
Abstract
Novel substituted series of aromatic copolymers was obtained by one-spot, metal-free super acid-catalyzed one-step polymerization of substituted isatin and phenothiazine. The polymerization reaction was performed at room temperature in the presence of Bronsted superacid (trifluoromethanesulfonic acid) and methylene chloride which condenses the compounds consists of carbonyl group (aldehydes and ketones) and aromatic rings to yield the polymers. Super acid catalyst has several advantages including the reaction proceeding at room temperature and great synthetic versatility. The obtained polymers have good solubility in common organic solvents. The polymers were purified by repeated precipitations with methanol. Polymers (P1–P3) were completely characterized by FT-IR, 1H NMR, TGA, and UV–visible, fluorescence and cyclic voltammetry techniques. Polymers (P1–P3) possessed excellent thermal stability up to 300 °C and have absorbance and emission maximum at 557 and 630 nm, respectively. The optical and electrochemical properties of these polymers revealed that it could be one of the capable materials for applications in optoelectronic device and in the area of proton exchange membrane fuel cell.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The development and establishment of various polymerization methods had their own merits and demerits in synthesizing polymers using different monomers and conditions. Synthesis of polymers for a particular application may go for selective polymerization method. For instance, Suzuki/Stille polycondensation technique is mostly chosen for optoelectronic device applications [1, 2]. Polymerization of monomers using different methodologies such as free radical polymerization [3], cationic polymerization [4], anionic polymerization [5], nitroxide-mediated polymerization (NMP) [6], and atom transfer radical polymerization (ATRP) [7] was extensively studied and many review articles are well documented in that arena [8,9,10,11,12]. In this context, we have also reported ultrasound and phase transfer-catalyzed polymerization of different alkyl methacrylate monomers [13,14,15,16,17]. These polymerization methods had advantages and disadvantages in their regime. Among the various polymerization techniques, the super acid-catalyzed polymerization method is an efficient and facile route to polymerize the compounds with carbonyl group and aromatic rings, the resulted polymers used for various applications [18].
Super acid-catalyzed (Friedel–Crafts aromatic electrophilic substitution reaction) reaction has gaining remarkable interest in recent past [19,20,21,22,23,24] because of their myriad advantages associated with eco-friendliness. Super acid-catalyzed reactions have the following advantages: the reaction proceeds at room temperature, structural selection and easy availability of monomers, high yields and selectivity, short duration of reaction, more versatility in polymer architectures, easy purification, possessing good physical, chemical, and thermal properties of the synthesized polymers, and high molecular weights, etc. [25,26,27]. The perception and explanation of super acid-catalyzed reactions were made by Olah in 1970 [28,29,30]. Consequently, several reactions have been performed using super acids [mostly trifluoromethanesulfonic acid (TFSA)] and open up a new avenue in chemistry. The dramatic use of super acid catalyst in hydroxyalkylation reactions [29], polyhydroxyalkylation [20], and synthesis of hyper-branched polymers [19, 38] was progressively reported.
Phenothiazine and isatin are prolific heterocyclic compounds with inherent electron donor and acceptor nature. Polymer based on phenothiazine core moieties has been progressively investigated in light-emitting diodes [31, 32], photovoltaic devices [33], and chemiluminescence, etc. [34, 35]. Indoline-2,3-dione derivative is one of the attractive electron-deficient moieties and was used to make influential donor/acceptor copolymers for obtaining high performance in organic electronic devices [36, 37]. This may be due to unique features of indole moieties having low-lying frontier orbital energy levels, strongly absorbing in the visible region.
The goal of the present investigation is the synthesis of series of polymers by the reaction of phenothiazine with substituted isatin using super acid catalyst as shown in Scheme 1. The structure and properties of the obtained polymers (P1–P3) were analyzed using spectral and other techniques. The polymerization reaction mechanism and general properties such as solubility, thermal stability, optical and electrochemical behaviors were discussed in detail.

