
Abstract
Coronavirus disease 2019 (COVID-19) is associated with a high inflammatory burden that can induce severe respiratory disease among other complications; vascular and neurological damage has emerged as a key threat to COVID-19 patients. Risk of severe infection and mortality increases with age, male sex, and comorbidities including cardiovascular disease, hypertension, obesity, diabetes, and chronic pulmonary disease. We review clinical and neuroradiological findings in five patients with COVID-19 who suffered severe neurological disease and illustrate the pathological findings in a 7-year-old boy with COVID-19-induced encephalopathy whose brain tissue sample showed angiocentric mixed mononuclear inflammatory infiltrate. We summarize the structural and functional properties of the virus including the molecular processes that govern the binding to its membrane receptors and cellular entry. In addition, we review clinical and experimental evidence in patients and animal models that suggests coronaviruses enter into the central nervous system (CNS), either via the olfactory bulb or through hematogenous spread. We discuss suspected pathophysiological mechanisms including direct cellular infection and associated recruitment of immune cells and neurovirulence, at least in part, mediated by cytokine secretion. Moreover, contributing to the vascular and neurological injury, coagulopathic disorders play an important pathogenic role. We survey the molecular events that contribute to the thrombotic microangiopathy. We describe the neurological complications associated with COVID-19 with a focus on the potential mechanisms of neurovascular injury. Our thesis is that following infection, three main pathophysiological processes—inflammation, thrombosis, and vascular injury—are responsible for the neurological damage and diverse pathology seen in COVID-19 patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Highly pathogenic coronavirus infections in humans include severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS). On December 31, 2019, the WHO Country Office in China was first alerted about a cluster of cases of atypical pneumonia of unknown etiology originating in Wuhan, the capital of Central China’s Hubei province. The emerging pathogen was rapidly characterized as a novel β-coronavirus and temporarily designated 2019-nCoV by the WHO [1]. The International Committee on Taxonomy on Viruses would later rename it SARS-CoV-2, given the structural similarity and sequence conservation to SARS-CoV-1, the β-coronavirus responsible for the SARS epidemic that occurred between 2002 and 2004 [2]. Coronavirus disease 2019 (COVID-19) caused by this novel virus would quickly escalate into a pandemic, spreading to countries and regions in all continents, with over 37,686,000 confirmed cases and 1,078,000 deaths worldwide, exceeding by far the total of SARS and MERS in 2002 and 2012, respectively, and representing the greatest medical challenge in decades.
The severity of COVID-19 correlates with the presence of comorbidities including cardiovascular diseases, hypertension, obesity, diabetes mellitus, chronic pulmonary diseases, and cancer. SARS-CoV-2 primarily causes a mild respiratory tract infection, but can also progress to interstitial pneumonitis and severe acute respiratory distress syndrome (ARDS), conditions most frequently seen in older patients. However, it also affects other organs including the liver, kidney, and heart. Importantly, it has become evident that COVID-19 can cause severe vascular damage and serious neurological manifestations [3, 4]. Experimental evidence in animal models shows that coronavirus’ spread starts in the olfactory bulb and progressively invades the subcortical structures and cerebral cortex [5]. Other authors theorize that coronavirus can use a hematogenous route to penetrate the central nervous system (CNS) through infected dendritic or white blood cells [6]. Experimental evidence and clinical data also suggest that the pathogenesis of neurovascular disease due to SARS-CoV-2 rests on a multifactorial process that involves primary direct infection of the vascular or neuronal cells, with subsequent inflammatory responses causing vasculitis, intravascular coagulation, and thromboembolic events, resulting in secondary ischemic and hemorrhagic strokes. Thus, there is an urgent need for further investigation of the mechanisms underlying SARS-CoV-2 pathogenesis, the associated vascular and CNS pathology, and the neurological manifestations. We review clinical and neuroradiological findings in five COVID-19 patients with severe neurological disease and illustrate the pathological findings in a 7-year-old boy with COVID-19-induced encephalopathy. We then review important structural and functional properties of SARS-CoV-2 and of its cell surface receptor angiotensin-converting enzyme (ACE2) and summarize molecular mechanisms controlling the binding and cellular entry of SARS-CoV-2. We comprehensively review and illustrate SARS-CoV-2-induced vascular and neurological injury and describe mechanistic considerations linked to COVID-19 including CNS tropism. Finally, we discuss pathogenesis of COVID-19-associated cellular and molecular mechanisms and propose a novel pathophysiological mechanism that could explain the secondary brain involvement in COVID-19 patients. We intend to provide a rationale for the development of novel preventive and therapeutic strategies to improve patient outcome.
Illustrative Cases
Case 1
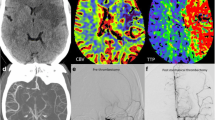
A 77-year-old female with a past medical history of hypertension presented with progressive left-sided hemiparesis for 2 days. She was afebrile but hypoxic to 89% on room air with positive SARS-CoV-2 nasopharyngeal PCR. Computerized tomography (CT) scan revealed a large acute right middle cerebral artery (MCA) infarction involving the right basal ganglia, insula, and frontal, parietal, and temporal lobes (Fig. 1). Her hospital course was complicated by rapidly progressive ARDS requiring mechanical ventilation, bilateral deep vein thromboses of lower extremities, and persistent hypotension requiring vasopressors. Given severe diffuse irreversible brain damage, the family agreed to transition to comfort care measures and palliative extubation.

