
Abstract
This review covers publications on siderophores applied for molecular imaging applications, mainly for radionuclide-based imaging. Siderophores are low molecular weight chelators produced by bacteria and fungi to scavenge essential iron. Research on these molecules has a continuing history over the past 50 years. Many biomedical applications have been developed, most prominently the use of the siderophore desferrioxamine (DFO) to tackle iron overload related diseases. Recent research described the upregulation of siderophore production and transport systems during infection. Replacing iron in siderophores by radionuclides, the most prominent Ga-68 for PET, opens approaches for targeted imaging of infection; the proof of principle has been reported for fungal infections using 68Ga-triacetylfusarinine C (TAFC). Additionally, fluorescent siderophores and therapeutic conjugates have been described and may be translated to optical imaging and theranostic applications. Siderophores have also been applied as bifunctional chelators, initially DFO as chelator for Ga-67 and more recently for Zr-89 where it has become the standard chelator in Immuno-PET. Improved DFO constructs and bifunctional chelators based on cyclic siderophores have recently been developed for Ga-68 and Zr-89 and show promising properties for radiopharmaceutical development in PET. A huge potential from basic biomedical research on siderophores still awaits to be utilized for clinical and translational imaging.
Similar content being viewed by others
Introduction
Progress in Molecular Imaging applications in particular in the context of radionuclide-based technologies is dependent on highly specific tracers aiming at an increasing number of available molecular targets. The development of radiopharmaceuticals is impressively advancing based on the progress in radiopharmaceutical chemistry embracing the increasing understanding of the molecular basis of pathophysiology in many clinical fields. Radiometals have been an essential part in this development, initially driven by technetium-99m based radiopharmaceutical developments, today overtaken by the interest in positron emission tomography (PET) with the implementation of gallium-68 in clinical routine and other radiometals entering the arena including zirconium-89, copper-64, scandium-44 and others. Integration of radiometals in “biomolecules” requires the attachment of a chelator binding the metal with high stability without impairing affinity to the target. Nature has designed specific chelators for a variety of metals; an important group is the so-called siderophores (from Greek translating to “Iron-Carrier”) for binding ferric ions, produced by bacteria, fungi and plants. This review summarizes applications of siderophores as chelators for general molecular imaging applications and in particular in the field of infection imaging.
Methods
Siderophores have been very widely investigated in biomedical research. A systematic search in PubMed was carried out, taking into account publications until August 2016. The search term “Siderophore” reveals 11,205 hits in PubMed (August 2016), starting from 1953 with first publications on Mycobactin [1]. Figure 1a shows the distribution of publication over the last 60 years indicating the constant interest of the scientific community in siderophores in biomedical research including preclinical and clinical applications. Combining the search term “Siderophore(s)” or the most widely used siderophore “Desferrioxamine” with key words related to imaging such as “Imaging”, “Radionuclide”, “PET”, “scintigraphy” or specific radionuclides all together 699 publications were found with relations of siderophores to imaging applications (Fig. 1b). Even though systematic search was carried out, the high number of publications made a selection of recent, up to date reviews on the general topic of siderophores or on 89Zr labelling based on siderophores necessary. This review also did not intend to analyse the clinical applications or outcomes; meta-analysis or risk assessment was, therefore, not applied.

Interest in siderophores in Biomedical Research over the last 60 years based on PubMed-listed publications; a search term using “Siderophore” presented in hits/decade; b publication hits combining search terms “Siderophore” with various Imaging key words
Microbial siderophores
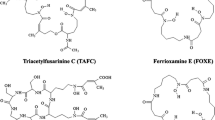
Iron is an essential cofactor for a variety of cellular processes in all eukaryotes and most prokaryotes including respiration, amino acid metabolism, and biosynthesis of DNA and sterols. Despite its high abundance in the Earth’s crust, the bioavailability of iron is extremely low owing to its oxidation by atmospheric oxygen into sparely soluble ferric hydroxides with a solubility of 10−18 M at pH 7.0. Moreover, for pathogens in both plant and animal hosts, iron is usually not freely available but tightly sequestered, e.g. in vertebrates associated with proteins such as transferrin, ferritin and haemoglobin. Blocking iron access to invading microbes represents a key pathway in host defence as a component of innate immunity, termed “nutritional immunity” [2, 3]. Consequently, pathogenic as well as non-pathogenic organisms had to evolve sophisticated strategies to ensure iron supply. Microorganisms are believed to lack mechanisms for iron excretion and, therefore, control of iron uptake is considered the major iron homeostatic mechanism. To satisfy the iron need in diverse niches, bacteria and fungi use different iron acquisition mechanisms, which are transcriptionally upregulated during iron limitation: (1) direct ferrous iron (Fe2+) uptake, (2) direct ferric iron (Fe3+) uptake, (3) siderophore-mediated ferric iron uptake, and (4) uptake and degradation of haeme. Most microbial species employ more than one system in parallel but not all species use all four strategies. With few exceptions, bacterial and fungal species secrete siderophores to scavenge extracellular iron. Siderophores, low molecular mass (≤1 kD), ferric iron-specific chelators, display a remarkable species-specific, structural diversity with >500 different siderophores being identified [4, 5]. Some bacteria possess plasma membrane-localized siderophores, e.g. mycobactins of mycobacteria. In contrast to bacteria, most fungi also possess intracellular siderophores for intracellular transport and storage of iron. Siderophores contain the most efficient iron-binding ligand types in nature, consisting of hydroxamate, catecholate or α-hydroxy-carboxylate ligands (Fig. 2). The most efficient siderophores form hexadentate complexes, satisfying the six co-ordination sites on ferric ions allowing iron-binding constants of 1020–1050. Examples are enterobactin in the catecholate class, triacetylfusarinine C (TAFC), ferrioxamines (FOX) E and G, as well as the ferrichromes in the hydroxamate class and staphyloferrin in the α-hydroxy-carboxylate class (Fig. 2) [6]. The majority of e.g. fungal siderophores belong to the hydroxamate class. Fungal hydroxamate siderophores can be grouped into four structural families: fusarinines, coprogens, ferrichromes and rhodotorulic acid [6]. The hydroxamate group is built by acylation of the non-proteinogenic amino acid N5-hydroxy-l-ornithine, which is derived by hydroxylation of l-ornithine, with acetyl or more complex groups such as anhydromevalonyl. Most fungal siderophores include three of these moieties linked by ester or peptide bonds to form the most efficient hexadentate structures. Cyclization of the siderophore is found in ferrichromes and some fusarinines. Although linear hexadentate siderophores are found in all siderophore classes, there is a tendency for cyclization, thereby enhancing complex and chemical stability.