Super acid catalyzed polycondensations of phenothiazine with modified isatin
Experimental
Materials
Phenothiazine (M1, 98%), isatin (M2, 97%), 5-fluroisatin (M3, 98%), 5-bromoisatin (95%) and trifluromethanesulfonic acid (TFSA, 98%) were purchased from Sigma-Aldrich, India. The common solvents such as methanol, ethanol, dichloromethane, DMSO, THF, NMP, Dioxane, cyclohexanone, DMA and DMF (obtained from Sigma-Aldrich, Avra, Acros, India) were also used with or without further purification.
Materials’ characterization
The FT-IR spectrum of polymers was recorded with FT-IR spectrometer (JASCO) in the spectral region from 4000 to 500 cm−1. 1HNMR spectra were recorded at Bruker 400 MHz using d-DMSO solvent and TMS as reference. The thermal analyses of polymers were carried out using Inkarp instrument. The sample weight of 3.5310 mg was loaded in alumina pans and ramped at a heating rate 10 °C per minutes from ambient to 800 °C at nitrogen atmosphere. UV–vis absorption spectra were obtained by a Perkin Elmer Lambda 25 UV/vis spectrometer. For the measurements of thin films, the polymer was drop casted at 90 °C onto pre-cleaned glass substrate from 10 mg/mL polymer solution in DMSO and dried slowly in a Petri dish for 3 h. Gel permeation chromatography (GPC) was performed in THF (HPLC grade, stabilized) at room temperature. GPC analyses were run on an Agilent Technologies system min using a pump and PSS columns (Polymer Standards Service, Germany). The inherent viscosity or intrinsic viscosity of the polymers was determined using a viscometer with NMP as a solvent. The viscosity of pure solvent and polymer solution (polymer dissolved in a solvent) is related to each other, which leads to a relative viscosity value. From this parameter, we can calculate different parameters such as specific viscosity, reduced viscosity and intrinsic viscosity [39]. The values of inherent or intrinsic viscosity of the polymers are presented in Table 1. Cyclic voltammetry measurements were carried out using a CH instrument with standard three-electrode configuration. Typically, a three-electrode cell equipped with a platinum working electrode, an Ag/AgNO3 (0.01 M in acetonitrile) reference electrode, and a Pt wire counter electrode were employed. The measurements were performed using anhydrous acetonitrile in tetrabutylammonium hexafluorophosphate (0.1 M) as the supporting electrolyte under nitrogen atmosphere at a scan rate of 100 mV/s. Polymers’ films were drop casted onto platinum working electrode from a 3 mg/mL in chloroform solution and dried under nitrogen stream prior to measurements. The potential of Ag/AgNO3 reference electrode was internally calibrated using the ferrocene/ferrocenium redox couple (Fc/Fc). The electrochemical onsets were determined at the position where the current starts to change from the baseline. The HOMO and LUMO in electron volts were calculated from the onset of the oxidation potential (Eox) and onset of the reduction potential (Ered) according to the following equation:
Results and discussion
The synthesis of targeted polymers (P1, P2 and P3) using phenothiazine and modified isatin in the presence of super acid catalyst is shown in Scheme 1. The structural, thermal, optical and electrochemical behaviors of obtained polymers were analyzed by different techniques. The monomer, 5-bromohexylisatin (M4), was synthesized by adopting the reported procedure [18, 21, 37, 38]. The detailed polymerization procedures and its characterization were discussed below.
