 2]. Homozygosity is rare with an average frequency of 1:640,000.
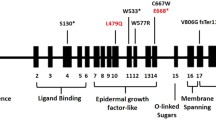
FH is mainly caused by mutations in the LDL receptor (LDLR) gene, encoding for a receptor that removes LDL-C particles from the circulation and facilitates uptake by the liver cell [3]. In approximately 7% of cases, the clinical phenotype of FH is the result of mutations in the region of the gene encoding for the LDLR-binding domain of apolipoprotein B (APOB) [4]. Also, gain-of-function mutations in the gene encoding for proprotein convertase subtilisin–kexin type 9 (PCSK9), a protein that regulates LDL receptor degradation, are a rare cause (<1%) of the FH phenotype.
More than 1000 different mutations, assumed to cause an FH phenotype, have been identified worldwide [5]. In the Netherlands, 552 different mutations have been identified so far. The specific mutations can vary with respect to clinical severity, as observed by LDL-C levels [6], atherosclerosis as assessed by carotid intima-media thickness [7] as well as by prevalence of CVD [8]. In some populations, the majority of cases of FH can be explained by only a few mutations, and a founder effect usually underlies a higher than expected frequency of certain mutations.
The importance of early diagnosis and management of FH is well established, and therefore, screening for FH is advocated [9]. In the Netherlands, approximately 20,000 subjects have been identified with a pathogenic FH mutation so far, mostly through a nationwide genetic cascade screening programme. Because FH has been studied this extensively in our country, this enables us to study some interesting aspects with regard to geographical distribution and prevalence of homozygous patients not directly related to consanguinity. In this study, we explored the geographical distribution, possible founder effects and clinical phenotype with regard to LDL-C levels of the 12 most prevalent FH mutations in the Netherlands.
Patients and Methods
Selection of Study Population
In the Netherlands, genetic cascade screening is carried out by the Foundation for Identification of Persons with Inherited Hypercholesterolaemia (in Dutch: StOEH). Physicians clinically diagnose patients suspected of having FH using uniform diagnostic criteria after which extensive DNA analysis is performed [10]. Once a mutation is identified, the patient is designated as an index case. Cascade screening is then performed to test all first-degree family members, and if positive for the familial mutation, more distant relatives are tested to expand the pedigree [11]. Since 2003, lipid levels are measured at the time of genetic testing with a Cholestech LDX analyser [12].
Patients with one of the 12 most prevalent FH mutations, index cases and those identified between 1994 and 2009 through cascade screening and whose postal code was known were included in the study. In order to prevent referral bias as a result of physicians being more or less active in referring cases for DNA analysis [13, 14], index cases and family members identified through the screening programme were studied separately. Mean LDL-C levels and mean percentile LDL-C for age and gender at the time of screening were only retrieved for patients identified though the screening programme, as these data were not routinely provided for index cases. When patients were on lipid-lowering medication, LDL-C levels were imputed by the estimated LDL-C lowering potency of a specific lipid-lowering drug and dose, as described by Huijgen et al. [15].
Index cases and carriers who were homozygous for a certain mutation were identified in the database of the DNA diagnostic laboratory of the Academic Medical Centre (AMC) at the University of Amsterdam.
Distribution Across Postal Code Areas
In the Netherlands, geographic areas are defined by so-called PC2 codes, in which the first two of four digits of the postal code determine a geographic region. The mean number of inhabitants of a postal code area is 180,000 (range, 35,170 to 652,685). The number of identified FH patients in each PC2 area was reproduced as prevalence per million, adjusted for inhabitant number of that area. The prevalence of the 12 mutations in each PC2 area has been visualised with the software of Mapinfo Professional (Mapinfo, Toronto, Canada). The locations where homozygous patients were identified were marked on the map for that specific mutation.
Results
Study Population
A total of 4861 index cases with 552 different pathogenic mutations were identified in the DNA database of the AMC. Of these, 528 were in LDLR (88%) and 25 in APOB (12%) mutations. Hitherto, mutations in PCSK9 that result in a FH phenotype have not been identified in the Netherlands. Between 1994 and 2009, 14,805 patients with FH were identified through the genetic cascade screening programme. The 12 most common mutations were responsible for 64% of all FH patients, including 2699 index cases and 9393 patients identified through the screening programme. The patients with a known postal code (2527 index cases and 8362 patients from the screening programme) were included in the study.
