
Abstract
As both reactive oxygen species (ROS) production and microRNA expression signature have been associated with tumor development, progression, metastasis, and therapeutic response, it is important to understand the crosstalk between ROS and microRNAs. Indeed, growing evidence suggests a reciprocal connection between ROS signaling and microRNA pathway, resulting in diverse biological effects in cancer cells. In this mini review, we discussed the ROS-responsive microRNAs that have implications in cancer and the possible mechanisms in which ROS regulate microRNAs. We also highlighted the microRNAs which are able to modify cellular ROS homeostasis during tumorigenesis, their biological targets and subsequent functions. As the use of antioxidants is limited due to the diverse or even opposing roles of ROS signaling in cancer, the discovery of ROS-responsive microRNAs provides a potential new strategy to specifically overcome ROS-mediated tumor progression or benefit from ROS-induced apoptosis.
Similar content being viewed by others

Introduction
Reactive oxygen species (ROS) are oxygen-containing and chemically reactive species formed by incomplete one-electron reduction of oxygen, which include superoxide (O2−), hydroxyl radical (OH−), hydrogen peroxide (H2O2), nitric oxide (NO), peroxynitrite (ONOO−), and nitrogen dioxide radical (NO2) [1]. ROS are naturally produced by cells through aerobic metabolism and are known to play a dual role in biological systems—with potentially beneficial or harmful effect [2]. Mitochondria respiratory chain, NADPH oxidase, and peroxisomes are the major endogenous sources of ROS, while environmental agents, ionized radiation, UV light, and chemical drugs contribute to the induction of exogenous sources of ROS. Low levels of ROS facilitate cell survival as a second messenger and mediate a plethora of intracellular signal transduction events, whereas high levels of ROS induce cell apoptosis and necrosis. ROS has been implicated in a number of pathologies, especially in cancer. Excessive and sustained ROS production has been found in a variety of cancer cells, which is strongly correlated with the tumorigenic potential of cancer cells [3]. However, ROS-mediated signaling pathways not only promote cell survival, oncogenic transformation, and metastasis, but also render cancer cells resistant to anticancer drugs [4]. Still, the mechanisms of ROS signaling and underlined redox adaptation in cancer cells remain elusive.
The past decade has witnessed a great progress on microRNA research. MicroRNAs are a small class of endogenous 18 ∼ 25 nucleotide long, non-coding RNA molecules that regulate gene expression at the posttranscriptional levels [5]. They can induce either mRNA degradation or translational suppression by binding to their target messenger RNAs, especially 3′untranslated region (UTR) based on the sequence complementarities. Alternatively, they can also cause the translational activation of their respective targets [6]. Up to one third of human mRNAs are potential microRNA targets. The observation that the frequent deletion of chromosome 13q14 causes the loss of miR-15a and miR-16-1 in chronic lymphocytic leukemia (CLL) is the first evidence of the involvement of microRNAs in human cancer [7]. It was further revealed that these non-coding genes are frequently located within cancer-associated genomic regions. Since that, increasing number of studies demonstrate that microRNAs may function as potential tumor suppressors or oncogenes in all stages of cancer development. Deregulations of microRNA expression have been associated with tumor development, progression, metastasis, and therapeutic responses, and have recently been known as one of the hallmarks of cancer [8].
Since both ROS and microRNAs are dysregulated in cancers, it is important to understand the crosstalk between ROS and microRNAs. Accumulative evidence suggests a reciprocal connection between ROS signaling and microRNA pathways. Some microRNAs, so-called ROSmirs, are regulated by oxidative stress to mediate the expression levels of their direct targets in response to ROS. For example, ionizing radiation, as a standard way to treat malignancy, causes severe DNA damage to kill cells, resulting in ROS accumulation and altered microRNA expression levels [9]. The addition of free radical scavenger cysteine can abrogate the microRNA response induced by radiation, suggesting ROS production is an upstream event of the microRNAs in response to exogenous genotoxic stress. On the other hand, as microRNAs may directly regulate 30 % of genes in a cell, it is not surprising that microRNAs are involved in all major cellular functions including modification of cellular redox homeostasis such as ROS production. In this review, we will highlight literatures regarding how ROS exert biological effects through microRNAs, and how microRNAs regulate cellular redox homeostasis in different types of cancers.