Synthesis of poly 3-(10H-phenothiazin-3-yl) indolin-2-one (P1)
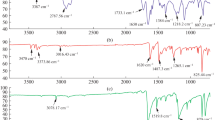
The mixture of phenothiazine (0.1990 g, 0.10 mmol) and isatin (0.1470 g, 0.10 mmol) and dichloromethane (5 mL) was taken in 100 mL two-necked round bottomed flask equipped with mechanical stirrers and ice bath. TFSA (0.5 mL) was added to the reaction mixture and the temperature was maintained around at 27° C and the reaction continued at this temperature until a reaction medium became more viscous in about 2 h. The dark-purple viscous solution was poured slowly into methanol and dark-purple precipitate was obtained. Then, the precipitate was filtered, washed, and re-precipitated with methanol and water. After that, the polymer was dried in vacuum oven at 80 °C for 12 h and purple powder was obtained; yield: 0.3050 g, 88%. The inherent viscosity (ηinh) of the 0.2% solution of the polymer in NMP was found to be 0.65 dL/g. 1H NMR (400 MHz, d-DMSO, δ ppm, Fig. 2a): 10.70 (br, 1H, N–H of isatin), 8.74 (br, 1H of N–H of phenothiazine), 7.21 (br, 2H of 4, 7 positions of isatin), 6.92–6.98 (br, 2H of 2, 10 positions of phenothiazine), 6.78 (br, 2H of 1, 6 aromatic positions of isatin), 6.59–6.62 (br, 4H of 1, 4, 8, 11 positions of phenothiazine). FT-IR (cm−1): 3265 (N–H bending), 3062 (C–H stretching), 1700 (C=O stretching), 1463 (C=C stretching) (Fig. 1).

FT-IR spectra of polymers (P1–P3)
Synthesis of poly (5-fluoro-3-(10H-phenothiazin-3-yl) indolin-2-one) (P2)
TFSA (0.5 mL) was added to ice-cooled mixtures of phenothiazine (0.1990 g, 0.10 mmol), 5-fluroisatin (0.16,510 g, 0.10 mmol) and dichloromethane (5 mL) in a 100 mL two-neck round bottomed flask equipped with mechanical stirrer. After addition, ice bath was removed. After that the temperature gradually increased to 27 °C and the reaction continued under stirring at this temperature until a reaction medium became highly viscous in about 2 h. The obtained dark-purpled mixture was slowly poured into methanol. The precipitate of polymer was filtered, washed and re-precipitated with methanol and water. The dark-purple solution was poured into methanol then filtered and washed with hot methanol. The polymer was dried overnight in a vacuum oven at 80 °C. After drying, dark-purple powder was obtained; yield: 0.328 g, 90%. The inherent viscosity (ηinh) of the 0.2% solution of the polymer in NMP was found to be 0.82 dL/g. 1H NMR (400 MHz, d-DMSO, δ ppm, Fig. 2b): 10.73 (br, 1H, of N–H of 5-fluroisatin), 8.48 (br, 1H of N–H of phenothiazine), 7.42–7.35 (br, 2H of 4, 8 positions of phenothiazine), 6.98–6.90 (br, 3H of 4, 6, 7 positions of 5-fluroisatin), 6.74–6.60 (br, 3H of 2, 10, 11 positions of phenothiazine), 6.21 (br, 1H of 1 positions of phenothiazine). FT-IR (cm−1): 3317 (N–H bending), 3072 (C–H stretching), 1706 (C=O stretching), 1473 (C=C stretching), 1177(C–F stretching) (Fig. 1).