Basic ferric-coordination units (top) and examples of natural siderophores (bottom)
Siderophore metabolism is highly specific to microbes: Siderophore production involves enzymes that are found exclusively in bacteria and fungi, e.g. nonribosomal peptide synthetases, and siderophore uptake is mediated by specific transporters. In bacteria and fungi, siderophore uptake is mediated by different transport systems. For example, in gram-negative bacteria siderophores have to cross both the outer membrane and the plasma membrane; e.g. ferrichrome type siderophores are transported through the outer membrane via the receptor FhuA, which is energized by the plasma membrane-localized TonB complex, and transported across the plasma membrane via ABC-transporter-dependent movement [7]. In contrast, cellular uptake of siderophore-iron complexes in fungi is mediated by “siderophore-iron transporters” (SITs), which belong to a subfamily of the major facilitator protein superfamily [8]. SITs act most likely as proton symporters energized by the plasma membrane potential. SIT-mediated iron uptake is universally conserved in the fungal kingdom, even in species not producing siderophores such as Saccharomyces cerevisiae, Candida spp. and Cryptococcus neoformans [9]. Moreover, most bacterial and fungal species are able to utilize not only the endogenous siderophores but also siderophore types that are produced by other bacterial or fungal species (so-called xenosiderophores).
Taken together, both siderophore biosynthesis and their specific cellular uptake are confined to the bacterial and fungal kingdoms. Moreover, there is overwhelming evidence that the siderophore system is active during infection; e.g. (1) siderophore biosynthesis and uptake are transcriptionally upregulated during iron starvation in vitro as well as in vivo in a murine model for pulmonary infection with the mold Aspergillus fumigatus (A. f.) [10, 11], and (2) genetic inactivation of siderophore biosynthesis attenuates virulence of A. f. in a murine infection model, which demonstrates that siderophore-mediated iron assimilation plays the major role for virulence [12, 13]. Moreover, the siderophore of A. f., triacetylfusarinine C (TAFC) was shown to be able to extract iron from host transferrin [14]. A scheme of TAFC-mediated iron uptake is shown in Fig. 3. Similarly, siderophore biosynthesis was shown to be crucial for the virulence of numerous bacterial species including, e.g. Yersinia pestis, Mycobacterium tuberculosis or Pseudomonas aeruginosa [e.g. 15]. As a result, siderophores were suggested as biomarkers in aspergillosis and tuberculosis [16, 17]. Due to the function of siderophores as virulence determinants, mammals evolved siderophore sequestering proteins, termed siderocalins, and pathogens evolved mechanisms to avoid recognition of their siderophores by siderocalins [18].

Siderophore-mediated iron uptake in the mold A. fumigatus. a The cyclic trihydroxamate siderophores FSC (R = H) and TAFC (R = acetyl) are shown in the ferri-form; for TAFC-based nuclear imaging, the iron (shaded in red) is replaced by 68Ga. b TAFC-mediated uptake of iron and gallium into fungal hyphae. TAFC is secreted by an unidentified exporter and the iron/gallium-siderophore complex is taken up by the siderophore transporter MirB. Within the cell, iron release from the siderophore is facilitated by TAFC hydrolysis by the esterase EstB [5]
Unequivocally, siderophores play a profound role in iron acquisition of most microorganisms. Nevertheless, there are evidences that siderophores can chelate also other metals with physiological relevance, e.g. the siderophore yersiniabactin was recently found to sequester extracellular copper to protect uropathogenic Escherichia coli from copper toxicity during human infection [19], while some siderophores appear to be involved in uptake of various non-iron metals such as yersiniabactin in zinc uptake by Yersina pestis [20, 21]. Due to the indispensability of siderophore-mediated iron acquisition, this system is hijacked during microbial competition, e.g. the outer membrane ferrichrome-type siderophore receptor of E. coli serves also as receptor for various bacteriophages [22] and naturally evolved siderophore-antibiotic conjugates, termed sideromycins, in which a bactericidal warhead is attached to a siderophore moiety [20, 21]. For instance, albomycins comprise a hydroxamate siderophore unit, reminiscent of those found in fungal ferrichromes, and bactericidal unit that inhibits seryl-tRNA synthetase. Albomycins display a broad-spectrum of antibiotic activity again both Gram-negative and Gram-positive bacteria because of the widespread nature of ferrichrome receptors. These natural “Trojan horses” inspired the development of designed synthetic conjugates [23]. Similarly, gallium salts have been described as potential anti-infectives. In this case, gallium is bound to siderophores and taken up by the pathogen via the siderophore transport system which negatively interferes as iron analogue with the pathogens’s iron homeostasis [24]. A human application of siderophores, which is not related to infectious diseases, is the use of desferrioxamine, a siderophore produced by Streptomycetes spp, in treatment of iron overload such as thalassemia to mobilize and decrease body iron stores [25].
Siderophores for molecular imaging of infection
The accurate localization and characterization of infection and its distinction from inflammation have emerged as one of the greatest challenges of modern medicine. Identification of patients at high risk and early and accurate diagnosis remains crucial for their successful therapy and underlines the urgent need for specific and sensitive diagnostic tools. Molecular imaging methods hold the potential to provide a more robust, non-invasive, selective and sensitive diagnosis of infections leading to improved clinical decisions and a fundamental change in patient management with better healthcare outcomes [26]. Radiological imaging techniques such as computed tomography (CT), magnetic resonance imaging (MRI) and ultrasonography (US) are widely used in clinical practice for identification of infection, although they have major limitations in specificity [27]. Optical imaging represents an interesting future approach to molecular imaging of infection, but no optical probes have been licensed for routine use in the clinic for microbial detection [28]. By contrast, nuclear imaging techniques including PET and SPECT have a rich history of different radiolabelled probes (radiopharmaceuticals) for imaging of infectious processes in patients. These include 111In- or 99mTc-labelled leucocytes, 99mTc-anti-granulocyte antibody, 99mTc-diphosphonates in the context of bone scanning, 67Ga-citrate and 2-[18F]-fluorodeoxyglucose [26]. These probes target predominantly secondary effects of infection such as increased blood flow and vascular permeability, activated endothelial cells or polymorphonuclear cell migration limiting their specificity or have other shortcomings related to blood manipulation or induction of immune response (HAMA) [29, 30]. Even though new developments are emerging especially for bacterial infections such as radiolabelled antimicrobial peptides [26], nuclear medicine clinicians are still awaiting improved radiopharmaceuticals overcoming these limitations.