ROS-Responsive MicroRNAs
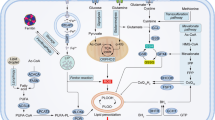
Cancer cells normally adapt to persistent oxidative stress in the cells. A global microRNA profiling analysis revealed that exposure to hydrogen peroxide (H2O2) causes changes of a set of microRNA contents, suggesting that the ROS-sensitive microRNAs may be important in cancer cells in response to ROS [9, 10]. Accumulative studies show that intracellular ROS can either inhibit or induce microRNA expression level, which generates subsequent biological effects through the regulation of their direct target genes. For example, macrophages are known to produce ROS during phagocytosis in response to stimuli [11]. Macrophage-derived ROS are not only critical for microbial killing but also may act as a carcinogen causing tumor initiation and promotion. Exposure to exogenous H2O2 in macrophage cell line RAW 246.7 alters the microRNA expression levels, including downregulation of miR-27a*, miR-27b*, miR-29b*, and miR-24-2, and upregulation of miR-21 [12]. In a context of cancer microenvironment, increased infiltrated macrophages in tumor stroma are found to suppress miR-328 expression level through ROS production, and then induced CD44 expression in gastric cancer cells for regulating tumor progression [13]. Our group previously showed that ROS are involved in insulin-regulated aerobic glycolysis in hepatocellular carcinoma cells [14]. Insulin-induced ROS inhibit miR-145 and miR-128 expression levels and upregulate the key enzyme M2 isoform of pyruvate kinase 2 (PKM2) to promote insulin-induced glucose consumption and lactate production. Although the cause-effect relationship between ROS and microRNAs has been proved through experiments by cell exposure to H2O2 or ROS scavengers, the mechanism underlying ROS-regulated microRNA expression is complicated and poorly understood. The major proposed mechanisms so far are showed in Fig. 1.

Schematic model showing mechanisms in which ROS regulate microRNA expression. ROS are involved in every step of miRNA biogenesis. ROS can induce epigenetic alterations of miRNA genes. For example, ROS inhibit and enhance expression of certain miRNA genes through DNMT1 and HDACs, respectively. ROS can also activate transcription factors to induce miRNA expression. Moreover, Drosha and Dicer, which are two essential enzymes for miRNA biogenesis, can be directly or indirectly regulated by ROS
ROS Regulate MicroRNA Biogenesis Enzymes
ROS can affect microRNA expression through the components of microRNA processing machinery. The biogenesis of miRNAs is controlled by two RNase-dependent processing steps.
MicroRNA genes are transcribed by RNA polymerase II into longer molecules (pri-microRNAs) which are further cleaved by RNA-specific RNAse III type ribonuclease Drosha into hairpin intermediates (pre-microRNAs). The pre-microRNAs then transport through nuclear pores into the cytoplasm with the aid of Exportin 5 and are cleaved by another ribonuclease Dicer into double-stranded mature microRNAs [15]. A study showed that aging-related oxidative stress is associated with downregulation of Dicer in cerebromicrovascular endothelial cells (CMVECs) [16]. Exogenous H2O2 exposure in endothelial cells causes significant decrease of Dicer expression resulting in downregulation of 89 % of microRNAs that are normally expressed in CMVECs. Interestingly, Dicer expression level can also affect ROS production as a negative feedback loop to maintain cellular homeostasis. Knockdown of Dicer in human microvascular endothelial cells (HMECs) decreased ROS production in the cells [17]. Further mechanism study showed that microRNA-deficient HMECs due to Dicer knockdown show higher levels of nuclear HBP1, a suppressor transcription factor of p47phox. The inhibition of p47phox by HBP1, a key subunit of NADPH oxidase, abolished ROS induction and impaired angiogenesis property of endothelial cells [17].
