
Abstract
Owl pellets have high potential as a source of DNA. However, this noninvasive method of collecting DNA is rarely used, and its methodological aspects are poorly understood. We investigated the methodology for DNA extraction and amplification from owl pellets containing the smallest European rodent—the Harvest mouse Micromys minutus—as an example. We used mandibles identified in owl pellets for mitochondrial and nuclear DNA amplification. For DNA extraction, we tested two commercial protocols and utilized a protocol being a combination of two commercial kits which ensured high efficiency of DNA extraction. Additionally, we recorded that the amount of DNA was five times higher in extracts from teeth as compared to DNA extracts from jawbones derived from the same mandible. The quantity of DNA was significantly positively correlated with biological sample weight; however, the age of the pellet remains had an impact on the level of inhibition. We recorded inhibition in 40 % of mtDNA extracts derived from pellets older than 150 months, whereas in DNA extracts from pellets younger than 80 months, we did not observe a negative impact of inhibition on PCR efficiency. The amplification success rate was 89.9 % for the mitochondrial fragment and 39.4 % in the case of the nuclear fragment. We observed partial degradation of DNA evidenced by the fact that the longest fragments that we were able to amplify in the case of mtDNA were 450 and 200 bp for nuDNA. The study shows that pellets can be considered as a source of DNA and have high potential for molecular research in the case of threatened species and species that are difficult to study using standard field techniques.
Similar content being viewed by others

Introduction
The efficiency of field sampling is one of the most basic problems in population genetic studies. Financial, time, and human resources necessary for the extended field sampling can seriously limit the effectiveness of research and application of results in conservation actions. Acquiring biological material for genetic research in the case of rare, cryptic, difficult-to-detect, and man-avoiding species can be a very serious logistical problem, is expensive, and is time consuming. Moreover, in the case of small organisms (e.g., invertebrates), sampling related with genetic studies is commonly associated with killing these animals for the purpose of DNA extraction (e.g., Wyngaard et al. 2010), which in the case of rare species should be avoided if possible. Additionally, some methods of sampling are unspecific, and many non-target animals can be affected (i.e., trapped and killed) in such studies. Pitfall traps in the case of invertebrates and snap traps in the case of small mammals are good examples of such methods. The destructive sampling—commonly applied not only in molecular ecology but also predominantly in the case of faunistic explorations—seems to receive increasing attention, and its consequences at the population level should be considered (Żmihorski et al. 2013). Therefore, methods of noninvasive sampling of biological materials collected for the purpose of molecular investigations have received much attention recently, and several such techniques have been proposed so far (Taberlet et al. 1999; Broquet and Petit 2004). Among the most popular and effective methods, one can mention DNA extraction form fecal pellets of ungulates, from feces of carnivores, from water samples in the case of aquatic fauna, from hairs collected in hair traps, from urine, and many others (Kohn et al. 1999; Valiere and Taberlet 2000; Kohn and Wayne 1997; Frantz et al. 2004; Balestrieri et al. 2011).
Among the methods of noninvasive sampling, pellets of diurnal and nocturnal birds of prey seem to fit well to molecular investigations. Pellets are formed from the bones, teeth, fur, feathers, and some other harder parts of the prey. Compared to pellets of diurnal birds of prey, owl pellets contain a higher proportion of skeletal remains, which are undigested in the owls' tract and rejected usually one to two times per day (Raczyński and Ruprecht 1974). Owl pellets have high potential as a source of DNA, especially in the case of rare and cryptic species, for several reasons. First of all, owls seem to be very effective samplers, especially generalists such as the Tawny owl (Strix aluco). The owls use various hunting techniques, utilize different habitats, and therefore kill a wide range of prey, both in the taxonomical and ecological sense (Mikkola 1983). The expected number of prey species per number of sampled individuals seems to be higher as compared to standard live-trapping (Żmihorski et al. 2011). Therefore, it is more probable to record some rare species in pellets than during standard trapping sessions (for the total number of recorded individuals kept constant). In Lithuania, it was found that the share of the European Harvest mouse in the prey remains of the Tawny owl was in the range 0.3–1.2 % (Balčiauskienė et al. 2005) and 2.2 % in pellets (Balčiauskienė et al. 2006). The skeletal material originating from owl pellets is widely available in a great number of zoological collections. Secondly, this method is absolutely noninvasive, as we use remains of preys that had already been killed by their natural predators, so there is no ecological footprint of studies based on pellets. Therefore, the method can be used in, e.g., protected areas. Despite all those advantages, the use of pellets as a source of DNA is surprisingly rare and has received relatively little attention up to now. In scientific literature, only few papers use this technique, and several methodological aspects remain poorly understood. It seems that Taberlet and Fumagalli (1996) proposed for the first time that owl pellets could be used as a source of DNA of their prey. Two recent papers used this method for analysis of spatial variability of the genetic structure of voles (Jaarola and Searle 2004) and shrews (Poulakakis et al. 2005). However, our knowledge on owl pellets as a source of DNA is still incomplete, and one may conclude that this method is strongly underrepresented among other techniques of noninvasive sampling of genetic material.
Methodological approach to DNA extraction from skeletal remains from owl pellets is similar to those applied in case of museum and ancient DNA studies. This kind of biological material constitutes a methodological challenge due to DNA limited quantity, degradation, presence of inhibitors, and risk of contamination increasing with age of sample (Wandeler et al. 2007). Additionally, museum and ancient material is usually scarce, and every sample is of high importance. Therefore, in case of such material, extraction constitutes the most important step, which has direct impact on quantity and quality of DNA and finally on genotyping results obtained during sequencing or microsatellite loci analyses (Wandeler et al. 2003; Morin and McCarthy 2007). Consequently, different standard methods of extraction and commercial protocols modifications have been developed for such material (Rohland et al. 2004; Morin et al. 2007; Casas-Marce et al. 2010). Nevertheless, improved approaches to DNA extraction from old samples are arising continuously (Shapiro and Hofreiter 2012; Buś et al. 2013) leading to reduction of costs and increases information gain from such material.