An interesting group of molecules, which could fulfill the requirements on the ‘optimal imaging agent’ for molecular imaging of infections, appears to be (radio)labelled siderophores. Table 1 summarizes applications of siderophores as imaging agents. They can be prepared either by the introduction of appropriate radiometal to the natural (iron-)siderophore complex via the exchange of iron or artificially by the modification of natural siderophore with a chromophore suitable for optical imaging [31–34]. Already in the 1970s and 1980s, first investigations of radiolabelled siderophores, including desferrioxamine (DFO), were already reported with gamma-emitting radionuclides—67Ga and 111In [35–38]. Gallium is an isosteric diamagnetic substitute for Fe(III) [39] and, thus, the affinity constants of many siderophores for gallium are in the range of their iron counterparts. At that time, it was also demonstrated that under reducing conditions, Ga(III) can rapidly displace Fe(III) from siderophores, whereas without concerted reduction of the iron no significant exchange was observed [40]. Emery and Hoffer [41] have used 67Ga to study the uptake mechanisms for different siderophores in Ustilago sphaerogena and found this energy-dependent process to be indistinguishable from that of its Fe(III) counterpart. They even postulated an involvement of siderophore binding in the accumulation of 67Ga-citrate in inflammatory lesions. A number of investigations were made with 3H, 14C, 55Fe and 59Fe labelled siderophores mainly to study iron transport or siderophore uptake mechanisms in microorganisms or plants [e.g. 42–44] unsuitable for molecular imaging and, therefore, cannot be used for detection of microbial infections in vivo. By contrast, radionuclides used in the studies of Moerlein and Emery [37, 40, 41]—67Ga and 111In—have found widespread use in nuclear medicine for SPECT imaging. Over the past decade, PET has experienced a significant increase applying a variety of positron emitting radiometals [45]. Recently, 68Ga use in particular is showing a dramatic growth because of the applicability in labelling of diverse range of compounds and because it is obtained from a long shelf-life and relatively inexpensive 68Ge/68Ga generator system [46].
More than 30 years after the first attempts of labelling siderophores with 67Ga [36–38, 40, 41], we evaluated the use of 68Ga labelled siderophores for PET imaging of fungal infections [47]. In proof of concept studies, which should confirm or refute the possibility of PET imaging of infections caused by Aspergillus fumigatus (A. f.) using 68Ga-siderophores [48], it was demonstrated that desferrisiderophores, particularly triacetylfusarinine C (TAFC), can be easily radiolabelled with 68Ga using a few micrograms of the siderophore and exhibit high chemical stability. Uptake of 68Ga-TAFC by A. f. was upregulated under iron starvation conditions and could be blocked with an excess of siderophore or NaN3, indicating specific and energy-dependent uptake. A variety of different siderophores such as fusarinine C (FSC), TAFC, coprogen, various ferrichrome and ferrioxamine-type-siderophores displayed excellent 68Ga-radiolabeling properties [49]. However, only 68Ga-TAFC and 68Ga-ferrioxamine E (FOXE), a siderophore produced by Streptomycetes, displayed a good combination of fungal uptake in culture, suitable pharmacokinetics for imaging (i.e. rapid clearance from organs and circulation with predominant renal excretion) and, in particular, excellent metabolic stability [50]. Significantly different in vivo behaviour compared to 68Ga-citrate (i.e. non-specific infection and inflammation PET imaging agent) was also found [51]. High contrast imaging of A. f. pulmonary infection in a rat model was achieved using micro-PET/CT technology [50, 52], exhibiting pronounced accumulation of 68Ga-TAFC in infected areas extremely early after onset of infection, which increased with severity of infection and correlated with abnormal CT images (Fig. 4). Significant accumulation of 68Ga TAFC was found neither in sterile inflammations nor in tumour cells [53], which also have a high iron metabolism. We also investigated the uptake of 68Ga-TAFC in a number of different fungal and bacterial species, which revealed high specificity for Aspergillus species, with no significant uptake by Candida and bacterial species, in particular. By comparison, FOXE displayed high in vitro uptake by Staphylococcus aureus, which was surprisingly not confirmed in vivo [53]. An interesting exception among Aspergillus species is Aspergillus terreus, which lacks the ability to take up TAFC but accumulates FOXE. Besides the investigations with siderophores labelled with 68Ga, we have also attempted to radiolabel siderophores with different radionuclides. So far we have succeeded to label TAFC, FOXE, desferrichrome A (FCHA) and DFO with zirconium-89 [54]. The interest in 89Zr has increased over the last years as it displays almost ideal properties allowing imaging of biological processes at late time points after the tracer application. Even though 89Zr has comparably low positron abundance and due to the long half-life (78.4 h) results in higher radiation dose, it allows long-term follow-up especially of slowly accumulating biomolecules such as antibodies, nanoparticles and other large biomolecules both for preclinical and clinical applications, thereby complementing 68Ga with its limitations of a very short half-life (67.7 min). Comparing the in vitro and in vivo characteristics of 68Ga-siderophores with their 89Zr counterparts, we found analogous properties with the potential for longitudinal Aspergillus infection imaging [54]. From all these studies, we concluded that 68Ga-labelled siderophores, in particular 68Ga-TAFC, have a high potential to be used as radiopharmaceuticals to specifically image Aspergillus infections in patients.

Micro-PET/CT (Albira PET/SPECT/CT small animal imaging system, Bruker Biospin Corporation, Woodbridge, CT, USA) imaging of A. fumigatus [coronal slices (a) and 3D images (b)] in a rat infection model and non-infected rat (c) 45 min post-intravenous injection of 68Ga-TAFC showing clear accumulation in infected [(a) and (b)] and no accumulation in healthy c lung tissue
Overall radiolabelled Siderophores certainly have the potential to be a highly specific tool for infection imaging, considering the essential role of the siderophore system for iron acquisition and virulence of microorganisms together with its upregulation during infection, whereas they are not utilized by mammals. This is also related to the low toxicity of siderophores exemplified by DFO, which is used safely in close to gram amounts for iron overload disease repeatedly. Selecting appropriate siderophores can also lead to a high specificity for particular microorganisms, e.g. being able to distinguish between certain fungal and bacterial infections. The requirement for upregulation of the siderophore transporters to accumulate the radiolabelled siderophore, however, will require a rather acute status of infection; therefore, it can be expected that its main role can be envisaged in a rather acute setting, such as detection and specific characterization of invasive Aspergillosis, with its live-threatening consequences rather than in a more chronic or less aggressive infection setting. This can only be revealed in a clinical setting; therefore, the first clinical studies of 68Ga-siderophores are currently eagerly awaited.
Besides radiolabelling, also other attempts have been made to use siderophores for pathogen detection. Several groups have developed strategies of synthesizing siderophore-chromophore conjugates for optical imaging [31–33, 55–58]. Siderophores (e.g. ferrichromes, pyochelin and DFO) derivatized with various fluorescent probes, such as fluorescein, rhodamine, 7-nitrobenz-2-oxa-1,3-diazole and anthracene, were used for the monitoring of siderophore transport in different microorganisms including bacteria (e.g. Pseudomonas spp.) [31, 32, 58] and fungi (Ustilago maydis, Saccharomyces cerevisiae, Candida albicans and Rhizzopus arrhizus) [33, 55, 57]. The microbial activity was not altered by the attachment of various functionalities and fluorescent siderophore analogues became invaluable tools in the investigation of molecular mechanisms involved in microbial iron transport and acquisition. Accordingly, these artificial siderophore analogues could also serve as a tool for in vivo diagnostic imaging or targeting of microbial pathogens [34].