Characteristics of the 12 most common mutations in patients identified through the screening programme are summarised in Table 1. Mean untreated LDL-C levels ranged from 3.9 to 5.6 mmol/l between mutations. Together with the estimated pre-treatment LDL-C levels in the patients on lipid-lowering medication, these levels were higher and the differences more pronounced, ranging from 4.4 to 6.4 mmol/l.
Geographical Distribution of FH Mutations in the Netherlands
The prevalence per PC2 area is illustrated for each mutation in Fig. 1 for the index cases and in Fig. 2 for patients identified through the screening programme. The most common mutation, the N543H/2393del9bp, was by far most frequent in West Friesland (a contemporary region in the North West of the Netherlands), with a prevalence of 2860 per million, which is similar to 1:349. The R3500Q mutation in APOB was the second most common mutation and was distributed about equally over the Netherlands; it was the only mutation present in each of the 90 PC2 areas.

Geographical distribution of the 12 most common FH mutations in the Netherlands, index cases only. Number of patients identified with the specific mutation, adjusted for resident number of concerning PC2 code area per million (between brackets, number of PC2 code areas)

Geographical distribution of the 12 most common FH mutations in the Netherlands, index cases excluded (arrows indicate homozygous patients). Number of patients identified with the specific mutation, adjusted for resident number of concerning PC2 code area per million (between brackets, number of PC2 code areas)
Mutations with a high prevalence (index cases included) in only one or two PC2 code areas included 1359-1 in the province of Brabant in the South (1548 per million ≈ 1:646 for the most dense area), 191-1 in the province of Zeeland in the south-western part (1151 per million ≈ 1:869 for the area known as Noord and Zuid Beveland), the E207K mutation concentrated in the western part of the Netherlands (498–707 per million in the two most dense areas) and the 2.5-kb deletion of exons 7–8 also in the province of Brabant (207–257 per million in the two most dense areas).
For the total prevalence of the 12 most frequent mutations together (Figs. 3 and 4), the PC2 areas with the highest prevalence were West Friesland (1:280), predominantly resulting from N543H/2393del9bp (82%), R3500Q (8%) and V408M (4%) mutation, and the islands Noord and Zuid Beveland in the province of Zeeland (1:329), mainly resulting from 191-2 (37%), 313 + 1/2 (18%), 1359-1 and G322S (both 5%) mutation. The lowest prevalence was found in the north of the province of Limburg in the south-eastern part of the Netherlands (1:6803) and in the east of the province of Overijssel, in the mid-eastern part of the Netherlands (1:6061).

Geographical distribution of the 12 most common FH mutations together, index cases only. Number of patients identified with one of the 12 most common mutations, adjusted for resident number of concerning PC2 code area per million (between brackets, number of PC2 code areas)

Geographical distribution of the 12 most common FH mutations together, index cases excluded. Number of patients identified with one of the 12 most common mutations, adjusted for resident number of concerning PC2 code area per million (between brackets, number of PC2 code areas)
Homozygous FH Patients
In total, 51 patients were identified as suffering from homozygous FH, of which 27 were compound heterozygotes and 24 were true homozygotes. Of the true homozygous patients, 13 were homozygous for one of the 12 most frequent mutations, and in 21 of the compound heterozygous patients, one of these mutations was involved. As shown in Fig. 2, almost all true homozygous patients were living in high-prevalence areas for that specific mutation.
Discussion
In the Netherlands, a national screening programme for FH is ongoing and has yielded the largest genotyped FH population worldwide. For this study, we determined the geographical distribution of the 12 most common mutations, which represent 64% of all Dutch FH patients. We demonstrated that almost all common mutations showed a clearly marked region of preference, suggesting the existence of a founder effect. This can be explained by the occurrence of a local founder mutation in combination with limited migration, possibly attributable to the geographical isolation in the past, as in West Friesland and the islands of Noord and Zuid Beveland, the two regions with the highest prevalence of mutations per se. The phenomenon of geographical preference of FH causing mutations can have several clinical implications.