ROS Regulate MicroRNA Expression Through Transcription Factors
ROS function as intermediates in the activation of stress-related transcription factors such as p53, nuclear factor (NF)-κB, c-jun, FOXO, and HIF in response to stress agents [18, 19]. Therefore, it is plausible to speculate that ROS can regulate certain microRNAs through these transcription factors. Tumor suppressor protein p53 is a central molecule in maintaining genomic integrity by inducing cell cycle arrest, senescence, and apoptosis via differential activation of target genes [20]. There are broad interactions between ROS and p53 pathway. As a cellular stress-related transcription factor, p53 expression can be induced by ROS to selectively activate p53 target genes for protecting the genome stability [21]. In addition, p53 is involved in microRNA processing pathway such as Drosha-mediated pri-microRNA processing and in directly transactivating some microRNA genes to promote the transcription of microRNAs such as miR-34a, miR-200c, and miR-17-92 clusters. p53 interacts with the Drosha processing complex through the association with DEAD-box RNA helicase p68 (also known as DDX5) and facilitates the processing of primary microRNAs to precursor microRNAs [22]. A number of stress-induced microRNAs are induced in a p53-dependent manner such as let-7s, miR-34s, and miR-200s. The miR-200 family has been implicated as a tumor suppressor by inhibition of epithelial-mesenchymal transition (EMT) process, an initial event for cancer metastasis [23]. Overexpression of the miR-200 family leads to a reversal of EMT in various cancers including bladder cancer, gastric cancer, ovarian cancer, pancreatic cancer, prostate cancer, and others [24]. It is reported that miR-200c is upregulated upon H2O2 exposure in vascular endothelial cells, which leads to cellular apoptosis and senescence through ZEB1 inhibition [25]. Knockdown of p53 remarkably reverses H2O2-induced miR-200c expression, an indicative of p53 regulation.
In addition to its pivotal role in the canonical Wnt pathway, β-catenin has been found to be involved in transcription regulation induced by ROS through interaction with the transcription factor FOXO [26, 27]. A recent study indicated that ROS-induced miR-182 is not regulated by p53 but by β-catenin in high-grade serous ovarian carcinoma [28]. They found that ROS greatly upregulate β-catenin resulting in its nucleus translocation. In p53 intact fallopian tube secretory epithelial (FTSE) cells, overexpression of miR-182 triggers cellular senescence by p53-mediated p21 upregulation. However, in cells with p53 mutations, miR-182 acts as an oncogene to promote tumorigenesis by impairing DNA damage responses mediated by ROS and β-catenin [28].
ROS are often generated within inflammatory environment to activate NF-κB in cells by various mechanisms [29]. Several microRNAs such as miR-9, miR-21, miR-143, and miR-146 and miR-224 have been validated to be directly transcriptionally regulated by NF-κB [30–35]. Among them, miR-21 is one of the most investigated oncogenic microRNAs, which is generally upregulated in cancer cells [36]. miR-21 possesses pro-survival and antiapoptotic properties by directly targeting tumor suppressor proteins PTEN, PDCD4, IGFBP3, and MKK3 [37–40]. It was reported that ROS induce miR-21 expression and functions, which contributes to the highly invasive and metastatic phenotype of prostate cancer cells [41]. NF-κB activation might be one of the mechanisms for ROS-mediated miR-21 induction. Environmental agents are one of the exogenous sources of ROS in the body. ROS-induced miR-21 is involved in arsenic-induced cell malignant transformation, and NF-κB mediates miR-21 induction upon ROS exposure by binding directly to the promoter of miR-21 gene [42]. In addition, NF-κB can also indirectly regulate microRNA expression through the synthesis of proteins that are responsible for microRNA biogenesis. For example, Lin28, a microRNA processing inhibitor, can be activated by NF-κB activation, resulting in rapid reduction of let-7 microRNA levels, which contributes to Src-induced cellular transformation [43].
ROS Regulate MicroRNA Expression Through Epigenetic Modification
Altered epigenomic patterns as major features of cancer are associated with aberrant microRNA expression profiles in cancer. The main epigenetic changes in mammals, particularly in humans, are DNA methylation and posttranslational histone modifications including acetylation, methylation, and phosphorylation [44]. Like protein-coding DNA sequences, microRNA genes in non-coding regions may undergo DNA methylation and histone modification.