The recognition of the role of siderophores as important microbial iron transporters has led to the exploitation of this pathway in a ‘Trojan Horse’ strategy not only for pathogen detection, but also for the development of therapeutic strategies [34, 59]. Banin et al. [60] have used siderophore-metal complex combining a strong siderophore, DFO with non-radioactive gallium for the treatment of Pseudomonas aeruginosa (P.a.) infection. The Ga-DFO complex was designed as an antioxidant that acts by ‘push and pull’ mechanism, sequestering ferric ions (the siderophore effect) and, in turn, releasing gallium ions that further compete with ferric ions at iron-binding sites of proteins. The Ga-DFO served as a ‘Trojan Horse’ that interferes with iron metabolism and delivers toxic gallium to P.a. cells. The antimicrobial effect of Ga-DFO to P.a. infections showed promising results; nevertheless, it warrants further investigation. Moreover, a number of studies on complex siderophore-drug conjugates have been made to test their potential as effective antimicrobial agents [23, 34, 61–63]. It could be speculated that these siderophore conjugates could be radiolabelled or derivatized and used for molecular imaging of infections.
Siderophores as bifunctional chelators
Table 2 summarizes applications of siderophores as bifunctional chelators, combining the two functions of metal coordination with the coupling property to a targeting vector.
Desferrioxamine and gallium
Already early in the development of targeted radiopharmaceuticals, siderophores were considered as chelators for radiometals. Initial studies focussed on gallium-67 as a gamma-emitting isotope with a half-life of 78.3 h for planar scintigraphy and SPECT imaging. 67Ga-Citrate was introduced for tumour studies and due to its similarities with Fe3+ DFO was proposed to enhance tumour to blood ratios in tumour imaging [64]. Three hydroxamate groups of DFO coordinate Ga3+ with fast kinetics and high affinity, forming a stable 1:1 chelate with high radiochemical yield. The free amino group can be used as coupling side to bioactive molecules. Already in 1982, the proof of principle was shown by coupling DFO to albumin for binding 67Ga, proposing DFO as bifunctional chelating agent [65]. A glutaraldehyde coupling reaction was applied and the authors showed a superior in vivo stability of 67Ga-DFO-HSA over 131I-labelled HSA and provided first images in patients. A first targeted application was reported in the same year by coupling DFO to fibrinogen [66]. A large number of DFO molecules were introduced to human fibrinogen using dialdehyde starch (DAS) as a spacer-functional polymer. Increased accumulation of 67Ga-fibrinogen in venous thrombi was depicted at 48 h after injection about 60 % of patients [67]. Other applications of DFO-conjugated macromolecules followed soon with radiolabelled lectins [68], which failed in tumour detection. A more successful approach was the development of DFO-conjugated monoclonal antibodies and antibody fragments. Motta-Hennessy C et al. [69] established conditions for the coupling of DFO with the bifunctional reagent glutaraldehyde to two rat IgG2b monoclonal antibodies M10/76 and 11/160, specific for the Hooded rat sarcomata MC 24 and HSN, respectively, which maintained their capacity for binding to their tumour-associated antigens. Koizumi et al. [70] compared the homocoupling reagent glutaraldehyde with two other heterocoupling reagents, N-succinimidyl-3-(2-pyridyldi-thio)propionate and succinimidyl-6-maleimidohexanoate, linking desferrioxamine to antibodies through alkylamine, disulphide, and thioether bonds, and showed superiority of thioether bonds in terms of tumour targeting and pharmacokinetics. Bartal et al. [71] compared the labelling of MAb 23H7, binding to human sarcoma, with 67Ga using glutaraldehyde-coupled DFO and 111In via DTPA, whereby higher specific activities were achieved with 67Ga. Amino-dextran-DFO was used to derivatise an anti-melanoma monoclonal antibody (TP41) for labelling with In-111 with promising results especially reduced liver uptake [72]. DFO as bifunctional chelator for antibodies was also proposed for radiotherapeutic applications using 67Ga Auger electrons. Govindan et al. [73] prepared different DFO-antibody conjugates and reported two main problems limiting further development. First, the stability was inadequate for the 3-day half-life of the nuclide. Second, the labels were poorly retained within cells after Ab internalization and catabolism. More recently, a novel bifunctional chelate (BFC) p-isothiocyanatobenzyl-DFO (Df-Bz-NCS), originally developed for 89Zr labelling, was used to prepare anti-EGF Nanobody conjugates of DFO for 68Ga labelling for PET applications [74]. Fast radiolabelling, high tumour uptake and tumour to normal tissue ratios in nude mice bearing A431 xenografts were obtained with the fast kinetics of the 68Ga-Nanobody conjugates, indicating a promising application of DFO conjugates with 68Ga.
Besides proteins also smaller molecules were conjugated to DFO for radiolabelling with 67/68Ga. Folic acid was covalently linked to DFO via an amide bond using a simple carbodiimide coupling reaction. 67Ga-DF-folate(gamma) exhibited specific uptake and was proposed as a diagnostic agent for noninvasive imaging of folate receptor-positing tumours [75]. 67/68Ga-DFO-Octreotide (SDZ 216-927), comprising DFO coupled to octreotide via a succinyl linker [76, 77], showed specific uptake in Somatostatin receptor expressing tumour models and was proposed as PET imaging agent. However, in patients 67Ga DFO-Octreotide radioactivity was detectable in the circulation even after 24 h; the blood clearance curve was much slower than the one of OctreoScan (111In-DTPA-Octreotide) due to relatively high protein binding in human serum [78]. So, overall a number of attempts have been made to develop siderophore-bioconjugates based on DFO for radiolabelling with 67/68Ga and to a limited extent with 111In, however, with inconclusive results in particular related to its stability especially at high specific activities [79], thereby being replaced mainly by aminocarboxylate-based chelators such as DOTA or NOTA.
Desferrioxamine and zirconium-89
In contrast to that in the past decade, DFO has established its role in the context of 89Zr-labelling [80–84]. 89Zr was proposed as a diagnostic radionuclide for quantitating the biodistribution of radiolabelled antibodies. The high affinity of zirconium for hydroxamic acid groups makes DFO a suitable and effective chelator for Zr4+. Meijs and co-workers initially reported that DFO exhibits rapid and efficient labelling with a 1:1 ratio of metal to chelate and demonstrates good stability with regard to demetallation, releasing less than 0.2 % of the metal in serum after 24 h [85]. Further evaluation of the complex by Holland and co-workers utilizing density functional theory (DFT) models exhibited Zr-DFO as an octadentate complex combining the six binding oxygens of DFO with two additional water molecules. Also, stability studies over longer periods of time indicated that still less than 2 % demetallation occurs after 7 days in serum [86].
The first clinical trial with an 89Zr-labelled antibody revealed the low immunogenicity of the DFO-conjugate [87] allowing repeated applications of the DFO immunoconjugate. For the coupling of DFO to antibodies, most widely 2,3,5,6 tetrafluorophenyl TFP-activated ester of N-succinyl-DFO-Fe forming stable amide bonds with free amines have been applied [88], or alternatively p-isothiocyanato-DFO forming a stable thiourea bond with lysine residues [89]. Standardized protocols have been established [90] making 89Zr labelling for Immuno-PET applications ever more widely applicable. Several reviews have summarized the latest progress of 89Zr-DFO-conjugated antibodies [82–84].