Firstly, knowledge of the prevalence of a certain mutation can be used in strategies for molecular testing; when a mutation predominates in a region, molecular tests can be designed to identify these specific variant alleles first. Secondly, knowledge of mutation distribution may explain the identification of homozygous patients in non-consanguineous families. This has been confirmed by our finding that most of the true homozygous patients are found in regions with a high prevalence of that specific mutation and the fact that the total number of identified homozygous patients (51) is higher than the expected number of 26 (1:640,000 in 16.5 million people in the Netherlands). Thirdly, our observations can influence the perception of physicians in different regions with regard to the prevalence of FH and the severity of the FH phenotype in their region. It is not unlikely that physicians in the north-western region where the relatively mild N543H/2393del9bp mutation is most prevalent have a different perception of CVD risk attributable to FH than colleagues in the south of the Netherlands where the severe 1359-1 mutation predominates. The latter is known for its significantly higher LDL-C levels and higher incidence of premature CVD. The relative risk of coronary artery disease in FH patients compared with unaffected relatives was found to be 7.83 (95% CI, 3.11–19.67) for carriers of the N543H/2393del9 mutation and 15.95 (95% CI, 5.52–46.14) for carriers of the 1359-1 mutation [16].
Dutch FH patients share mutations with FH populations in other countries, as discussed by Fouchier et al. [3, 17]. This can be partly explained by the migration of individuals over Europe during the last centuries, demonstrated by large numbers of similar mutations in neighbouring countries such as Germany, the UK, Belgium and Denmark [3]. However, detection rates of mutations depend on the intensity of screening in different countries, and the global array of mutations is therefore not complete.
Towns with harbours that served as an outpost for trading with other coastal communities may have been the source of the spread of the prevalent mutations in those regions. A mutation of special interest in that respect is the V408M mutation, which is most commonly found in West Friesland, close to the former Zuiderzee (now a lake, but it used to be connected to the North Sea). This is one of the regions from where settlers to South Africa departed in the seventeenth century, with important port towns as Hoorn and Enkhuizen. The V408M mutation is one of the founder mutations in South Africa, present in 15–20% of the South African Afrikaner FH patients [18]. It has been suggested that this was the result of Dutch migration, in times of the expeditions of the Dutch colonial administrator Jan Van Riebeeck, the founder of Cape Town in the seventeenth century. It was indeed established that some of the Dutch FH patients shared the same haplotype with an Afrikaner, who was homozygous for this mutation [19]. Furthermore, it has been suggested that this mutation was also introduced in Canada at the beginning of the twentieth century by the large immigration waves from (the northern part of) the Netherlands. The founder of a Canadian FH family with the V408M mutation was traced back to Andijk [20], which is also situated in West Friesland.
By comparing mutation frequencies within and between populations, insight into the evolutionary history of genes and populations can be gained. It has been found that almost all FH patients with R3500Q (showing no founder effect in the Netherlands and present in different populations over the world) share the same rare haplotype. Therefore, it is postulated that the first original R3500Q mutation occurred approximately 6750 years ago [21, 22]. However, it is also possible that some mutations occurred on multiple occasions, referred to as recurrent mutations. For instance, ‘Dutch’ mutations are also found in countries as Japan, which is geographically isolated and Japanese people are believed to be uniracial [23]. The same applies for some mutations found in rural China [24].