We previously described that endogenous ROS inhibit miR-199a and miR-125b expression in ovarian cancer cells, and overexpression of these microRNAs inhibit tumor-induced angiogenesis associated with the decreased expression of HIF-1α and VEGF [45]. We further found that the promoter regions of both miR-199a and miR-125b genes are hypermethylated upon H2O2 exposure analyzed through methylation-specific PCR and bisulphite sequencing assays, which is mediated by DNMT1 upregulation [46]. DNMT1 functions in a way of DNMT1/transcription factor-including complex, which is recruited to the oxidative stress-damaged DNA in order to silence gene transcription temporarily for the DNA repair and/or microRNA expression [47]. Redox-sensitive transcription factors in the DNMT1 complex such as NF-κB may act as cofactors to promote direct binding of DNMT1 into certain microRNA promoters.
Histone acetylation refers to the acetylation of lysine residues, and the levels of histone acetylation play a crucial role in chromatin remodeling to regulate gene transcription. The deacetylation of lysine residues by histone deacetylases (HDACs) is associated with a more condensed chromatin state and transcriptional gene silencing [48]. Several studies show the reduced activities of HDACs under oxidative stress may cause alteration of microRNA expression levels [49, 50]. For example, miR-466h-5p is shown to have a pro-apoptotic role through targeting several antiapoptotic genes including bcl2l2, birc6, dad1, smo, and stat5a [51]. Accumulation of ROS caused by glucose deprivation increases miR-466h-5p expression levels through inhibition of HDAC2, a direct target of miR-466h-5p, which increases acetylation and induction of the microRNA expression, resulting in increased apoptosis [49].
MicroRNAs Regulate ROS Production
In addition to as essential regulators of the ROS-mediated stress response across multiple species, microRNAs are capable of regulating redox homeostasis by modulating genes that are either ROS activators or scavengers. MicroRNAs that are associated with ROS levels and related signaling pathways in cancer cells are listed in Table 1.
Inhibition of ROS Production
Certain microRNAs may inhibit ROS levels through targeting genes that are responsible for ROS synthesis. NADPH oxidases are considered as the major source of ROS production in epithelial cells [66]. NADPH oxidase-derived ROS may elevate the risk for genomic instability and cancer [67]. NOX2 subunit is the catalytic core of NADPH oxidase complex. A study showed that NOX2 is the direct target of miR-34a, and restoration of miR-34a in glioma cells induces apoptosis through NOX2-derived ROS generation [52]. Proline oxidase (POX), as a mitochondria suppressor, is a p53-induced redox gene that can generate ROS and mediate apoptosis in tumor cells [68]. Upregulation of miR-23b* is found in renal cancer [53]. As a negative regulator of POX, miR-23b* knockdown increases apoptosis through induction of mitochondria-derived ROS and inhibition of HIF signaling, thus suppresses kidney tumor growth.
Enhancement of ROS Production
The enzymatic and non-enzymatic antioxidant defense systems including superoxide dismutase (SOD), glutathione peroxidase (GPX), catalase, ascorbic acid (vitamin C), and α-, glutathione (GSH) act against ROS accumulation in cells in order to maintain cellular oxygen radical homeostasis [69]. Several microRNAs increase cellular ROS levels through targeting antioxidants. SOD is the antioxidant enzyme that catalyzes the dismutation of the highly reactive superoxide anion to O2 and to the less reactive species H2O2. In humans, there are three forms of SOD: cytosolic Cu/Zn-SOD (SOD1), mitochondrial Mn-SOD (SOD2), and extracellular SOD (SOD3) [70]. A study showed that miR-21 potentiates ionizing irradiation (IR)-induced ROS production by its direct target SOD2 and an indirect target SOD3 through TNFα, resulting in an increase of IR-induced cell transformation [54]. Hydrogen peroxide (H2O2) is eliminated by antioxidant enzyme catalase and GPX. miR-551b expression level is high in apoptosis-resistant lung cancer cells. Further study indicates that higher miR-551b expression levels inhibit catalase expression and enhance ROS accumulation, which activates a redox-active oncogene Mucin-1 (MUC1) for survival and chemoresistance in cancer cells [55]. Upregulation of miR-155 is involved in K-Ras-induced oncogenic transformation in pancreatic cancer through redox regulation [56]. The study suggests that miR-155 promotes ROS levels through directly targeting a transcription factor Foxo3a that induces SOD2 and catalase transcription [56]. Like miR-21, the miR-200 family not only responds to oxidative stress as a downstream effector of ROS, but also regulates intracellular ROS levels through antioxidants. For instance, miR-200c negatively regulates three redox proteins, PRDX2, GABPA/Nrf2, and SESN1, all of which contribute to antioxidant defense system in cancer cells. Overexpression of miR-200c augments ROS and p21 expression levels, leading to increased radiosensitivity in lung cancer cells [57].