The use of 89Zr-labelled bioactive molecules using siderophores is not limited to the antibodies. Beyond antibodies, 89Zr-DFO conjugated to peptides and peptide multimers [91], nanoparticles [92, 93], carbon nanotubes [94], Albumin nanocolloids [95], and proteins [96, 97] has also been investigated.
Improvement of DFO for 89Zr
Despite the prevalent use of 89Zr-DFO-conjugated antibodies for preclinical studies and clinical applications, several preclinical studies reported bone accumulation of dissociated 89Zr ranging from 3 to 15 % after 3–7 days [86, 98, 99]. This insufficient stability of the 89Zr-DFO complexes is attributed to the incomplete coordination of 89Zr4+ by DFO and the linear structure of DFO. Based on the knowledge of DFO, Patra et al. developed an octadentate DFO analogue termed DFO*, which fully saturates the coordination sphere of Zr4+, by coupling an additional hydroxamic acid entity to DFO [100]. DFT calculations predicted the expected molecular structure involving coordination through the eight oxygen atoms of all four hydroxamic acid moieties. Coupling the model peptide bombesin ([Nle14]BBS(7–14)), DFO*-bombesin showed a remarkably improved stability in comparison to the DFO analogue when challenged with 300- to 3000-fold molar excess DFO over the course of 1 day. The in vitro experiment demonstrated that the new chelator did not influence the properties of the peptidic vector. Based on those results, DFO* holds promise to provide new PET imaging agents with superior stability profiles; applications on DFO* coupled antibodies are awaited soon.
Other siderophores as bifunctional chelators
Recently, we reported that Fusarinine C (FSC), a representative of the class of hydroxamate siderophores, is a promising 68Ga and 89Zr bifunctional chelator [101–104]. FSC, possessing three hydroxamic acid groups for binding 68Ga or 89Zr similar to DFO embedding an additional cyclic structure, offers a potential advantage with respect to the stability of 68Ga/89Zr complexes. FSC not only allows fast and highly selective labelling with 68Ga in a wide pH range and results in high specific activities, but also shows very high stability of 68Ga-FSC complexes at low concentration demonstrating the superiority over DFO which was reported to be unstable at low ligand concentrations (<50 nM) [79]. Compared to 89Zr-DFO, 89Zr-FSC derivatives showed excellent in vitro stability and resistance against transchelation in phosphate-buffered saline (PBS), ethylenediaminetetraacetic acid solution (EDTA) and human serum for up to 7 days making it an alternative as 89Zr BFC [103]. The three primary amines of FSC facilitate the derivatization of FSC with targeting biomolecules in a number of ways, also applying the concept of multivalency. By attaching a cyclic RGD peptide, binding to integrin αvβ3 expressed during angiogenesis, via a succinic acid linker (FSC-(RGD)3), high stability 68Ga complexes with excellent receptor-binding properties and in vivo targeting were prepared (Fig. 5), superior to monomeric [68Ga]NODAGA-RGD [104]. Currently, monovalency- and divalency FSC for 68Ga/89Zr labelling are under investigation and different coupling strategies e.g. click chemistry are being investigated.

Structure of the siderophore FSC as bifunctional chelator for 68Ga and 89Zr, three-dimensional volume projections of fused microPET/CT images of M21/M21-L tumor xenograft bearing nude mouse ([68Ga]FSC(succ-RGD)3 at 1 h, [89Zr]FSC(succ-RGD)35 MBq) at 24 h p.i. Red arrow αvβ3 integrin-positive M21 tumor; blue arrow αvβ3 integrin-negative M21-L tumor (from [103, 104])
Conclusion
Extensive publications from the last decades have described a wide variety of Fe3+ binding siderophores produced by bacteria and fungi. Their role in iron acquisition and human diseases has been reported and methods for chemical modification, chemical synthesis and even radiolabelling with a variety of radiometals are available. This knowledge has been translated towards radiopharmaceuticals for molecular imaging in general and specific imaging of infection in particular. There are many opportunities to further use this knowledge towards development of new, improved radiopharmaceuticals for molecular imaging in PET, but also towards theranostics and optical imaging applications.
References
Francis J, Macturk HM, Madinaveitia J, Snow GA (1953) Mycobactin, a growth factor for Mycobacterium johnei. I. Isolation from Mycobacterium phlei. Biochem J 55:596–607
Ganz T (2009) Iron in innate immunity: starve the invaders. Curr Opin Immunol 21:63–67
Weinberg ED (2009) Iron availability and infection. Biochim Biophys Acta 1790:600–605
Hider RC, Kong X (2010) Chemistry and biology of siderophores. Nat Prod Rep 27:637–657. doi:10.1039/b906679a
Haas H (2014) Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat Prod Rep 31:1266–1276
Winkelmann G (2002) Microbial siderophore-mediated transport. Biochem Soc Trans 30:691–696
Krewulak KD, Vogel HJ (2011) TonB or not TonB: is that the question? Biochem Cell Biol 89:87–97. doi:10.1139/o10-141
Philpott CC, Protchenko O (2008) Response to iron deprivation in Saccharomyces cerevisiae. Eukaryot Cell 7:20–27
Haas H, Eisendle M, Turgeon BG (2008) Siderophores in fungal physiology and virulence. Annu Rev Phytopathol 46:149–187
McDonagh A, Fedorova ND, Crabtree J, Yu Y, Kim S, Chen D, Loss O, Cairns T, Goldman G, Armstrong-James D, Haynes K, Haas H, Schrettl M, May G, Nierman WC, Bignell E (2008) Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog 4:e1000154
Schrettl M, Kim HS, Eisendle M, Kragl C, Nierman WC, Heinekamp T, Werner ER, Jacobsen I, Illmer P, Yi H, Brakhage AA, Haas H (2008) SreA-mediated iron regulation in Aspergillus fumigatus. Mol Microbiol 70:27–43
Schrettl M, Bignell E, Kragl C, Joechl C, Rogers T, Arst HN Jr, Haynes K, Haas H (2004) Siderophore biosynthesis but not reductive iron assimilation is essential for Aspergillus fumigatus virulence. J Exp Med 200:1213–1219