Currently, more than 550 different FH mutations have been identified in the Netherlands, from which 471 have been used in cascade screening. These numbers are different because not all patients had given consent for family screening. In populations where one or two mutations predominate, founder effects are much more pronounced than in the Netherlands. In Finland, four different mutations are responsible for 75% of FH patients, which can be explained by the unique geographical position of Finland between Eastern and Western cultures as well as by linguistic barriers [25]. Other examples of populations with a clear founder effect are the French-Canadians, with the 10-kb deletion in 60% of patients and a total FH prevalence of about 1:200 [26], and the earlier mentioned South African Afrikaners, where three founder mutations are responsible for 95% of all Afrikaner FH patients (including the ‘Dutch’ V408M mutation), and an estimated prevalence for FH of 1:72 [27]. As in these populations, this study showed that also in the Dutch population, founder effects result in high prevalence in certain regions, in particular in the area of West Friesland, where a prevalence of 1:280 was found, which is higher than the overall estimated prevalence for the Netherlands [2]. It has to be taken into account that patients without a known postal code were excluded from our analysis (approximately 7% for index cases and 12% for other patients), that only the data of the 12 most frequent mutations were used, and that all patients might not have been traced yet, so in fact the real prevalence for this area could be considerably higher. For instance, the area with the highest prevalence of index cases (825 per million) is the islands of Noord and Zuid Beveland. From the experience of the screening programme, we know that through one index case, approximately eight family members with FH can be traced [14]. This would mean that by adding up the 7% with an unknown postal code, the prevalence for the 12 most common mutations in this area can be estimated to be 1:142.
Efficacy of identification of FH patients through a cascade screening programme such as in the Netherlands is dependent on several factors: regional referral rate, regional coverage of the screening programme, family size and willingness of relatives to participate. All these factors may have influenced the results of our study. We attempted to reduce referral bias of physicians by analysing index cases and identified family members separately, but as almost similar patterns can be observed in the two figures, this appears to play a minor role. Our results show different distributions for all 12 mutations with dense areas in all parts of the country, which reflects nationwide coverage. Estimations of the percentage of traced FH patients differ among provinces [14], but these are based on a presumed even distribution. Furthermore, we assume that family size and willingness for screening are similar for different mutations. Although these assumptions may not be completely correct and therefore may have biased our results to some extent, we have found a clear, unique distribution for the 12 most prevalent mutations.
Conclusion
Although more than 550 different FH mutations are identified in the Netherlands, for most of the 12 most prevalent Dutch mutations a founder effect can be observed, resulting in differences in geographical prevalence across the Netherlands. This can be explained by local founder effects plus limited migration, which is also reflected by the fact that neighbouring countries and countries were Dutch immigrants used to go, share the same mutations. The high prevalence and typical distribution of Dutch homozygous patients can also be understood by these founder effects. Molecular screening can be more efficient if it is tailored to the allele frequency distribution of the Dutch population. Furthermore, physician’s perception of FH could be influenced by the severity of the phenotype of the most prevalent mutation in a certain area, but further research is needed to confirm this.
References
Goldstein JL, Hobbs HH, Brown MS. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001.
Lansberg PJ, Tuzgol S, van de Ree MA, et al. Higher prevalence of familial hypercholesterolemia than expected in adult patients of four family practices in Netherlands. Ned Tijdschr Geneeskd. 2000;144:1437–40.
Fouchier SW, Defesche JC, Umans-Eckenhausen MW, et al. The molecular basis of familial hypercholesterolemia in the Netherlands. Hum Genet. 2001;109:602–15.
Innerarity TL, Mahley RW, Weisgraber KH, et al. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res. 1990;31:1337–49.
Leigh SE, Foster AH, Whittall RA, et al. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet. 2008;72:485–98.
Huijgen R, Kindt I, Fouchier SW, et al. Functionality of sequence variants in the genes coding for the low-density lipoprotein receptor and apolipoprotein B in individuals with inherited hypercholesterolemia. Hum Mutat. 2010;31:752–60.
Junyent M, Gilabert R, Jarauta E, et al. Impact of low-density lipoprotein receptor mutational class on carotid atherosclerosis in patients with familial hypercholesterolemia. Atherosclerosis. 2010;208:437–41.
Umans-Eckenhausen MA, Sijbrands EJ, Kastelein JJ, et al. Low-density lipoprotein receptor gene mutations and cardiovascular risk in a large genetic cascade screening population. Circulation. 2002;106:3031–6.
Wonderling D, Umans-Eckenhausen MA, Marks D, et al. Cost-effectiveness analysis of the genetic screening programme for familial hypercholesterolemia in the Netherlands. Semin Vasc Med. 2004;4:97–104.