CT scan in a 77-year-old COVID-19 patient. An acute infarct is present in the right MCA territory
Case 2
A 62-year-old male with remote history of resected colon cancer presented to the hospital with respiratory symptoms and was found to be positive for SARS-CoV-2 by nasopharyngeal PCR. He developed COVID-19 pneumonia and, despite medical therapy, required intubation 4 days later. He was placed on unfractionated heparin due to bilateral deep vein thrombosis and elevated d-dimer levels. He developed hypotension and acute renal failure requiring continuous renal replacement therapy/hemodialysis. Due to persistent encephalopathy over the next week, a CT scan of the head was performed and showed (Fig. 2) multiple acute intraparenchymal hemorrhages involving both cerebral hemispheres, with marked edema, mass effect, and herniation. He developed bilateral fixed and dilated pupils with diminished brain stem reflexes and was unresponsive to stimulation. The patient died on hospital day 17.

CT scan in a 62-year-old COVID-19 patient. Multiple acute parenchymal hemorrhages involving both cerebral hemispheres, leading to marked swelling, mass effect, and brain herniation
Case 3
A 72-year-old male with a past medical history of hypertension presented with acute onset of dry cough and dyspnea, and was found to be hypoxic to 86% on room air with severe patchy bilateral opacities on chest x-ray and positive SARS-CoV-2 nasopharyngeal PCR. On the fifth hospital day, he fell out of bed and was noted to have left facial droop with left hemiparesis. Magnetic resonance imaging (MRI) showed acute infarction within the right MCA territory (Fig. 3). He was administered tPA and underwent mechanical thrombectomy. Further investigation also revealed the presence of an aortic arch thrombus.

MRI in a 72-year-old COVID-19 patient. Diffusion-weighted imaging shows a large acute infarction in the right MCA territory
Case 4
A 40-year-old male with a past medical history of hypertension and type 2 diabetes mellitus presented with acute global aphasia and right hemiparesis. CT scan showed early ischemic changes in the left insula and frontal operculum and hyperdensity involving the distal M1 and proximal M2 segments of the left MCA, concerning for acute thrombus (Fig. 4). Despite not having respiratory symptoms, he was swabbed and was found to be SARS-CoV-2-positive. He underwent mechanical thrombectomy and was discharged to rehabilitation after 8 days of hospital stay.