Schrettl M, Bignell E, Kragl C, Sabiha Y, Loss O, Eisendle M, Wallner A, Arst HN Jr, Haynes K, Haas H (2007) Distinct roles for intra- and extracellular siderophores during Aspergillus fumigatus infection. PLoS Pathog 3:1195–1207
Hissen AH, Wan AN, Warwas ML, Pinto LJ, Moore MM (2005) The Aspergillus fumigatus siderophore biosynthetic gene sidA, encoding l-ornithine N5-oxygenase, is required for virulence. Infect Immun 73:5493–5503
Cornelis P, Dingemans J (2013) Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front Cell Infect Microbiol 3:75
Carroll CS, Amankwa LN, Pinto LJ, Fuller JD, Moore MM (2016) Detection of a serum siderophore by LC–MS/MS as a potential biomarker of invasive aspergillosis. PLoS One 11:e0151260
Pan SJ, Tapley A, Adamson J, Little T, Urbanowski M, Cohen K, Pym A, Almeida D, Dorasamy A, Layre E, Young DC, Singh R, Patel VB, Wallengren K, Ndung’u T, Wilson D, Moody DB, Bishai W (2015) Biomarkers for tuberculosis based on secreted, species-specific, bacterial small molecules. J Infect Dis 212:1827–1834
Sia AK, Allred BE, Raymond KN (2013) Siderocalins: siderophore binding proteins evolved for primary pathogen host defense. Curr Opin Chem Biol 17:150–157
Koh EI, Henderson JP (2015) Microbial copper-binding siderophores at the host-pathogen interface. J Biol Chem 290:18967–18974
Perry RD, Bobrov AG, Fetherston JD (2015) The role of transition metal transporters for iron, zinc, manganese, and copperin the pathogenesis of Yersinia pestis. Metallomics 7:965–978
Johnstone TC, Nolan EM (2015) Beyond iron: non-classical biological functions of bacterial siderophores. Dalton Trans 44:6320–6339
Braun V (2009) FhuA (TonA), the career of a protein. J Bacteriol 191:3431–3436
Ji C, Juárez-Hernández RE, Miller MJ (2012) Exploiting bacterial iron acquisition: siderophore conjugates. Future Med Chem 4:297–313
Kelson AB, Carnevali M, Truong-Le V (2013) Gallium-based anti-infectives: targeting microbial iron-uptake mechanisms. Curr Opin Pharmacol 13:707–716
Kontoghiorghe CN, Kontoghiorghes GJ (2016) Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des Dev Ther 10:465–481
Signore A, Glaudemans AWJM (2011) The molecular imaging approach to image infections and inflammation by nuclear medicine techniques. Ann Nucl Med 25:681–700
Enggelston H, Panizzi P (2014) Molecular imaging of bacterial infections in vivo: the discrimination between infection and inflammation. Informatics 1:72–99
Mills B, Bradley M, Dhaliwal K (2016) Optical imaging of bacterial infections. Clin Transl Imaging 4:163–174
Lupetti A, de Boer MGJ, Erba P, Campa M, Nibbering PH (2011) Radiotracers for fungal infection imaging. Med Mycol 49:62–69
Auletta S, Galli F, Lauri C, Martinelli D, Santino I, Signore A (2016) Imaging bacteria with radiolabelled quinolones, cephalosporins and siderophores for imaging infection: a systematic review. Clin Transl Imaging 4:229–252
Weizman H, Ardon O, Mester B, Libman J, Dwir O, Hadar Y, Chen Y, Shanzer A (1996) Fluorescently-labeled ferrichrome analogs as probes for receptor-mediated, microbial iron uptake. J Am Chem Soc 118:12368–12375
Nudelman R, Ardon O, Hadar Y, Chen Y, Libman J, Shanzer A (1998) Modular fluorescent-labeled siderophore analogues. J Med Chem 41:1671–1678
Ouchetto H, Dias M, Mornet R, Lesuisse E, Camadro JM (2005) A new route to trihydroxamate-containing artificial siderophores and synthesis of a new fluorescent probe. Bioorg Med Chem 13:1799–1803
Szebesczyk A, Olshvang E, Shanzer A, Carver PL, Gumienna-Kontecka E (2016) Harnessing the power of fungal siderophores for the imaging and treatment of human diseases. Coord Chem Rev. doi:10.1016/j.ccr.2016.05.001 (in press)
Hoffer PB, Samuel A, Bushberg JT, Thakur M (1979) Desferoxamine mesylate (Desferal): a contrast-enhancing agent for gallium-67 imaging. Radiology 131:775–779
Oberhaensli RD, Mueller RM, Fridrich R (1984) Different actions of deferoxamine and iron on Ga-67 abscess detection in rats. J Nucl Med 25:668–672
Moerlein SM, Welch MJ, Raymond KN et al (1981) Tricatecholamide analogs of enterobactin as gallium- and indium-binding radiopharmaceuticals. J Nucl Med 22:710–719
Chandra R, Pierno C, Braunstein P (1978) 111In Desferal: a new radiopharmaceutical for abscess detection. Radiology 128:697–699
Llinas M, Klein MP, Neilands JB (1970) Solution conformation of ferrichromes a microbial iron transport cyclohexapeptide, as deduced by high resolution proton magnetic resonance. J Mol Biol 52:399–414
Emery T (1986) Exchange of iron by gallium in siderophores. Biochemistry 25:4629–4633
Emery T, Hoffer PB (1980) Siderophore-mediated mechanism of gallium uptake demonstrated in the microorganism Ustilago sphaerogena. J Nucl Med 21:935–939
Schalk IJ, Kyslik P, Prome D, van Dorseelaer A, Poole K, Abdallah MA, Pattus F (1999) Copurification of the FpvA ferric pyoverdin receptor of Pseudomonas aeruginosa with its iron-free ligand: implications for siderophore-mediated iron transport. Biochemistry 38:9357–9365
Hantke C, Nicholson G, Rabsch W, Winkelman G (2003) Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc Natl Acad Sci 107:3677–3682
Crowley ED, Reid CPP, Szaniszlo PJ (1988) Utilization of microbial siderophores in iron acquisition by oat. Plant Physiol 87:680–685
Conti M, Eriksson L (2016) Physics of pure and non-pure positron emitters for PET: a review and a discussion. EJNMMI Physics 3:1–17
Velikyan I (2014) Prospective of 68Ga-radiopharmaceutical development. Theranostics 4:47–80
Haas H, Petrik M, Decristoforo C (2015) An iron-mimicking, trojan horse-entering fungi-has the time come for molecular imaging of fungal infections? PLoS Pathog 11:e1004568
Petrik M, Haas H, Dobrozemsky G et al (2010) 68Ga-Siderophores for PET imaging of invasive pulmonary aspergillosis: proof of principle. J Nucl Med 51:639–645
Petrik M, Haas H, Schrettl M, Helbok A, Blatzer M, Decristoforo C (2012) In vitro and in vivo evaluation of selected 68Ga-siderophores for infection imaging. Nucl Med Biol 39:361–369
Petrik M, Franssen GM, Haas H et al (2012) Preclinical evaluation of two 68Ga-siderophores as potential radiopharmaceuticals for Aspergillus fumigatus infection imaging. Eur J Nucl Med Mol Imaging 39:1175–1183