Nuclear factor erythroid2-related factor2 (Nrf2) is a redox-sensitive transcription factor serving as a “master regulator” of cell survival through the coordinated induction of antioxidants and phase II detoxification enzymes via antioxidant-response element (ARE) in the promoter of target genes [71]. Activation of Nrf2 will increase the transcription of antioxidant defense genes such as catalase and SOD. As a result, microRNAs that are capable of regulating Nrf2 would have an impact on intracellular ROS levels. Nrf2 pathway seems to play dual roles in cancer cell biology, which depends on the activation of Nrf2-dependent pro-survival genes or antioxidant genes [72]. A study reported a role of miR-28 in regulation of Nrf2 expression in breast cancer. miR-28 not only targets the 3′UTR region of Nrf2 mRNA but also reduces the stability of Nrf2 mRNA and protein. As a result, loss of Nrf2 increases colony formation in breast cancer cells [58].
Mitochondria respiratory complex is the major source of free radicals, and compromised mitochondria function can augment ROS production. The hypoxia-inducible miR-210 is known to regulate cancer cell metabolism by reducing mitochondria respiratory activity and activating superoxide production [73]. The possible mechanism involves inhibition of mitochondrial iron-sulfur cluster scaffold homologue (ISCU), a target gene of miR-210. ISCU is essential for the assembly of Fe-S cluster, and loss of function of ISCU can decrease mitochondria activity and disrupt iron homeostasis [59]. miR-210 increases ROS generation by downregulation of ISCU [60]. In addition, inhibition of ISCU by miR-210 causes ROS induction in hypoxia, resulting in a shift to glycolysis in normoxia and enhanced cell survival [61]. Another group also found that miR-210 increases ROS levels in colon cancer cells under hypoxia condition, but argued that the upregulation of ROS caused by miR-210 may be due to other identified targets because knockdown of ISCU does not show a significant change of ROS levels [74]. The polycomb gene Bim1 has functions in maintaining mitochondrial activities and redox homeostasis. Cells derived from Bmi1-/- mice show impaired mitochondrial activities, a marked increase in the intracellular levels of reactive oxygen species and subsequent engagement of DNA damage response pathway [75]. It is reported that miR-128a increases ROS levels via the specific inhibition of Bim1, thus represses medulloblastoma cancer cell growth [62].
MicroRNAs can regulate ROS levels not only through ROS synthesis, but also by redox-active signaling pathways such as p38α MAPK and NF-κB. The p38α MAPK usually acts as a sensor of oxidative stress and suppresses tumorigenesis by promoting apoptosis [76]. Inactivation of p38α is associated with ROS accumulation and the subsequent activation of antioxidant defense system [77]. Two members of the miR-200 family, miR-141 and miR-200a, are identified to directly target p38α and modulate the oxidative stress response in ovarian cancer [63]. Interestingly, p38α deficiency achieved by upregulation of miR-141 and miR-200a potentiates tumor growth, but at the same time, sensitizes cancer cells in response to chemotherapy evidenced by the improved survival in patients. It further indicates the diverse and even opposing effects of ROS on cell behavior. NF-κB activity can inhibit ROS by activation of antioxidant enzymes. Upregulation of miR-506 occurs in 83 % lung cancer; however, miR-506 is considered as a tumor suppressor instead of an oncomir. It is known that miR-506 negatively regulates NF-κB p65 expression and thus increases ROS production, which, in turn, activates p53 to kill cancer cells through increased apoptosis [64].