Defesche JC. Familial hypercholesterolaemia. In: Betteridge DJ, editor. Lipids and vascular disease. London: Martin Dunitz; 2000. p. 65–76.
Huijgen R, Vissers MN, Defesche JC, et al. Familial hypercholesterolemia: current treatment and advances in management. Expert Rev Cardiovasc Ther. 2008;6:567–81.
Cobbaert C, Boerma GJ, Lindemans J. Evaluation of the Cholestech L.D.X. desktop analyser for cholesterol, HDL-cholesterol, and triacylglycerols in heparinized venous blood. Eur J Clin Chem Clin Biochem. 1994;32:391–4.
Hofman N, Postema PG, van Langen IM, et al. Genetic identification of patients and families with a long-QT syndrome: large regional differences in the result of 10 years. Ned Tijdschr Geneeskd. 2007;151:644–8.
StOEH. Annual report 2008. Amsterdam: StOEH; 2009.
Huijgen R, Kindt I, Verhoeven SB, et al. Two years after molecular diagnosis of familial hypercholesterolemia: majority on cholesterol-lowering treatment but a minority reaches treatment goal. PLoS ONE. 2010;5:e9220.
Umans-Eckenhausen MA, Sijbrands EJ, Kastelein JJ, et al. Low-density lipoprotein receptor gene mutations and cardiovascular risk in a large genetic cascade screening population. Circulation. 2002;106:3031–6.
Fouchier SW, Kastelein JJ, Defesche JC. Update of the molecular basis of familial hypercholesterolemia in the Netherlands. Hum Mutat. 2005;26:550–6.
Kotze MJ, Langenhoven E, Warnich L, et al. The molecular basis and diagnosis of familial hypercholesterolaemia in South African Afrikaners. Ann Hum Genet. 1991;55:115–21.
Defesche JC, Van Diermen DE, Lansberg PJ, et al. South African founder mutations in the low-density lipoprotein receptor gene causing familial hypercholesterolemia in the Dutch population. Hum Genet. 1993;92:567–70.
Defesche JC, Van Diermen DE, Hayden MR, et al. Origin and migration of an Afrikaner founder mutation FHAfrikaner-2 (V408M) causing familial hypercholesterolemia. Gene Geogr. 1996;10:1–10.
Austin MA, Hutter CM, Zimmern RL, et al. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160:407–20.
Myant NB, Forbes SA, Day IN, et al. Estimation of the age of the ancestral arginine3500–>glutamine mutation in human apoB-100. Genomics. 1997;45:78–87.
Yu W, Nohara A, Higashikata T, et al. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis. 2002;165:335–42.
Khoo KL, van Acker P, Defesche JC, et al. Low-density lipoprotein receptor gene mutations in a Southeast Asian population with familial hypercholesterolemia. Clin Genet. 2000;58:98–105.
Vuorio AF, Turtola H, Piilahti KM, et al. Familial hypercholesterolemia in the Finnish north Karelia. A molecular, clinical, and genealogical study. Arterioscler Thromb Vasc Biol. 1997;17:3127–38.
Betard C, Kessling AM, Roy M, et al. Molecular genetic evidence for a founder effect in familial hypercholesterolemia among French Canadians. Hum Genet. 1992;88:529–36.
Steyn K, Goldberg YP, Kotze MJ, et al. Estimation of the prevalence of familial hypercholesterolaemia in a rural Afrikaner community by direct screening for three Afrikaner founder low density lipoprotein receptor gene mutations. Hum Genet. 1996;98:479–84.
den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12.
Acknowledgements
The authors thank Pieter Postema for his help with making the maps, the National Institute for Public Health and Environment (RIVM) for executing the FH screening programme and participants of the programme in the Netherlands.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kusters, D.M., Huijgen, R., Defesche, J.C. et al. Founder mutations in the Netherlands: geographical distribution of the most prevalent mutations in the low-density lipoprotein receptor and apolipoprotein B genes. Neth Heart J 19, 175–182 (2011). https://doi.org/10.1007/s12471-011-0076-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-011-0076-6