Petrik M, Vlckova A, Novy Z, Urbanek L, Haas H, Decristoforo C (2015) Selected 68Ga-siderophores versus 68Ga-colloid and 68Ga-citrate: biodistribution and small animal imaging in mice. Biomed Pap Med Fac Univ Palacky Olomouc 159:60–66
Pluhacek T, Petrik M, Luptakova D, Benada O, Palyzova A, Lemr K, Havlicek V (2016) Aspergillus infection monitored by multimodal imaging in a rat model. Proteomics 16:1785–1792
Petrik M, Haas H, Laverman P, Schrettl M, Franssen GM, Blatzer M, Decristoforo C (2014) 68Ga-Triacetylfusarinine C and 68Ga-Ferrioxamine E for Aspergillus infection imaging: uptake specificity in various microorganisms. Mol Imaging Biol 16:102–108
Petrik M, Zhai C, Novy Z, Urbanek L, Haas H, Decristoforo C (2016) In vitro and in vivo comparison of selected Ga-68 and Zr-89 labelled siderophores. Mol Imaging Biol 18:344–352
Ardon O, Nudelman R, Caris C, Libman J, Schanzer A, Chen Y, Hadar Y (1998) Iron uptake in Ustilago maydis: tracking the iron path. J Bacteriol 180:2021–2026
Lytton SD, Cabantchik ZI, Libman J, Shanzer A (1991) Reversed siderophores as antimalarial agents. II. Selective scavenging of Fe(III) from parasitized erythrocytes by a fluorescent derivative of desferal. Mol Pharmacol 40:584–590
Larcher G, Dias M, Razafimandimby B, Bomal D, Bouchara JP (2013) Siderophore production by pathogenic Mucorales and uptake of deferoxamine B. Mycopathologia 176:319–328
Noel S, Guillon L, Schalk IJ, Mislin GLA (2011) Synthesis of fluorescent probes based on the pyochelin siderophore scaffold. Org Lett 13:844–847
de Carvalho CC, Fernandes P (2014) Siderophores as “Trojan Horses”: tackling multidrug resistance? Front Microbiol 5:290
Banin E, Lozinski A, Brady KM, Berenshtein E, Butterfield PW, Moshe M, Chevion M, Greenberg EP, Banin E (2008) The potential of desferrioxamine-gallium as an anti-Pseudomonas therapeutic agent. Proc Natl Acad Sci 105:16761–16766
Roosenberg JM, Lin YM, Lu Y, Miller MJ (2000) Studies and syntheses of siderophores, microbial iron chelators, and analogs as potential drug delivery agents. Curr Med Chem 7:159–197
Page MGP (2013) Siderophore conjugates. NY Acad Sci 1277:115–126
Mislin GLA, Schalk IJ (2014) Siderophore-dependent iron uptake systems as gates for antibiotic Trojan horse strategies against Pseudomonas aeruginosa. Metallomics 6:408–420
Koizumi K, Tonami N, Hisada K (1982) Deferoxamine mesylate enhancement of 67Ga tumor-to-blood ratios and tumor imaging. Eur J Nucl Med 7:229–233
Yokoyama A, Ohmomo Y, Horiuchi K, Saji H, Tanaka H, Yamamoto K, Ishii Y, Torizuka K (1982) Deferoxamine, a promising bifunctional chelating agent for labeling proteins with gallium: Ga-67 DF-HSA: concise communication. J Nucl Med 23:909–914
Ohmomo Y, Yokoyama A, Suzuki J, Tanaka H, Yamamoto K, Horiuchi K, Ishii Y, Torizuka K (1982) 67Ga-labeled human fibrinogen: a new promising thrombus imaging agent. Eur J Nucl Med 7:458–461
Yamamoto K, Senda M, Fujita T, Kumada K, Fukui K, Yonekura Y, Yokoyama A, Torizuka K (1988) Positive imaging of venous thrombi and thromboemboli with Ga-67 DFO-DAS-fibrinogen. Eur J Nucl Med 14:60–64
Kojima S, Jay M (1987) Comparisons of labeling efficiency, biological activity and biodistribution among 125I-, 67Ga-DTPA-and 67Ga-DFO-lectins. Eur J Nucl Med 13:366–370
Motta-Hennessy C, Eccles SA, Dean C, Coghlan G (1985) Preparation of 67Ga-labelled human IgG and its Fab fragments using desferoxamine as chelating agent. Eur J Nucl Med 11:240–245
Koizumi M, Endo K, Kunimatsu M, Sakahara H, Nakashima T, Kawamura Y, Watanabe Y, Saga T, Konishi J, Yamamuro T et al (1988) 67Ga-labeled antibodies for immunoscintigraphy and evaluation of tumor targeting of drug–antibody conjugates in mice. Cancer Res 48:1189–1194
Bartal AH, Lavie E, Boazi M, Weininger J, Bitton M, Iosilevsky G, Front D, Hirshaut Y, Robinson E (1987) Human sarcoma-associated murine monoclonal antibody labeled with indium-111, gallium-67, and iodine-125. NCI Monogr 3:153–155
Wang TS, Fawwaz RA, Van Heertum RL (1993) Amino-dextran-deferoxamine: a potential polymeric heterobifunctional agent for high-level 111In-labeling of anti-melanoma monoclonal antibody TP41.2. J Nucl Biol Med 37:97–103
Govindan SV, Michel RB, Griffiths GL, Goldenberg DM, Mattes MJ (2005) Deferoxamine as a chelator for 67Ga in the preparation of antibody conjugates. Nucl Med Biol 32:513–519
Vosjan MJ, Perk LR, Roovers RC, Visser GW, Stigter-van Walsum M, van Bergen En Henegouwen PM, van Dongen GA (2011) Facile labelling of an anti-epidermal growth factor receptor Nanobody with 68Ga via a novel bifunctional desferal chelate for immuno-PET. Eur J Nucl Med Mol Imaging 38:753–763
Wang S, Lee RJ, Mathias CJ, Green MA, Low PS (1996) Synthesis, purification and tumor cell uptake of 67Ga–deferoxamine–folate, a potential radiopharmaceutical for tumor imaging. Bioconjug Chem 7:56–62
Smith-Jones PM, Stolz B, Bruns C et al (1994) Gallium-67/gallium-68-[DFO]-octreotide—a potential radiopharmaceutical for PET imaging of somatostatin receptor-positive tumors: synthesis and radiolabeling in vitro and preliminary in vivo studies. J Nucl Med 35:317–325
Stolz B, Smith-Jones P, Albert R, Reist H, Maecke H, Bruns C (1994) Biological characterisation of [67Ga] or [68Ga] labelled DFO-octreotide (SDZ 216-927) for PET studies of somatostatin receptor positive tumors. Horm Metab Res 26:453–459
Heppeler A, Froidevaux S, Eberle AN, Maecke HR (2000) Receptor targeting for tumor localisation and therapy with radiopeptides. Curr Med Chem 7:971–994
Caraco C, Aloj L, Eckelman W (1998) The gallium–deferoxamine complex: stability with different deferoxamine concentrations and incubation conditions. Appl Radiat Isot 49:1477–1479
Zhang Y, Hong H, Cai W (2011) PET tracers based on Zirconium-89. Curr Radiopharm 4:131–139
Severin GW, Engle JW, Barnhart TE, Nickles RJ (2011) Zr-89 radiochemistry for positron emission tomography. Med Chem 7:389–394
Nayak TK, Brechbiel MW (2009) Radioimmunoimaging with longer-lived positron-emitting radionuclides: potentials and challenges. Bioconjug Chem 20:825–841