ROS and miRNAs in Carcinogenesis
Cancer development is a complex and multiphase process with three major stages: initiation, promotion, and progression. Oxidative stress inducing the accumulation of DNA damage or mutation is the critical step in cancer initiation [78]. In addition, the endogenous elevated ROS production found in many tumor cells activates oncogenic signaling pathways, which results in cancer promotion and progression [78, 79]. As for miRNAs, it contributes to carcinogenesis through functional interactions with oncogenes or tumor suppressor genes. Overexpression, deletion, epigenetic silencing, or mutation of mature miRNAs may inhibit or stimulate oncogenic activity through regulation of the expression level of the target protein [80]. Therefore, ROS may affect carcinogenesis through miRNAs by alteration of methylation status of miRNA genes, miRNA biogenesis, and oxidative DNA damage-induced mutation of miRNA genes or mature miRNA sequences. Similarly, miRNAs are capable of regulation of intracellular ROS levels by targeting proteins responsible for ROS generation and elimination, thus indirectly are involved in ROS-mediated carcinogenesis. It is worth noting that ROS and miRNAs can act either synergistically or antagonistically to influence cancer development. A few targets or pathways cannot fully explain the biological effect; it depends on the net result of multiple pathways and the dominant molecules in the specific context.
Therapeutic Implications and Challenges
Growing studies have demonstrated a dual role of ROS in cancer. Excessive and sustained ROS production promotes carcinogenesis by inducing DNA damage causing genomic instability. ROS can positively respond to mitogenic stimulation such as growth factors to active receptor tyrosine kinases, which are necessary in cancer progression and maintenance. On the contrary, intracellular ROS are also critical to activate apoptosis in response to exogenous agents such as chemical drugs and ionized radiation through activation of pro-apoptotic signaling molecules including p53 and p38 MAPK, which sensitize tumor cells to the treatment. Therefore, the use of antioxidants in cancer treatment has a limited effect [81–83]. In some cases, antioxidant treatments such as N-acetylcysteine (NAC) and vitamin E even speed cancer progression in an animal model [84]. The discovery of ROS-responsive microRNAs provides a potential new strategy to specifically overcome ROS-mediated tumor progression or benefit from ROS-induced apoptosis. As discussed in the majority of studies in this review, the roles of microRNAs in cellular adaptation to ROS are different in cells based on cell types or tumors. This raises the possibilities to apply specific microRNAs as therapeutic target(s) in different contexts. Theoretically, microRNA-target therapy has several potential advantages. First, microRNAs are short and much conserved across multiple species with known sequences. Secondly, an individual microRNA is able to target multiple genes within defined pathways; it would be possible to regulate the entire oncogenic or tumor suppressor network via pharmacological modulation of cancer-associated microRNAs [85]. Several clinical trials based on microRNA-based therapy have been on the way [86]. The locked nucleic acid (LNA)-modified anti-miR inhibitor against miR-122 called miravirsen is the first miRNA-targeted drug to enter human clinical trials to treat HCV [85, 87]. Clinical data from the phase IIa trial show that miravirsen is well-tolerated and generally provides antivirus activity in HCV patients. Although the great progress has been made, challenges are remaining in several aspects. Firstly, new techniques are needed to design molecules with appropriate modifications to specifically inhibit or “mimic” mature microRNAs in vivo. Second, the application of microRNAs in humans is also hindered by lack of approaches to achieve high efficiency in vivo delivery for microRNA mimic or anti-miR inhibitor. In the context of ROS signaling, further understanding of ROS-microRNA network is of importance for the application of microRNAs to ameliorate ROS-mediated oxidative stress or enhance ROS-stimulated apoptosis in response to cancer therapy.
References
D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–24.
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40.
Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205.
Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008;266:6–11.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Simone NL, Soule BP, Ly D, Saleh AD, Savage JE, DeGraff W, et al. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One. 2009;4:e6377.
Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C. Involvement of microRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J Biol Chem. 2009;284:7903–13.
Weitzman SA, Gordon LI. Inflammation and cancer: role of phagocyte-generated oxidants in carcinogenesis. Blood. 1990;76:655–63.
Thulasingam S, Massilamany C, Gangaplara A, Dai H, Yarbaeva S, Subramaniam S, et al. miR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in RAW 264.7 cells. Mol Cell Biochem. 2011;352:181–8.
Ishimoto T, Izumi D, Watanabe M, Yoshida N, Hidaka K, Miyake K, et al. Chronic inflammation with Helicobacter pylori infection is implicated in CD44 overexpression through miR-328 suppression in the gastric mucosa. J Gastroenterol. 2015;50:751–7.