NMR spectra of polymers (P1–P3): a1H NMR spectra of P1 in d-DMSO, b1H NMR spectra of P2 in d-DMSO, c1H NMR spectra of P3 in d-DMSO
Synthesis of poly (5-bromo-1-hexyl-3-(10H-phenothiazin-3-yl) indolin-2-one) (P3)
The mixtures of phenothiazine (0.1990 g, 0.10 mmol), 5-bromo-1-hexylindoline-2,3-dione (0.3101 g, 0.10 mmol) and dichloromethane (5 mL) were taken in 100 mL two-necked round bottomed flask placed in mechanical stirrer and ice bath. TFSA (5 mL) was added slowly to the ice-cooled reaction mixture. After addition, ice bath was removed. Then, the temperature gradually increased to 27 °C and the reaction continued under stirring at this temperature until a reaction medium became highly viscous in about 2 h. The highly viscous dark-purple solution was slowly poured into methanol. The precipitate, dark-purple polymer, was filtered and washed with methanol and water. After that, the polymer was dried in vacuum at 80 °C for 24 h to obtain polymer as in purple powder; yield: 0.428 g, 84%. The inherent viscosity (ηinh) of the 0.2% solution of the polymer in NMP was found to be 0.45 dL/g. The chemical structure of P3 was confirmed by 1HNMR in Fig. 2c. The peak of aromatic ring of isatin and phenothiazine is appeared at 6.0–8.0 ppm and the peak of N–H proton appeared at 9.0–9.5 ppm. The alkyl group in the isatin was appeared at 0.80–2.0 ppm and the N–CH2 protons located at 3.5–4.0 ppm. All the protons are well positioned and it was labeled clearly in the spectra. FT-IR (cm−1): 3319 (N–H bending), 3062 (C–H stretching), 2926 (CH2 stretching), 1705 (C=O stretching), 1472 (C=C stretching), 535 (C–Br stretching) (Fig. 1).
Solubility, molecular weights and thermal properties
There is always a good challenging interest in the synthesis of polymers with fair solubility in common solvents. The solubility is the key parameter in polymerization, purification, characterization and processability (via solution route) and also polymers should be free from defects and impurity which favors the formation of good films. Generally, conjugated polymers are poorly soluble in common organic solvents due their inherent chemical structure. The solubility and processability of polymers were effectively improved by tuning their chemical structure with introduction of solubilizing side substituents (like alkyl group or other functional substituents) in the core moiety. The solubilities of polymers (P1–P3) were tested using different solvents and the results of relative solubilities of polymers are summarized in Table 2. In general, all polymers were soluble in polar aprotic organic solvents such as DMF, DMSO, NMP, THF and DMA. P3 is easily soluble in solvents on comparison with P1 and P2. This is because P3 has hexyl chain in isatin core.
The thermal properties of polymers were explored with the help of thermogravimetric curves as shown in Fig. 3 and the corresponding data are listed in Table 3. The polymers showed no weight loss at low temperature, but exhibited weight loss approximately 10% at 348, 305 and 276 °C for P1, P2 and P3, respectively. Hence, it can be concluded that the three polymers possess good thermal stability under nitrogen atmosphere which is more desirable for application in optoelectronic devices and it may be helpful to avoid device degradation during the long-term operation.

TGA curves of polymers (P1–P3) at a heating rate of 10 °C/min under nitrogen atmosphere
Molecular weight is one of the vital characteristics of polymers and the properties of polymers are significantly dependent on their molecular weight. The optoelectronic properties and device performances were much influenced by degree of polymerization (molecular weight value) and purity. The molecular weights [number average molecular weight (Mn) and weight average molecular weights (Mw)] and polydispersity of polymers are considered to be a fundamental core to investigate property of polymers for various applications [40]. The weight average molecular weight (Mw) and number average molecular weight (Mn) with the polydispersity index (PDI = Mw/Mn) of polymers (P1–P3) are listed in Table 3 and GPC curve is shown in Fig. 4. The molecular weight of the polymers (P1–P3) was in the range of 28,000 to 33,000 and the number of repeating units (degree of polymerization) is from 17 to 62. P1 has high molecular weight compared with P2 and P3 and it may be due to variation in the average number of repeating units. Further, it was found that Mw, Mn and degree of polymerization were reduced with the addition of alkyl side chain in P3. The possible reason may be because the presence of alkyl chain length (P3) will generate steric hindrance in the polymeric chain and or during polymerization reaction. As suggested by the reviewer, the PDI value of P1 and P3 of peak 2 (from Fig. 4) was found to be 1.53 (3775/2456) and 1.54 (3724/2416), respectively.