In this study, we made an attempt to extract DNA from the remains of the Harvest mouse Micromys minutus (Rodentia, Muridae) found in owl pellets, in the context of investigations into the methodology for using pellets as a source of DNA of small mammals. We used the Harvest mouse as it is the smallest rodent species in Central Europe—we expect therefore that DNA extraction from its remains should be more difficult as compared to other, larger, species. We concentrated on the laboratory methodology and factors limiting the effectiveness of DNA extraction and amplification of mitochondrial and nuclear DNA fragments. More specifically, we aimed to (1) test a protocol of DNA extraction from mandibles, (2) compare the efficiency of DNA extraction from jawbones and teeth separately, (3) check the level of inhibition in extracts, (4) measure the impact of weight and age of samples on the amplification success rate, and (5) assess the amplification success rate and level of fragmentation of DNA in the case of selected fragments of mtDNA and nuDNA. In general, we wanted to test and select the most effective methodology for using owl pellets as a source of DNA of prey species in the hope that description of the methodological protocol of this noninvasive sampling can facilitate acquisition of new data on the molecular characteristics of populations of many mammal species.
Materials and methods
Characteristics of biological samples
In the analyses, we used mandibles defined as a jawbone with teeth (one lower incisor and three molars, Fig. 1) of the Harvest mouse originating from pellets of Tawny owls, S. aluco, and Barn owls, Tyto alba, found in Poland and Lithuania. Pellets were collected during all seasons and in different environments—majority of them were found below trees used by owls for breeding and roosting, and some were collected in buildings (barns, churches, etc.). Unfortunately, as the oldest pellets were collected over 16 years ago, exact environmental conditions of collection of some of them are not exactly known. Date of collection of each pellet was used for age assessment. After the collection, the pellets were stored in dry place and used for owl's diet analysis according to standard protocols (Raczyński and Ruprecht 1974). For the purpose of this study, mandibles of the Harvest mouse were selected from previously examined material. The mandibles were identified to the species level on the basis of morphological traits (Pucek 1981; Yalden and Morris 1990, reference collection) and weighted with accuracy of 0.001 g. The mandibles were ca. 11 mm in length and, in almost all cases except one, contained molars and an incisor (Fig. 1). Hereafter, the mandibles with teeth used for all further analyses described in the manuscript are referred to as samples. In cases when more than one mandible from a given locality were available, we used only right or left ones to avoid including one individual twice. Mandibles that were not used for the analyses were used for preliminary testing of the method. To prevent moisture and to slowdown the degradation process, mandibles were packed in separate paper envelopes and stored in a ventilated, dry place. DNA was stored at 4 °C during performing of molecular analyses and at −20 °C after completing the study.

Mandible of the Harvest mouse Micromys minutus used for the molecular analyses with three molars and an incisor marked. A and B visualize the lateral and top view of the molars, respectively
Prevention of contamination during molecular analyses
Extraction of DNA and PCR preparation was performed in physically isolated facilities. Furthermore, all working areas and laboratory equipment between each experiment were regularly UV-irradiated and cleaned using sodium hypochlorite (bleach). Additionally, appropriate protective clothing like lab full protective coats, facemasks, and gloves were used to avoid contamination. Only one extract was obtained from a given sample, but at least two independent PCR reactions were conducted from each extract. Additionally, independent sequencing was carried out for each PCR product, which allowed confirmation of the reliability of the observed haplotypes. All analyses (extraction, amplification, quantification, and sequencing setup) were performed with numerous negative controls.
DNA extraction and quantification
In general, we performed extraction of DNA on three types of biological samples: (1) mandible, jawbone with teeth; (2) teeth, separated from jawbone; and (3) jawbone, bone with teeth removed. The two last types of samples (i.e., teeth and jawbones) were derived from the same mandible (i.e., one individual). At the beginning, two extraction protocols were tested separately: extraction of genomic DNA from bones using the QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) and the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA). Twenty mandibles characterized by a similar mass varying from 0.010 to 0.012 g were chosen for the test. Due to the fact that the extraction and amplification yield in the case of both protocols was unsatisfied, a new protocol as a combination of the two with own modifications was created and tested on 109 mandibles. To eliminate surface contamination, the biological samples were soaked in 15 % bleach for 10 min. Next, each mandible was rinsed twice with deionized water (dH2O), dried with a powder-free tissue, and powdered in a mortar. Total powder obtained from an individual mandible was used for DNA extraction. The powdered mandible was placed into 1.5-ml Eppendorf tubes. Before the extraction procedure, 500 μl of 0.5-M, pH 7.5, EDTA (ethylenediaminetetraacetic acid) was added to each sample and incubated overnight. Next, 300 μl of buffer ATL (QIAamp DNA Micro Kit) and 30 μl of proteinase K was added to each sample and incubated at 56 °C for 12 h. Then, the salting-out procedure was employed for extraction of DNA, according to the animal tissue (Wizard® Genomic DNA Purification Kit) protocol with minor modifications like overnight precipitation of DNA with the isopropanol. Dried genomic DNA was dissolved in 200 μl of a rehydration solution and subsequently purified according to the purification of genomic DNA from bones commercial protocol, the QIAamp DNA Micro Kit (Qiagen). Incubation of DNA bound to QIAamp MinElute column (Qiagen) with the elution buffer AE lasted 3 min, and finally, elution step was conducted in 50 μl of buffer AE. Finally, using the new protocol, we measured DNA extraction efficiency from jawbones and teeth obtained from the same mandibles (n = 7). For this purpose, we weighted each mandible and then the teeth were removed. Next, we weighted jawbone and teeth separately, and both types of material were used for DNA extraction.