Li Q, Liu X, Yin Y, Zheng JT, Jiang CF, Wang J, et al. Insulin regulates glucose consumption and lactate production through reactive oxygen species and pyruvate kinase M2. Oxidative Med Cell Longev. 2014;2014:504953.
Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res. 2005;65:3509–12.
Ungvari Z, Tucsek Z, Sosnowska D, Toth P, Gautam T, Podlutsky A, et al. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci. 2013;68:877–91.
Shilo S, Roy S, Khanna S, Sen CK. Evidence for the involvement of miRNA in redox regulated angiogenic response of human microvascular endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:471–7.
Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008;18:421–9.
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–16.
Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604.
Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–35.
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–33.
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–9.
Feng X, Wang Z, Fillmore R, Xi Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014;344:166–73.
Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarra-Ferraris G, et al. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011;18:1628–39.
Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–6.
Hoogeboom D, Burgering BM. Should I stay or should I go: beta-catenin decides under stress. Biochim Biophys Acta. 2009;1796:63–74.
Liu Y, Qiang W, Xu X, Dong R, Karst AM, Liu Z, et al. Role of miR-182 in response to oxidative stress in the cell fate of human fallopian tube epithelial cells. Oncotarget. 2015;6(36):38983–98.
Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86.
Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A. 2009;106:5282–7.
Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol. 2011;3:159–66.
Niu J, Shi Y, Tan G, Yang CH, Fan M, Pfeffer LM, et al. DNA damage induces NF-kappaB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J Biol Chem. 2012;287:21783–95.
Zhang X, Liu S, Hu T, Liu S, He Y, Sun S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology. 2009;50:490–9.
Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–6.
Scisciani C, Vossio S, Guerrieri F, Schinzari V, De IR, de D’Onorio MP, et al. Transcriptional regulation of miR-224 upregulated in human HCCs by NFkappaB inflammatory pathways. J Hepatol. 2012;56:855–61.
Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13:39–53.
Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–36.
Li Z, Deng X, Kang Z, Wang Y, Xia T, Ding N, et al. Elevation of miR-21, through targeting MKK3, may be involved in ischemia pretreatment protection from ischemia-reperfusion induced kidney injury. J Nephrol 2015.
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–58.
Yang CH, Yue J, Pfeffer SR, Fan M, Paulus E, Hosni-Ahmed A, et al. MicroRNA-21 promotes glioblastoma tumorigenesis by down-regulating insulin-like growth factor-binding protein-3 (IGFBP3). J Biol Chem. 2014;289:25079–87.
Jajoo S, Mukherjea D, Kaur T, Sheehan KE, Sheth S, Borse V, et al. Essential role of NADPH oxidase-dependent reactive oxygen species generation in regulating microRNA-21 expression and function in prostate cancer. Antioxid Redox Signal. 2013;19:1863–76.
Ling M, Li Y, Xu Y, Pang Y, Shen L, Jiang R, et al. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-kappaB in arsenite-induced cell transformation. Free Radic Biol Med. 2012;52:1508–18.
Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706.
Gronbaek K, Hother C, Jones PA. Epigenetic changes in cancer. APMIS. 2007;115:1039–59.
He J, Jing Y, Li W, Qian X, Xu Q, Li FS, et al. Roles and mechanism of miR-199a and miR-125b in tumor angiogenesis. PLoS One. 2013;8:e56647.
He J, Xu Q, Jing Y, Agani F, Qian X, Carpenter R, et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012;13:1116–22.
O’Hagan HM, Wang W, Sen S, Destefano SC, Lee SS, Zhang YW, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG islands. Cancer Cell. 2011;20:606–19.
Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25.
Druz A, Betenbaugh M, Shiloach J. Glucose depletion activates mmu-miR-466h-5p expression through oxidative stress and inhibition of histone deacetylation. Nucleic Acids Res. 2012;40:7291–302.
Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123:2921–34.
Druz A, Chu C, Majors B, Santuary R, Betenbaugh M, Shiloach J. A novel microRNA mmu-miR-466h affects apoptosis regulation in mammalian cells. Biotechnol Bioeng. 2011;108:1651–61.