GPC curves of polymers P1, P2 and P3
Optical properties
The optical properties of the polymers (P1–P3) in DMSO solution and thin films were studied using UV–vis near-infrared (UV–vis NIR) spectroscopy (Fig. 5a–c). All the polymers exhibited typical donor–acceptor absorption behaviors with two absorption bands corresponding to localized π–π* transition and ICT bands, respectively. The polymers showed red shift (16, 36 and 8 nm) and band broadening occurred when transitioning from the solution to film state, especially for P3, reflecting a relatively stronger aggregation of their backbone chains in the films. This may be due to extended π–c conjugation of phenothiazine and isatin [36]. The absorption and emission data along with their optical band gap are given in Table 4.

Normalized absorption and emission spectra of polymers (P1–P3): a P1 in DMSO solution (1 × 10−5 M) and thin film (10 mg/ml) on quartz substrate, b P2 in DMSO solution (1 × 10−5 M) and thin film (10 mg/ml) on quartz substrate, c P3 in DMSO solution (1 × 10−5 M) and thin film (10 mg/ml) on quartz substrate
Electrochemical properties
The electrochemical behavior of the polymers was investigated by cyclic voltammetry (CV). Figure 6 shows the cyclic voltagrammograms of three polymers (P1–P3). It exhibits that all the polymers (P1–P3) showed reversible reduction and oxidation trends with stronger oxidative peaks than reduction. The oxidations was observed with an onset potential at about 1.22, 0.60 and 1.24 V, respectively, for P1, P2 and P3 versus standard and the reductions were observed with an onset potential at about − 0.60, − 0.74 and − 0.58 V, respectively, for P1, P2 and P3 versus standard. The HOMO and LUMO levels were estimated to be − 5.62/− 3.80, − 5.00/− 3.66 and − 5.64/− 3.82 eV for P1, P2 and P3. The detailed electrochemical data of all polymers are listed in Table 5. The slight differences between optical and electrochemical band gaps may be attributed to the interface barrier between polymer film and electrode surface, charge injection and different range of errors. The band gap obtained from electrochemical method should be more significant for optoelectronic device applications [41].

Cyclic voltammograms of P1, P2 and P3 films on Pt electrode in 0.1 M TBAPF6/CH3CN solution at 100 mV s−1
XRD pattern of polymers
The powder XRD patterns of the polymers were measured over the angular range of 5° to 65° at ambient temperature to understand the polymer morphology. The P1 exhibited crystalline nature with more intense and sharper diffraction peaks with smaller π–π* stacking indicating intermolecular interactions between main backbone chains at the same time that P2 and P3 showed amorphous nature in powder form (Fig. 7). From that we can assume that the relative degree of crystallinity and film roughness of the polymers may be able to influence the variations in mobility and performance in device.

X-ray diffraction of P1, P2 and P3
Mechanism of super acid-catalyzed polycondensations
The acid-catalyzed reaction mechanism was one of the focused subjects of theoretical studies [18, 42, 43]. The reaction step such as complex formation between electrophile, aromatic hydrocarbon, and σ intermediate was well explained. On the basis of reported investigation and the generalized polymerization, mechanism of phenothiazine with modified isatin by super acid catalyst was proposed as shown in Scheme 2. The reaction between carbonyl compounds and aromatic hydrocarbon (hydroxyalkylation) continued as electrophilic aromatic substitutions. The process can be catalyzed by protic or Lewis acids. Depending on the monomer structures and reaction conditions, an alcohol, a diaryl compound or a mixture of these can be obtained [20]. The protonation of carbonyl group occurred in the presence of TFSA leading to the attack at carbonyl carbon (1) by electrophilic aromatic substitution of M1 (3). The formed alcohol was protonated and led to another attack of Ar–H (M1), which is produced by diarylisatin derivatives [43].