Quantification was performed with the help of the Qubit® 2.0 Fluorometer with Qubit® dsDNA HS Assay Kit (Life Technologies, Carlsbad, CA, USA). The concentration of DNA in particular extracts was expressed in nanograms. In three cases, the fluorometer recorded too low concentrations of DNA; therefore, for such samples, we assumed the lowest limit of detection, which is 0.0005 ng/μl, following the lowest detectability level declared by the producer.
Amplification of mitochondrial and nuclear DNA fragments
Fragments of the control region of mitochondrial genome (mtDNA) and nuclear (nuDNA) RAG1 gene were amplified. To take into account potential degradation of the DNA strand, several primer pairs generating short and longer fragments were tested to obtain a product of PCR amplification (Table 1). Primers were designed based on sequences derived from NCBI (http://www.ncbi.nlm.nih.gov/), control region accession number: AB202062.1, and for RAG1 gene: EU349904.1 using Primer 3 software (Rozen and Skaletsky 2000). All the fragments were tested in a wide range of melting temperature from 48 °C to 60 °C, and the longest amplified fragment was chosen for further analysis. For both mtDNA and nuDNA, the PCR reaction mixture contained 2.5 U of HotStarTaq Plus Master Mix Kit (Qiagen, Hilden, Germany), 0.2 μM of each primer, 1 mg/ml BSA, and 25 % glycerol in a total volume of 50 μl. To take into account potential presence of inhibitors in the DNA extracts, at the beginning, we did not normalize the amount of DNA in each PCR reaction. First, the same volume of 7 μl of the extract containing from 0.003 to 11.6 ng of DNA was added to each reaction. Subsequently, in samples in which the lowest concentration of DNA was detected, we gradually increased the volume of DNA until a strong band of PCR product guaranteeing a good result of sequencing was obtained. In general, two to four PCR reactions from the same extract were set up at the same time together with several negative controls. Samples where no result of amplification was observed, even after increasing the amount of DNA and several repetitions of PCR, were subsequently diluted to check if presence of inhibitors could prevent the amplification. PCR was performed in a Veriti® Thermal Cycler (Life Technologies, Carlsbad, CA, USA) by 10-min initial denaturation at 95 °C, followed by 40 cycles of 45 s at 95 °C, 45 s at 48 °C in the case of control region and at 53 °C for RAG1, and 60 s at 72 °C. The program was completed by a final extension step at 72 °C for 10 min and a final hold at 4 °C. All products of amplification were visualized in 2 % agarose gel with Midori Green DNA Stain (Nippon Genetics Europe, Düeren, Germany).
Sequences analysis
Purification of the PCR products was performed using the Clean Up (A&A Biotechnology, Poland). Each product was eluted in 50 μl of dH2O. Forward and reverse sequencing was performed using the Big Dye Terminator v3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA, USA) and the PCR primers as sequencing primers. Sequence analysis was performed on a 3500xL Genetic Analyzer (Life Technologies, Carlsbad, CA, USA). Data were analyzed using the Sequencing Analysis v5.4 (Life Technologies, Carlsbad, CA, USA) and BioEdit 7.1.3 (Hall 1999). BLAST (http://blast.ncbi.nlm.nih.gov/) was used for verification and confirmation if the sequences obtained belong to appropriate species and for elimination of the suspicion of contamination.
Statistical analyses
We compared the quantity of DNA extracted from the teeth and jawbones with the help of paired samples t test.
We modeled the quantity of DNA with the help of the general linear model (GLM). We used the GLM with Gaussian error distribution and used the sample age and weight as two linear predictors of the DNA quantity.
As we did not measure the presence of inhibitors directly, we used an indicator of inhibition. The samples that did not amplify after several repetitions of PCR were diluted (1:10, 1:20, 1:30, 1:50, and 1:60). If products of amplification appeared after the dilution, we assumed that in such samples, inhibitors were present. Following this assumption, we divided all the samples that were successfully amplified to those inhibited (i.e., diluted and then successfully amplified) and remaining (amplified without dilution). We used GLM with binomial error distribution implemented in R (R Core Team 2012) to study which characteristics of the samples determine the presence of inhibitors. We used presence of inhibitors as a binomial response variable (i.e., present vs. absent), whereas the age of the samples, the weight of the samples, and the quantity of DNA were used as explanatory variables.
Finally, we made an attempt to study the success of amplification of mitochondrial and nuclear DNA as a function of sample age and weight. For this purpose, we used the GLM with binomial error distribution implemented in R (R Core Team 2012). We conducted the modeling for mitochondrial DNA and nuclear DNA independently (two GLMs were computed).
Results
Quantity of DNA
The test of the extraction protocols indicated that in the case of Qiagen Kit, on average, 8.15 ng (SE = 0.39, i.e., 815 ng/g of mandible) of DNA was obtained per sample, whereas the Promega protocol provided on average 19.20 ng (SE = 0.455, i.e., 1,920 ng/g of mandible) of DNA, and the difference was highly significant (t test, t = 18.35, df = 17.65, p < 0.0001; Table 1). A higher level of inhibition was observed with the use of the Promega protocol, where in all the cases PCR products were obtained only after dilution of all the extracts 1:50. In the case of Qiagen, only in three extracts the 1:30 dilution was sufficient to receive results of amplification. After combination of both protocols (see “Materials and methods” section for details), the mean efficiency of the isolation using the new protocol reached 1,481.8 ng/g of the mandible, and the extracts were characterized by a lower level of inhibition. Generally, the amount of extracted DNA varied between 0.025 and 79.0 ng per mandible, but that it is worth to emphasize that the mass of particular mandibles was variable (from 0.001 to 0.035 g).