Li SZ, Hu YY, Zhao J, Zhao YB, Sun JD, Yang YF, et al. MicroRNA-34a induces apoptosis in the human glioma cell line, A172, through enhanced ROS production and NOX2 expression. Biochem Biophys Res Commun. 2014;444:6–12.
Liu W, Zabirnyk O, Wang H, Shiao YH, Nickerson ML, Khalil S, et al. miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene. 2010;29:4914–24.
Zhang X, Ng WL, Wang P, Tian L, Werner E, Wang H, et al. MicroRNA-21 modulates the levels of reactive oxygen species by targeting SOD3 and TNFalpha. Cancer Res. 2012;72:4707–13.
Xu X, Wells A, Padilla MT, Kato K, Kim KC, Lin Y. A signaling pathway consisting of miR-551b, catalase and MUC1 contributes to acquired apoptosis resistance and chemoresistance. Carcinogenesis. 2014;35:2457–66.
Wang P, Zhu CF, Ma MZ, Chen G, Song M, Zeng ZL, et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget. 2015;6:21148–58.
Cortez MA, Valdecanas D, Zhang X, Zhan Y, Bhardwaj V, Calin GA, et al. Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. Mol Ther. 2014;22:1494–503.
Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat. 2011;129:983–91.
Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–84.
Kim JH, Park SG, Song SY, Kim JK, Sung JH. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via PTPN2. Cell Death Dis. 2013;4:e588.
Favaro E, Ramachandran A, McCormick R, Gee H, Blancher C, Crosby M, et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One. 2010;5:e10345.
Venkataraman S, Alimova I, Fan R, Harris P, Foreman N, Vibhakar R. MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS One. 2010;5:e10748.
Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O, et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med. 2011;17:1627–35.
Yin M, Ren X, Zhang X, Luo Y, Wang G, Huang K, et al. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-kappaB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene. 2015;34:691–703.
Kwon JE, Kim BY, Kwak SY, Bae IH, Han YH. Ionizing radiation-inducible microRNA miR-193a-3p induces apoptosis by directly targeting Mcl-1. Apoptosis. 2013;18:896–909.
Dworakowski R, Anilkumar N, Zhang M, Shah AM. Redox signalling involving NADPH oxidase-derived reactive oxygen species. Biochem Soc Trans. 2006;34:960–4.
Weyemi U, Redon CE, Parekh PR, Dupuy C, Bonner WM. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anticancer Agents Med Chem. 2013;13:502–14.
Rivera A, Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem. 2005;280:29346–54.
Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95.
Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–49.
Cheng X, Ku CH, Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic Biol Med. 2013;64:4–11.
Kwak MK, Kensler TW. Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol. 2010;244:66–76.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97.
Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29:4362–8.
Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–92.
Dolado I, Swat A, Ajenjo N, De VG, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205.
Gutierrez-Uzquiza A, Arechederra M, Bragado P, Aguirre-Ghiso JA, Porras A. p38alpha mediates cell survival in response to oxidative stress via induction of antioxidant genes: effect on the p70S6K pathway. J Biol Chem. 2012;287:2632–42.
Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–214.
Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–30.
Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–91.
Fortmann SP, Burda BU, Senger CA, Lin JS, Whitlock EP. Vitamin and mineral supplements in the primary prevention of cardiovascular disease and cancer: an updated systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2013;159:824–34.
Greenberg ER, Baron JA, Stukel TA, Stevens MM, Mandel JS, Spencer SK, et al. A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med. 1990;323:789–95.
Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361:2017–23.
Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra15.
Thorsen SB, Obad S, Jensen NF, Stenvang J, Kauppinen S. The therapeutic potential of microRNAs in cancer. Cancer J. 2012;18:275–84.
Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143–59.
Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–94.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work has been supported by the National Institutes of Health grants R01ES020868, R21CA175975, and R01CA193511.
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Free Radicals Medicine
Rights and permissions
About this article

Cite this article
He, J., Jiang, BH. Interplay Between Reactive Oxygen Species and MicroRNAs in Cancer. Curr Pharmacol Rep 2, 82–90 (2016). https://doi.org/10.1007/s40495-016-0051-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-016-0051-4