The mechanism of TFSA catalyzed polycondensations of phenothiazine with isatin. Comparison: reports on the synthesis of series of phenylene-based polyelectrolytes and copolymers were obtained using isatin, 2,2ʹ-biphenol and biphenyl in the presence of super acid catalyst and the resulting polymeric materials were used in the fabrication of proton exchange membrane fuel cell (PEMFC) applications [22, 23, 44, 45]. In this background, herein reported polymers (P1–P2) could be suitable materials for PEMFC and organic electronic device applications
Conclusions
In summary, we reported the synthesis and characterization of a series of linear copolymers of phenothiazine with modified isatin using simple, one-pot, metal-free super acid catalyst. Polymerization was carried out with stoichiometric and direct slow addition of monomers at room temperature in the presence of Brønsted superacid (TFSA). The obtained polymers were soluble in most common organic solvents, preferably polar aprotic solvents and form smooth films on glass substrate by spin casting. The polymer structures were confirmed by 1H NMR and FT-IR analyses. The optical, electrochemical properties and molecular weights of polymers were ascertained. The synthesized new functional polymers exhibited good processability, thermal stability and favorable band gap. The resulted polymer materials could be a good candidate for applications in organic electronic device and in proton membrane exchange fuel cell.
References
Leclerc M, Morin J-F (2017) Synthetic methods for conjugated polymers and carbon materials. In: Stille polycondensation: a versatile synthetic approach to functional polymers, Chap 1, pp 1–58. Suzuki polycondensation, Chap 2, pp 59–95, 1st edn. Wiley, Weinheim
Murugesan V, Bettignies R, Mercier Régis, Guillerez S, Perrin L (2012) Synthesis and characterizations of benzotriazole based donor–acceptor copolymers for organic photovoltaic applications. Synth Met 162:1037–1045
Diniakhmetova DR, Frizen AK, Yumagulova RK (2018) Simulation of potentially possible reactions at the initial stages of free-radical polymerization of styrene and methyl methacrylate in the presence of fullerene C60. Polym Sci Ser B 60:414
Bilbao E, Rodriguez M, Leon LM (1983) Cationic polymerization of N-vinylcarbazole initiated by trityl salts in nitrobenzene. Polym Bull 10:483–486
Hongyuan B, Zhi Z, Hongwei M, Li H, Xiaochun M, Wei H, Pibo L, Yibo Wu (2018) Investigation of the features in living anionic polymerization with styrene derivatives containing annular substituents. Polym Chem 10:1140–1149
Hawker CJ, Bosman AW, Harth E (2001) New polymer synthesis by nitroxide mediated living radical polymerizations. Chem Rev 101(12):3661–3688
Thomas GR, Francesca L, Marco F, Krzysztof M (2019) Atom transfer radical polymerization: billion times more active catalysts and new initiation systems. Macromol Rapid Commun 40:1800616
Bisht HS, Chatterjee AK (2007) Living free-radical polymerization—a review. J Macromol Sci Polymer Rev 41:139–173
Hirao A, Goseki R, Ishizone T (2014) Advances in living anionic polymerization: from functional monomers, polymerization systems, to macromolecular architectures. Macromolecules 47(6):1883–1905
Aoshima S, Kanaoka S (2009) A renaissance in living cationic polymerization. Chem Rev 109(11):5245–5287
Matyjaszewski Krzysztof (2012) Atom transfer radical polymerization (ATRP): current status and future perspectives. Macromolecules 45(10):4015–4039
Grubbs RB (2011) Nitroxide-mediated radical polymerization: limitations and versatility. Polym Rev 51:104–137