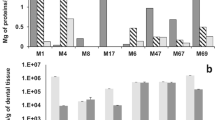
The mean mass of the teeth derived from the seven mandibles used in the comparison was 0.0037 g, whereas the mass of the jawbone (i.e., the mandible with teeth removed) was 0.0060 g, and the difference was significant (paired samples t test, t = 2.94, df = 6, p = 0.0260). The mean DNA quantity extracted from the jawbone was on average 1.086 ng per sample (ranging from 0.250 to 3.200 ng for particular samples), whereas the quantity of DNA extracted from the sole teeth was on average 5.000 ng per sample (from 4.550 to 5.800 ng), and the difference was highly significant (paired samples t test, t = 6.75, df = 6, p = 0.0005; Fig. 2). The jawbones were heavier than the teeth; therefore, the efficiency of the isolation per standardized unit of the sample was much higher for the latter—it amounted to 1,351.3 ng/g of the teeth sample and 181.0 ng/g of the jawbone sample (Table 1).

Mean (and 95 % CI for the mean) quantity of DNA per sample extracted from seven jawbones and seven teeth of the Harvest mouse Micromys minutus found in owl pellets
We found that the quantity of DNA did not depend on the age of the samples (GLM, estimate = −0.02; SE = 0.02; t = 1.40; p = 0.166); however, a highly significant effect of sample weight was recorded (GLM, estimate = 1,341.2, SE = 267.66; t = 5.01, p < 0.0001; Fig. 3). The weight effect was still highly significant after excluding the three outliers (marked in Fig. 3 with asterisks; GLM, estimate = 1,688.0, SE = 247.5; t = 6.82, p < 0.0001).

Quantity of DNA (nanograms) per sample extracted from mandibles of the Harvest mouse Micromys minutus found in owls pellets in relation to the weight of sample. The three outlying observations are marked with asterisks
Presence of inhibition
In amplification of the RAG1 gene fragment, dilution (1:10) was necessary for two samples (this value is not indicated in Table 1). Therefore, we assumed presence of inhibition in the case of these extracts. It is worth to emphasize that in the case of these two samples, dilution was also necessary in mtDNA (i.e., control region) amplification. However, the success of amplification in the case of nuDNA was low; therefore, we analyzed the effect of inhibition on the basis of mtDNA (see Table 1 and GLM procedure in the following paragraph). To receive products of control region amplification, even after purification of DNA with the Qiagen column, dilution 1:10 was still necessary in ten extracts. However, for 11 samples, no amplification was observed even after dilution 1:60, which suggested that a huge level of inhibitors was still present in the extracts.
Presence of inhibition in samples where the control region fragment was amplified did not depend on the quantity of DNA (GLM, estimate = −0.031, SE = 0.034, z = 0.91, p = 0.3615) nor on the weight of the sample (GLM, estimate = 139.646, SE = 88.487, z = 1.58, p = 0.1145). However, the effect of the sample age displayed a statistically significant positive correlation with the presence of inhibition (estimate = 0.014, SE = 0.005, z = 2.81; p = 0.005). There were no cases, except one, of inhibition in samples younger than 80 months, but in samples older than 150 months, the share of samples with inhibitors constituted 40 % (Fig. 4).

Presence of inhibitors in the samples in relation to the sample age. The curve shows logistic fit from the GLM; random noise along the y-axis was added to avoid symbol overplotting
Success of amplification
We did not observe amplification products longer than 450 bp in the case of mtDNA and 200 bp in the case of nuDNA, which indicated that fragmentation of the DNA strand occurred as a result of degradation. The amplification success rate amounted to 89.9 % for the control region fragment and 39.4 % in the case of the RAG1 fragment (Table 1). Comparison of the results obtained to sequences of the control region and the RAG1 gene of M. minutus deposited in BLAST showed up to 99 % identity.
In the case of mitochondrial DNA, all three predictors of success of amplification were insignificant (p > 0.149 in all cases); however, after removing the least significant explanatory variable, i.e., the weight of the sample, the quantity of DNA appeared to be significant (GLM, estimate = 0.086, SE = 0.042, z = 2.045, p = 0.0408; Fig. 5), whereas the age of the samples remained insignificant (GLM, estimate = −0.006, SE = 0.006, z = 1.108, p = 0.2680).

Probability of amplification success in relation to the quantity of DNA. The curve shows logistic fit from GLM; random noise along the y-axis was added to avoid symbol overplotting
In the case of the nuclear fragment of the RAG1 gene, none of the three hypothetical predictors appeared to be significant (p > 0.471 in all cases)—the success of amplification of the nuclear gene did not depend of the sample weight, sample age, or the quantity of DNA.