Fischer G, Seibold U, Schirrmacher R, Wangler B, Wangler C (2013) 89Zr, a radiometal nuclide with high potential for molecular imaging with PET: chemistry, applications and remaining challenges. Molecules 18:6469–6490
Deri MA, Zeglis BM, Francesconi LC, Lewis JS (2013) PET imaging with 89Zr: from radiochemistry to the clinic. Nucl Med Biol 40:3–14
Meijs WE, Herscheid JD, Haisma HJ, Pinedo HM (1992) Evaluation of desferal as a bifunctional chelating agent for labeling antibodies with Zr-89. Int J Rad Appl Instrum A 43:1443–1447
Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS (2010) 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med 51:1293–1300
Börjesson PK, Jauw YW, Boellaard R, de Bree R, Comans EF, Roos JC, Castelijns JA, Vosjan MJ, Kummer JA, Leemans CR, Lammertsma AA, van Dongen GA (2006) Performance of immuno-positron emission tomography with zirconium-89-labeled chimeric monoclonal antibody U36 in the detection of lymph node metastases in head and neck cancer patients. Clin Cancer Res 12:2133–2140
Verel I, Visser GW, Boellaard R, Stigter-van Walsum M, Snow GB, van Dongen GA (2003) 89Zr immuno-PET: comprehensive procedures for the production of 89Zr-labeled monoclonal antibodies. J Nucl Med 44:1271–1281
Perk LR, Vosjan MJ, Visser GW, Budde M, Jurek P, Kiefer GE, van Dongen GA (2010) p-Isothiocyanatobenzyl-desferrioxamine: a new bifunctional chelate for facile radiolabeling of monoclonal antibodies with zirconium-89 for immuno-PET imaging. Eur J Nucl Med Mol Imaging 37:250–259
Vosjan MJ, Perk LR, Visser GW, Budde M, Jurek P, Kiefer GE, van Dongen GA (2010) Conjugation and radiolabeling of monoclonal antibodies with zirconium-89 for PET imaging using the bifunctional chelate p-isothiocyanatobenzyl-desferrioxamine. Nat Protoc 5:739–743
Jacobson O, Zhu L, Niu G, Weiss ID, Szajek LP, Ma Y, Sun X, Yan Y, Kiesewetter DO, Liu S, Chen X (2011) MicroPET imaging of integrin alphavbeta3 expressing tumors using 89Zr-RGD peptides. Mol Imaging Biol 13:1224–1233
Keliher EJ, Yoo J, Nahrendorf M, Lewis JS, Marinelli B, Newton A, Pittet MJ, Weissleder R (2011) 89Zr-labeled dextran nanoparticles allow in vivo macrophage imaging. Bioconjug Chem 22:2383–2389
Miller L, Winter G, Baur B, Witulla B, Solbach C, Reske S, Lindén M (2014) Synthesis, characterization, and biodistribution of multiple 89Zr-labeled pore-expanded mesoporous silica nanoparticles for PET. Nanoscale 6:4928–4935
Ruggiero A, Villa CH, Holland JP, Sprinkle SR, May C, Lewis JS, Scheinberg DA, McDevitt MR (2010) Imaging and treating tumor vasculature with targeted radiolabeled carbon nanotubes. Int J Nanomed 5:783–802
Heuveling DA, Visser GWM, Baclayon M, Roos WH, Wuite GJL, Hoekstra OS, Leemans CR, de Bree R, van Dongen GAMS (2011) Zr-89-Nanocolloidal albumin-based PET/CT lymphoscintigraphy for sentinel node detection in head and neck cancer: preclinical results. J Nucl Med 52:1580–1584
Evans MJ, Holland JP, Rice SL, Doran MG, Cheal SM, CamposC Carlin SD, Mellinghoff IK, Sawyers CL, Lewis JS (2013) Imaging Tumor Burden in the Brain with Zr-89-Transferrin. J Nucl Med 54:90–95
Holland JP, Evans MJ, Rice SL, Wongvipat J, Sawyers CL, Lewis JS (2012) Annotating MYC status with Zr-89-transferrin imaging. Nat Med 18:1586–1597
Chang AJ, DeSilva R, Jain S, Lears K, Rogers B, Lapi S (2012) 89Zr-radiolabeled trastuzumab imaging in orthotopic and metastatic breast tumors. Pharmaceuticals 5:79–93
Perk LR, Visser GW, Vosjan MJ, Stigter-van Walsum M, Tijink BM, Leemans CR, van Dongen GA (2005) 89Zr as a PET surrogate radioisotope for scouting biodistribution of the therapeutic radiometals 90Y and 177Lu in tumor-bearing nude mice after coupling to the internalizing antibody cetuximab. J Nucl Med 46:1898–1906
Patra M, Bauman A, Mari C, Fischer CA, Blacque O, Häussinger D, Gasser G, Mindt TL (2014) An octadentate bifunctional chelating agent for the development of stable zirconium-89 based molecular imaging probes. Chem Commun (Camb) 50:11523–11525
Knetsch PA, Zhai C, Rangger C, Blatzer M, Haas H, Kaeopookum P, Haubner R, Decristoforo C (2015) [68Ga] FSC-(RGD)3 a trimeric RGD peptide for imaging αvβ3 integrin expression based on a novel siderophore derived chelating scaffold—synthesis and evaluation. Nucl Med Biol 42:115–122
Zhai C, Summer D, Rangger C, Haas H, Haubner R, Decristoforo C (2015) Fusarinine C, a novel siderophore-based bifunctional chelator for radiolabeling with Gallium-68. J Label Comp Radiopharm 58:209–214
Zhai C, Summer D, Rangger C, Franssen GM, Laverman P, Haas H, Petrik M, Haubner R, Decristoforo C (2015) Novel bifunctional cyclic chelator for (89)Zr labeling-radiolabeling and targeting properties of RGD conjugates. Mol Pharm 12:2142–2150
Zhai C, Franssen GM, Petrik M, Laverman P, Summer D, Rangger C, Haubner R, Haas H, Decristoforo C (2016) Comparison of Ga-68-Labeled Fusarinine C-Based Multivalent RGD Conjugates and [68Ga]NODAGA-RGD-In Vivo Imaging Studies in Human Xenograft Tumors. Mol Imaging Biol. doi: 10.1007/s11307-016-0931-3. [Epub ahead of print]
Acknowledgments
Open access funding provided by University of Innsbruck and Medical University of Innsbruck. We gratefully acknowledge the financial support of National Programme of Sustainability (Project No. LO1304 to M.P.) and Technology Agency of the Czech Republic (Project No. TE01020028 to M.P.), of the Austrian Science Foundation (FWF) Grant P 25899-B23 (to C.D.) and the China Scholarship Council (to C.Z.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors (Milos Petrik, Hubertus Haas, Chuangyan Zhai and Clemens Decristoforo) declare to have no conflict of interest
All institutional and national guidelines for the care and use of laboratory animals were followed.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Petrik, M., Zhai, C., Haas, H. et al. Siderophores for molecular imaging applications. Clin Transl Imaging 5, 15–27 (2017). https://doi.org/10.1007/s40336-016-0211-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40336-016-0211-x