Vajjiravel M, Umapathy MJ (2009) Kinetics and mechanism of multi-site phase transfer catalyzed radical polymerization of ethyl methacrylate. Int J Polym Mater 58:61–76
Vajjiravel M, Umapathy MJ (2008) Multi-site phase transfer catalyst assisted radical polymerization of glycidyl methacrylate using potassium peroxydisulphate as initiator—a kinetic study. J Polym Res 15:235–240
Murugesan V, Marimuthu E (2018) Comparative investigation on radical polymerization of methyl and ethyl methacrylate under multi-site phase transfer catalytic conditions. Appl Petrochem Res 8:1–11
Elumalai M, Vajjiravel M (2017) Influence of ultrasonic condition on phase transfer catalyzed radical polymerization of methyl methacrylate in two phase system—a kinetic study. Ultrason Sonochem 38:560–569
Elumalai M, Vajjiravel M (2017) Ultrasonic condition boosts up the rate of phase transfer catalyzed polymerization of acrylonitrile in two-phase system. Appl Petrochem Res 7:85–96
Carmen G, Hernandez M, Mikhail GZ (2009) A high molecular weight aromatic PhOLED matrix polymer obtained by metal-free, superacid-catalyzed polyhydroxyalkylation. Macromolecules 42:9225–9230
Olver LI, Zolotukhin MG, Olivia HC, Fomine S, Cárdenas J, Gaviño-Ramírez RL, Ruiz-Trevino FA (2015) Linear, single-strand heteroaromatic polymers from super acid-catalyzed step-growth polymerization of ketones with bisphenols. ACS Macro Lett 4:492–494
Maria TG, Daniel RN, Serguei F, Salvador LM, Mikhail GZ, Carmen G, Hernandez M, Hans K, Edward SW (2011) Dramatic enhancement of superacid-catalyzed polyhydroxyalkylation reactions. Macromolecules 44:194–202
Hernandez M, Carmen G, Zolotukhin MG, Fomine S, Cedillo G, Morales SL (2010) Novel, metal-free, superacid-catalyzed “click” reactions of isatins with linear, nonactivated, multiring aromatic hydrocarbons. Macromolecules 43:6968–6979
Taewook R, Sabuj SC, Faiz A, Nasrin SL, Soojin Y, Hanmo Y, Seungchan L, Inhwan C, Whangi K (2018) Synthesis and characterization of fluorosulfonyl imide isatin biphenylene block copolymer for PEMFC. Int J Hydrog Energy 43:11803–11810
Taewook R, Hohyoun J, Faiz A, Nasrin SL, Sujin Y, Inhwan C, Whangi K (2018) Synthesis and characterization of polymer electrolyte membrane containing methylisatin moiety by polyhydroalkylation for fuel cell. Int J Hydrog Energy 43:5398–5404
Dong Y, Jie K (2016) 100% Hyperbranched polymers via acid-catalyzed friedel–crafts aromatic substitution reaction. Polym Chem 7:5226–5232
Kricheldorf HR, Zolotukhin MG, Cardenas J (2012) Non-stoichiometric polycondensations and the synthesis of high molar mass polycondensates. Macromol Rapid Commun 33:1814–1832
Cruz AR, Carmen M, Hernandez G, Guzmán-Gutiérrez MT et al (2012) Precision synthesis of narrow polydispersity, ultrahigh molecular weight linear aromatic polymers by A2 + B2 nonstoichiometric step-selective polymerization. Macromolecules 45:6774–6780
Olvera LI, Guzmán-Gutiérrez MT, Zolotukhin MG et al (2013) Novel high molecular weight aromatic fluorinated polymers from one-pot, metal-free step polymerizations. Macromolecules 46:7245–7256
Olah GA (1993) Superelectrophiles. Angew Chem Int Ed Eng 32:767–788
Olah GA, Klumpp DA (2008) Super electrophiles and their chemistry, Chap 5, Wiley, Hoboken, pp 501–750
Olah GA, Surya Prakash GK, Molnar A, Sommer J (2009) Superacid chemistry, Chap 5, 2nd edn. Wiley, Hoboken, pp 501–750
Hwang DH, Kim SK, Park MJ, Lee JH, Koo BW, Kang IN, Kim SH, Zyung T (2004) Conjugated polymers based on phenothiazine and fluorene in light-emitting diodes and field effect transistors. Chem Mater 16:1298–1303