Discussion
There are many time-consuming points in molecular analyses based on samples containing a low amount of degraded DNA; therefore, an extraction method providing a satisfying yield within a short time is of high importance, especially in the case of population genetic research where many samples should be examined. The use of commercial kits for extraction with the manufacturer's protocol is time efficient and is very often applied to extract DNA even from such difficult material as museum specimens (Rohland et al. 2004) or ancient DNA (Rohland et al. 2009). Additionally, this is a simple technique for removal of PCR inhibitors from bone extracts, which are considered as one of the most difficult material for molecular analysis because of the presence of many substances blocking the PCR reaction (Kemp et al. 2006). Unfortunately, it has been frequently observed that column-based extracting methods are fast in comparison with, e.g., the isopropanol protocol but yield a rather low concentration of DNA (Rohland et al. 2004). Our preliminary test of extraction efficiency confirmed this observation. Promega kit provided a definitely higher concentration of DNA in comparison to the Qiagen protocol, but the inhibition level was much higher for the former. This could be explained by the fact that the approaches to extraction in both protocols are completely different. The Promega procedure is based on alcohol precipitation, whereas the Qiagen method on silica membrane purification and elution of DNA. These differences could have an important impact on the yield of extracted DNA. In the case of the column-based method, binding of DNA to silica is performed only for a few seconds (Rohland and Hofreiter 2007). This could be enough to recover the high yield of extraction when fresh material serves as a source of DNA. In the case of degradation or small number of copies, there are some limitations of this method, e.g., copies still remain in silica matrix even after the elution step, which could have a huge impact on the final concentration of DNA. In the precipitation method, most of the DNA copies contained in the alcohol mixture form a strong pellet, which is subsequently washed using ethanol or isopropanol; this ensures that the loss of DNA copies is much lower in comparison to the silica column method (Rohland et al. 2004). Nevertheless, in the case of our test, it was not the extraction yield that was problematic but the presence of inhibitors. Qiagen silica columns yielded a lower number of copies but, in comparison to Promega method, were more effective and by far better in elimination of inhibitors from the extracts. Finally, we combined the best points of the two commercial kits and purified DNA extracted with the Promega protocol using a silica column from the Qiagen kit. One of the most important advantages of this solution is the ability to concentrate of the low copy number in the extract using a silica column. The volume of the final elution can be decreased without fear that inhibitors will have significantly higher concentration as well. The new combined protocol was characterized by good efficiency of amplifiable DNA and a low level of inhibition. There was no need to dilute most of the samples to obtain mtDNA PCR products; however, in the case of some extracts, we still had to apply dilution 1:10. Curiously, we observed inhibition mainly in extracts older than 150 months. In very old, for example, ancient bones or teeth, numerous substances such as humic and fulvic acids, tannins, porphyrin products, phenolic compounds, hematin, and collagen type I are commonly present acting as strong PCR inhibitors (Eilert and Foran 2009). Our result indicates that the quantity of inhibition level increases with the age of the sample and reaches a relatively high level for the owl pellets samples older than 150 months.
An obvious advantage of using noninvasively collected material as a source of DNA is its availability in the field and the fact that it can be collected relatively frequently. Frequent collection of biological material for molecular study heightens the probability that the isolated DNA will be characterized by high quality. However, many molecular researchers claim that amplification of DNA extracted from this kind of biological material is less efficient than of DNA isolated from freshly sampled tissues (Frantzen et al. 1998; Gagneux et al. 1997). Numerous factors influence the amplification rate, among which the amount and quality of the isolated DNA are the most predominant. Usually, from 1 ng to 1 μg of DNA, i.e., from 300 to 300,000 copies, are used in PCR. Sometimes, only a few picograms of DNA could be obtained from noninvasively collected samples (Taberlet et al. 1996). Taberlet et al. (1996) and Taberlet and Luikart (1999) reported that in order to obtain a high amplification success rate and to determine animal genotypes with 99 % reliability, one should have at least eight diploid cells, i.e., over 56 pg of mammalian DNA. It is worth to emphasize that we extracted a relatively high amount of DNA, i.e., 1,481.8 ng/g of a mandible. Our favorable results could be explained by the rather young age of the samples. For example, Pajnič et al. (2012) extracted up to 131 ng DNA per gram of powder from bones and teeth, but the authors investigated ca. 80-year-old human biological material excavated from mass graves. In other research, the average yield of DNA from mammal remains was <50 ng of DNA per gram of bone used for extraction, but the results were obtained from ancient remains of a horse and wolf mandible, dated by radio-thermoluminescence to 40,000–50,000 years before present (BP) (Blow et al. 2008).
Our results indicate that DNA is much better preserved in teeth, although we demonstrated that jawbones are a good source of DNA as well, which is important information in the case of incomplete remains present in pellets. The result suggests that identification of species on the basis of bones (e.g., jawbones without teeth) is still possible, which may be important in the case of rare species and/or incomplete pellet samples.
One should also take into account the fact that the small amount of DNA in the biological material found in the field is caused by degrading factors. The higher the exposition of genetic material to such factors, the worse the quality of the isolated DNA is. Fortunately, skeletal remains in owl pellets are rather excellently protected against one of the most potent and directly affecting environmental degradation factors as UV irradiation. This leads to a conclusion that there is a high probability that DNA extracted from bones or teeth derived from pellets should be characterized by far better quality in comparison with scats, feathers, urine, etc. The results obtained in the present study support this claim partially because the success of amplification was different depending on the type of molecular markers (mtDNA vs. nuDNA) used. Firstly, amplification of the 450-bp mtDNA fragment and the high success of amplification (almost 90 %), irrespective of the sample age, indicate only a partial and slow pace of mtDNA degradation in the analyzed material. For example, in the case of the most frequently tested noninvasive samples, i.e., feces, Vigilant (1999) obtained amplification in the case of 80 % of the samples, but only when the mtDNA fragments did not exceed 300 bp; for the 400-bp fragments, amplification was observed in 60 % of the extracts, whereas for 500 bp, the amplification success rate was 15 %. However, it was demonstrated that degradation was still observed even in the case of well-protected DNA from remains derived from pellets. Poulakakis et al. (2005) examined the DNA of the shrew from barn owl pellets and amplified only 300 bp fragments. Using owl pellets as a source of DNA of small mammals, Taberlet and Fumagalli (1996) demonstrated that it was possible to amplify DNA fragments longer than 1 kb, but the amplification success rate was by far lower than that observed in the case of the 300-bp mtDNA fragment. The above-mentioned results are not surprising since, as claimed by Pääbo (1989), fragments of degraded DNA are usually 40–500 bp long, but most frequently they do not exceed 200 bp. This principle has especially applied to nuclear DNA degrading faster than mitochondrial DNA. Following this assumption, we did not receive RAG1 fragments longer than 200 bp. Additionally, the differences in the success of amplification between mtDNA and nuDNA are not a surprising result either. Degradation of DNA begins during apoptosis, but this process together with environmental factors and specific features of mtDNA and nuDNA can result in considerably different patterns of double strand fragmentation in both cases. Two copies of nuclear DNA are present in a single cell, whereas the mitochondrion contains 1–15 copies of mitochondrial DNA, and in a single somatic cell, up to 107 of these organelles could be present (Satoh and Kuroiwa 1991). This main characteristic makes mtDNA much more resistant to the impact of degradation factors; hence, by choosing mitochondrial markers, a higher amplification success rate and longer fragments can be obtained in extracts from material collected in the field.
We admit that in case of other noninvasively collected samples, amplification success rate, especially in the case of nuDNA, could be higher as well as longer fragments can be amplified (e.g., Zemanová et al. 2011; Reddy et al. 2012) as compared to our results. However, in case of the studies mentioned above, only fresh feces (<12 h old) and/or collected at low temperatures, usually from snow cover, were used for DNA extraction. Höss et al. (1996) studies show that cold environment could have critical importance for the rate of DNA degradation. A decrease in environment temperature of 20 °C can cause the 10- to 25-fold reduction in the rate of chemical reactions such as the decay of nucleotide bases. Hence, it must be taken into account that many various factors (especially environmental) can affect extraction and amplification at once. The most important in our opinion could be the type of tissues and their resistance to various degrading factors. For example, in case of feces samples, fresh cells (derived from exfoliated epithelial intestinal tract) could be a source of well-preserved DNA, which may result in high efficiency of DNA extraction and in low level of double strand fragmentation. Here again, of course, one should remember that this holds on for fresh (or frozen) stool because environmental factors (e.g., UV radiation and humidity) can greatly modify the efficiency of both isolation of DNA and amplification (Shapiro 2008). In turn, in our research, other factors could have led to fragmentation of DNA because pellets pass through the bird's gastrointestinal tract where they are subjected to the digestive enzymes. However, the impact of digestion process on DNA degradation level can vary significantly. Carnivores ground prey bones, which makes the hard tissue more susceptible to enzymatic juices, whereas in case of owls, whole bones pass through the digestive track, and are additionally protected by the mix of hairs and feathers. Nevertheless, it is difficult to definitely say how the digestive enzymes affect the degradation of DNA in the owl's pellets. It seems that the environmental factors could have the greatest impact on the success of the amplification and length of amplified DNA fragments in the case of our results. Moreover, the possible impact of bacteria and fungi on degradation of double strand should be also taken into account. Studies of ancient and museum specimens show that all of the factors mentioned may cause that samples do not contain DNA useful for amplification, or genetic material occurs only in the form of short-length fragments (see Willerslev and Cooper 2005; Rowe et al. 2011 for thorough review). In case of our study, the pellets collected in the field were stored in dry conditions, which probably caused the slowing of the degradation process.
Interestingly, in our study, the age of the pellets (up to 190 months, i.e., nearly 16 years old) had no significant effect on the quantity of DNA. This result could be rather hardly achievable in the case of other noninvasively collected samples as feces or hair where hydrolytic and oxidative damage could act quickly and cause total destruction of the DNA structure even after a couple of months (Piggot 2004). Furthermore, the effect of weight was highly significant—the quantity of DNA increased by 20 ng with the increase in the sample weight from 0.001 to 0.016 g. This means that the general conclusion drawn from our study is simple—the higher the amount of powdered bone will be used for DNA extraction, the higher the efficiency of extraction could be expected. However, it seems that this source of variation was not taken into account in some studies. For example, Taberlet and Fumagalli (1996) compared the efficiency of two extraction methods (chelex and silica), but in the case of chelex, they added <5 mg of bone powder to the extraction protocol, whereas in the case of the silica method, <10 mg powdered bone was used. Possibly, this is the reason why in the case of the quoted research the silica procedure gave 32 positive results of PCR out of 33 attempts, whereas the chelex procedure gave only eight positive results out of 14 attempts. Additionally, the authors emphasized that they were not be able to amplify mtDNA fragments in three cases out of eight using the chelex procedure, despite the fact that strong PCR products were obtained in the same samples when a given sample was extracted with the silica protocol. Obviously, the results observed by Taberlet and Fumagalli (1996) could in fact depend on the chemical properties of the reagents used in the particular protocol, but it cannot be excluded either that the principle concerning weight of powdered bone vs. the extraction efficiency correlation also contributed to the final result in this case. Definitely, the principle could be applied rather when the extraction protocol proposed in this research is employed. Moreover, in the case of the presented protocol, EDTA demineralization was applied, which may have decreased the yield of extraction. The EDTA both decalcifies the bone (to an extent dependent on the EDTA concentration and the volume of extraction buffer) and inactivates DNAses by chelating bivalent cations such as Mg++ or Ca++ (Loreille et al. 2007).
Our studies seem to be very important from the point of view of surveys based on DNA extracted from museum specimens or ancient bones samples. In such studies, the quantity of DNA frequently has crucial significance for the amplification success. Our study suggests that, if possible, more bone should be used for extraction. On the other hand, some research proved that using 200 mg of powdered bone had an impact on the decrease in the yield of extraction and amplification (Rohland et al. 2009).