Cho NS, Park JH, Lee SK, Lee J, Shim HK, Park MJ, Hwang DH, Jung BJ (2006) Saturated and efficient red light-emitting fluorene-based alternating polymers containing phenothiazine derivatives. Macromolecules 39:177–183
Kim G, Yeom HR, Cho S, Seo JH, Kim JY, Yang C (2012) Easily attainable phenothiazine-based polymers for polymer solar cells: advantage of insertion of S, S-dioxides into its polymer for inverted structure solar cells. Macromolecules 45:1847–1857
Lai RY, Kong X, Jenekhe SA, Bard AJ (2003) Synthesis, cyclic voltammetric studies, and electro generated chemiluminescence of a new phenylquinoline–biphenothiazine donor–acceptor molecule. J Am Chem Soc 125:12631–12639
Sun D, Rosokha SV, Koich JK (2004) Donor-acceptor (electronic) coupling in the precursor complex to organic electron transfer: intermolecular and intramolecular self-exchange between phenothiazine redox centers. J Am Chem Soc 126:1388–1401
Jian W, Jingde C, Hao H, Shengxia L, Hongwei W, Chao H, Jianxin T, Qing Z (2016) (Z)-(Thienylmethylene)oxindole-based polymers for high-performance solar cells. Macromolecules 49:2145–2152
Zhen L, Cuihong L, Tao F, Guangwu L, Zhishan B (2013) Triindole-cored star-shaped molecules for organic solar cells. J Mater Chem A 1:7657–7665
Yu F, Chantal VO, Annelies V, Agnieszka KB, Xi Z, Andrzej D, Wim D, Mario S (2008) Hyperbranched poly(arylene oxindole)s with a degree of branching of 100% for the construction of nanocontainers by orthogonal modification. Macromolecules 41:2388–2393
Murugesan V, Marimuthu E, Yoganand KS, Umapathy (2017) Multi-site phase transfer catalyzed radical polymerization of methyl methacrylate in mixed aqueous–organic medium: a kinetic study. Int J Ind Chem 8:241–251
Tian Y, Kuzimenkova MV, Halle J, Wojdyr M, Mendaza ADZ, Larsson PO, Muller C, Scheblykin IG (2015) Molecular weight determination by counting molecules. J Phys Chem Lett 6:923–927
Misra A, Kumar P, Srivastava R, Dhawan SK, Kamalasanan MN, Chandra S (2005) Electrochemical and optical studies of conjugated polymers for three primary colours. Indian J Pure Appl Phys 43:921–925
Jiménez Castillo U, Zolotukhin MG, Fomina L, Romero Nieto D, Olivera Garza L, Fomine S (2013) Reactions of ketones with aromatics in acid media. The effect of trifluoromethyl groups and the acidity media. A theoretical study. J Mol Model 19:793–801
Estrella RP, Mikhail Z, Serguei F (2004) Factors enhancing the reactivity of carbonyl compounds for polycondensations with aromatic hydrocarbons. A computational study. Macromolecules 37:6227–6235
Lee S, Lim Y, Hossain MA, Jang H, Jeon Y, Lee S, Jin L, Kim W (2015) Synthesis and properties of grafting sulfonated polymer containing isatin by super-acid catalyzed polyhydroxyalkylation reaction for PEMFC. Renew Energy 79:72–77
Lim Y, Lee S, Jang H, Hossain MA, Choi S, Cho Y, Lim J, Kim W (2015) Synthesis and characterization of pendant propane sulfonic acid on phenylene based copolymers by super-acid catalyzed reaction. Renew Energy 79:85–90
Acknowledgements
This work was supported by the Science and Engineering Research Board (DST-SERB), New Delhi under the start-up research Grant No.SB/FT/CS-008/2014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest regarding the publications of this research article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kasi, B., Murugesan, V. & Kaliaperumal, N. Super acid-catalyzed polymerization of phenothiazine and modified isatin. Appl Petrochem Res 9, 91–100 (2019). https://doi.org/10.1007/s13203-019-0229-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-019-0229-x