The quantity of DNA may not be the only factor affecting the PCR efficiency. As a source of DNA, bones are regarded as one of the most challenging biological materials mainly due to naturally occurring inhibitors such as collagen, Ca++, or humic acids (Eilert and Foran 2009). Inhibitors can inactivate polymerase, compete with, or bind other PCR reaction components; hence, no amplification results are observed beside the presence of DNA in the extract. From this point of view, it is possible that a high level of inhibition was present in the case of our nine extracts. There are a couple of methods to overcome inhibition that can be applied at different stages beginning from the extraction protocol through optimization of PCR parameters. Using a dilution is one of the simplest solutions (Loreille et al. 2007; King et al. 2009).
From our point of view, mandibles from pellets could constitute an additional of noninvasively collected source of DNA. The biological material is quite well preserved and protected against degradation factors, which results in high efficiency of PCR, especially in the case of mitochondrial DNA. Additionally, the age of the sample did not have an impact on the amount of DNA extracted and success of amplification; however, the age of the sample had an impact on inhibition level, which may affect the success of amplification in older samples. This result is important as it shows that bones or teeth deposited, for example, in museum collections could be potentially used as a source of DNA and provide genetic information about historical populations, extinct species, etc. Obviously, it is much better to investigate mitochondrial DNA rather than nuclear DNA. The latter is characterized by a higher degradation rate, but we can still obtain information from long fragments applying a strategy of amplification of short overlapping fragments (Alonso et al. 2003). We believe, however, that obtained results of amplification of nuclear DNA constitute some kind of guidelines to help us to choose appropriate length of additional nuclear markers, which we plan to analyze in the next stages of DNA analysis extracted from mandibles. The present research clearly shows that in the case of old samples, there could appear a problem with inhibitors, but again, it is possible to overcome this problem by application of a dilution and enhancers of PCR such as BSA or/and glycerol proposed by other authors (Eilert and Foran 2009). All the methodology and results presented could constitute some kind of instructions how to deal with problems occurring during molecular analyses, especially if only partial remains are found in a pellet, but also during the extraction and amplification of DNA from biological material collected in the field.
References
Alonso A, Albarran C, Martin P, Garcia P, Garcia O, de la Rua C, Alzualde A, Fernandez de Simon L, Sancho M, Piqueras JF (2003) Multiplex–PCR of short amplicons for mtDNA sequencing from ancient DNA. Int Congr Ser 1239:585–588
Balčiauskienė L, Juškaitis R, Atkočaitis O (2005) The diet of the Tawny owl (Strix aluco) in South-Western Lithuania during the breeding period. Acta Zool Lit 15:13–20
Balčiauskienė L, Jovaišas A, Naruševičius V, Petraška A, Skuja S (2006) Diet of Tawny owl (Strix aluco) and Long-eared owl (Asio otus) in Lithuania as found from pellets. Acta Zool Lit 16:37–45
Balestrieri A, Remonti L, Ruiz-González A, Vergara M, Capelli E, Gómez-Moliner BJ, Prigioni C (2011) Food habits of genetically identified pine marten (Martes martes) expanding in agricultural lowlands (NW Italy). Acta Theriol 56:199–207
Blow MJ, Zhang T, Woyke T, Speller CF, Krivoshapkin A, Yang DY, Derevianko A, Rubin EM (2008) Identification of ancient remains through genomic sequencing. Genome Res 18:1347–1353
Broquet T, Petit E (2004) Quantifying genotyping errors in noninvasive population genetics. Mol Ecol 13:3601–3608
Buś MM, Bodganowicz W, Voldros D (2013) DNA analysis of ancient bone and tooth samples by using PrepFiler® Express BTA Forensic DNA Extraction and AmpFℓSTR® NGM SElect™ PCR Amplification Kits. Forensic News. Available from http://www.lifetechnologies.com/ForensicNews
Casas-Marce M, Revilla E, Godoy JA (2010) Searching for DNA in museum specimens: a comparison of sources in a mammal species. Mol Ecol Resour 10:502–507
Eilert KD, Foran DR (2009) Polymerase resistance to polymerase chain reaction inhibitors in bone. J Forensic Sci 54:1001–1007
Frantz AC, Schaul M, Pope LC, Fack F, Schley L, Muller CP, Roper TJ (2004) Estimating population size by genotyping remotely plucked hair: the Eurasian badger. J Appl Ecol 41:985–995
Frantzen MAJ, Silk JB, Ferguson JWH, Wayne RK, Kohn MH (1998) Empirical evaluation of preservation methods for faecal DNA. Mol Ecol 7:1423–1428
Gagneux P, Boesch C, Woodruff DS (1997) Microsatellite scoring errors associated with noninvasive genotyping based on nuclear DNA amplified from shed hair. Mol Ecol 6:861–868
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Höss M, Jaruga P, Zastawny TH, Dizdaroglu M, Pääbo S (1996) DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res 24:1304–1307
Jaarola M, Searle JB (2004) A highly divergent mitochondrial DNA lineage of Microtus agrestis in southern Europe. Heredity 92:228–234
Kemp BM, Monroe C, Smith DG (2006) Repeat silica extraction: a simple technique for the removal of PCR inhibitors from DNA extracts. J Archaeol Sci 33:1680–1689
King C, Debruyne R, Kuch M, Schwarz C, Poinar H (2009) A quantitative approach to detect and overcome PCR inhibition in ancient DNA extracts. Biotechniques 47:941–949
Kohn MH, Wayne RK (1997) Facts from feces revisited. Trends Ecol Evol 12:223–227
Kohn MH, York EC, Kamradt DA, Haugt G, Sauvajot RM, Wayne RK (1999) Estimating population size by genotyping faeces. Proc R Soc Lond Ser B 266:657–663
Loreille OM, Diegoli TM, Irwin JA, Coble MD, Parsons TJ (2007) High efficiency DNA extraction from bone by total demineralization. Forensic Sci Int Genet 1:191–195
Mikkola H (1983) Owls of Europe. T & AD Poyser, London
Morin PA, McCarthy M (2007) Highly accurate SNP genotyping from historical and low-quality samples. Mol Ecol Notes 7:937–946
Morin PA, Hedrick NM, Robertsom KM, Leduc CA (2007) Comparative mitochondrial and nuclear quantitative PCR of historical marine mammal tissue, bone, baleen, and tooth samples. Mol Ecol Notes 7:404–411
Pääbo S (1989) Ancient DNA: extraction, characterization, molecular cloning and enzymatic amplification. Proc Natl Acad Sci USA 86:1939–1943
Pajnič IZ, Gornjak Pogorelc B, Balažic J, Zupanc T, Štefanič B (2012) Highly efficient nuclear DNA typing of the World War II skeletal remains using three new autosomal short tandem repeat amplification kits with the extended European Standard Set of loci. Croat Med J 53:17–23
Piggot MP (2004) Effect of sample age and season of collection on the reliability of microsatellite genotyping of faecal DNA. Wildl Res 31:485–493
Poulakakis N, Lymberakis P, Paragamian K, Mylonas M (2005) Isolation and amplification of shrew DNA from barn owl pellets. Biol J Linn Soc 85:331–340
Pucek Z (1981) Keys to vertebrates of Poland. Mammals. PWN, Warszawa
R Core Team (2012) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, http://www.R-project.org/. ISBN 3-900051-07-0
Raczyński J, Ruprecht AL (1974) The effect of digestion on the osteological composition of owl pellets. Acta Ornithol 14:25–38
Reddy PA, Bhavanishankar M, Bhagavatula J, Harika K, Mahla RS, Shivaji S (2012) Improved methods of carnivore faecal sample preservation, DNA extraction and quantification for accurate genotyping of wild tigers. PLoS One 7:e46732
Rohland N, Hofreiter M (2007) Comparison and optimization of ancient DNA extraction. Biotechniques 42:343–352
Rohland N, Siedel H, Hofreiter M (2004) Nondestructive DNA extraction method for mitochondrial DNA analyses of museum specimens. Biotechniques 36:814–821
Rohland N, Siedel H, Hofreiter M (2009) A rapid column-based ancient DNA extraction method for increased sample throughput. Mol Ecol Resour 10:677–683
Rowe KC, Singhal S, Macmanes MD, Ayroles JF, Morelli T, Rubidge EM, Bi K, Moritz CC (2011) Museum genomics: low-cost and high-accuracy genetic data from historical specimens. Mol Ecol Resour 11:1082–1092
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana, Totowa, pp 365–386, Source code available at http://fokker.wi.mit.edu/primer3/
Satoh M, Kuroiwa T (1991) Organization of multiple nucleids and DNA molecules in mitochondria of a human cell. Exp Cell Res 196:137–140
Shapiro B (2008) Engineered polymerases amplify the potential of ancient DNA. Trends Biotechnol 26:285–287
Shapiro B, Hofreiter M (eds) (2012) Ancient DNA: methods and protocols. Methods in molecular biology, Vol. 840, Springer protocols. Humana, Totowa
Taberlet P, Fumagalli L (1996) Owl pellets as a source of DNA for genetic studies of small mammals. Mol Ecol 5:301–305
Taberlet P, Luikart G (1999) Non-invasive genetic sampling and individual identification. Biol J Linn Soc 68:41–55
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194
Taberlet P, Waits LP, Luikart G (1999) Noninvasive genetic sampling: look before you leap. Trends Ecol Evol 14:323–327
Valiere N, Taberlet P (2000) Urine collected in the field as a source of DNA for species and individual identification. Mol Ecol 9:2150–2152
Vigilant L (1999) An evaluation of techniques for the extraction and amplification of DNA from naturally shed hairs. Biol Chem 380:1329–1331
Wandeler P, Smith S, Morin PA, Pettifor RA, Funk SM (2003) Patterns of nuclear DNA degeneration over time—a case study in historic teeth samples. Mol Ecol 12:1087–1093
Wandeler P, Hoeck PEA, Keller LF (2007) Back to the future: museum specimens in population genetics. Trends Ecol Evol 22:634–642
Willerslev E, Cooper A (2005) Ancient DNA. Proc R Soc B 272:3–16
Wyngaard GA, Holynska M, Schulte JA II (2010) Phylogeny of the freshwater copepod Mesocyclops (Crustacea: Cyclopidae) based on combined molecular and morphological data, with notes on biogeography. Mol Phylogenet Evol 55:753–764
Yalden DW, Morris PA (1990) The analysis of owl pellets. Occasional publication No. 13. The Mammal Society, London
Zemanová B, Hájková P, Bryja J, Zima J Jr, Hájková A, Zima J (2011) Development of multiplex microsatellite sets for noninvasive population genetic study of the endangered Tatra chamois. Folia Zool 60:70–80
Żmihorski M, Gryz J, Krauze-Gryz D, Olczyk A, Osojca G (2011) The Tawny owl Strix aluco as a material collector in faunistic investigations: the case study of small mammals in NE Poland. Acta Zool Lit 21:185–191
Żmihorski M, Sienkiewicz P, Tryjanowski P (2013) Never ending story: a lesson in using sampling efficiency methods with ground beetles. J Insect Conserv 17:333–337
Acknowledgments
We are grateful to P. Ślipiński for the picture of the mandible, and to two anonymous reviewers for their helpful comments on the manuscript. A. Zon kindly improved the English.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Cino Pertoldi
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Buś, M.M., Żmihorski, M., Romanowski, J. et al. High efficiency protocol of DNA extraction from Micromys minutus mandibles from owl pellets: a tool for molecular research of cryptic mammal species. Acta Theriol 59, 99–109 (2014). https://doi.org/10.1007/s13364-013-0144-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-013-0144-y