CT scan in a 40-year-old COVID-19 patient. A large acute infarct in the left MCA territory involves a large part of the left cerebral hemisphere with associated swelling, mass effect, and mild midline shift. Also noted is a small acute infarct in the right temporal lobe
Case 5
A previously healthy 7-year-old male, whose parents had been sick with COVID-19 1 month prior, presented to the emergency department with abdominal pain, vomiting, headache, dizziness, and 3 days of fever. His nasopharyngeal swab was SARS-CoV-2-positive. CT venogram of his head was normal at that time. The following evening, he developed worsening headache and his mental status began to fluctuate. He then became unresponsive with left gaze deviation. Head CT and CT angiography again showed no intracranial pathology or thrombosis. He was loaded with levetiracetam and an electroencephalogram (EEG) showed no epileptiform activity. During that night, the EEG developed left frontotemporal slowing followed by a loss of fast activity on the right with an increase in delta waves on the left. Several hours later, the EEG suddenly showed bilateral voltage attenuation. He developed fixed and dilated pupils, and lost his brainstem reflexes. Head CT at that time demonstrated diffuse cerebral edema with loss of gray-white differentiation (Fig. 5). His inflammatory markers, including procalcitonin, C-reactive protein, and d-dimer, were significantly elevated, though his white blood cell count remained normal. An emergently placed intracranial pressure monitor measured pressures up to 100 mmHg which were refractory to aggressive medical therapy. Two hours after death, a brain biopsy from the right frontal region was obtained. Pathological evaluation showed a mixed mononuclear inflammatory infiltrate in leptomeningeal space and cortical tissue (Figs. 6 and 7). Ultrastructural analysis, PCR testing, and specific immunohistochemistry for SARS-CoV-2 performed on the central nervous system tissue were all negative.

CT scan in a 7-year-old COVID-19 patient. Non-contrast images show diffuse loss of gray-white matter differentiation and diffuse cerebral edema

a Lymphocytes and histiocytes in the leptomeninges. b, c Perivascular infiltrates of lymphocytes and histiocytes in the right superior frontal cortex. d Shrunken and hyperchromatic pyramidal neurons in an edematous neuropil (hematoxylin and eosin; original magnification a, c, d × 400, b × 200)

a Perivascular inflammatory infiltrates composed partly of CD3-immunopositive lymphocytes in the cerebral cortex. b Lymphocytes are also immunopositive for CD5. c Leptomeningeal infiltrates of CD3-immunopositive lymphocytes. d Lymphocytes are also immunopositive for CD5. e, f Leptomeningeal infiltrate of mostly perivascular CD163-immunopositive histiocytes. g, h Perivascular infiltrate of CD163-immunopositive histiocytes and numerous microglia in the cerebral cortex. (a and c CD3 immunostains, b and d CD5 immunostains, e–h CD163 immunostains; original magnification a and h × 400; b × 800; c, d, and f × 200; e × 40; g × 100)
Characteristics of SARS-CoV-2
SARS-CoV-2 is a newly discovered member of the broad coronavirus family and the seventh of these viruses known to infect humans after HCoV-229E, HCoV-NL63 (both α-coronaviruses), and the β-coronaviruses HCoV-HKU1, HCoV-OC43, SARS-CoV-1, and MERS-CoV. It is an enveloped positive-sense single-stranded linear RNA virus, and, similarly to other coronaviruses, it contains four essential structural proteins: the Spike (S) glycoprotein, responsible for its crown-like appearance on electron microscopy, the Envelope (E) protein, the Membrane (M) protein, and the Nucleocapsid (N) protein, which binds the RNA genome (Fig. 8) [7]. SARS-CoV-2 entry into host cells is mediated by the S glycoprotein, which is composed of two functional subunits responsible for binding to the host cell (S1 subunit) and fusion of the viral and host cell membranes (S2 subunit). The S1 subunit contains the receptor-binding domain (RBD), which determines the virus host range and cellular tropism. As with SARS-CoV-1, the RBD of this novel coronavirus binds to angiotensin-converting enzyme 2 (ACE2) to enter cells [7]. Membrane fusion and viral entry into the host cell are dependent on host proteases including transmembrane protease serine 2 (TMPRSS2) [8]. Cathepsin L, a lysosomal protease, has also been implicated in cell entry through endocytosis [9]. The S glycoprotein is cleaved at the S1/S2 and at the S2’ sites, exposing the S2 fusion peptides [8]. It is known that several viruses including filoviruses (Marburg and Ebola), HIV, highly pathogenic forms of influenza, and dengue fever must have their envelope proteins cleaved by furin or furin-like proteases to become fully functional. In HIV for example, furin is one of the proteases responsible for the proteolytic cleavage of the polyprotein precursor gp160 into gp120 and gp41 [10]. A novel furin cleavage site at the S1/S2 boundary of SARS-CoV-2 S glycoprotein was identified, setting it apart from SARS-CoV-1 and other SARS-related viruses [11, 12]. This novel polybasic cleavage site could potentially expand SARS-CoV-2 cell and tissue tropism and enhance its transmissibility, given the near-ubiquitous distribution of furin-like proteases [11, 12].

Diagram of the structure of SARS-CoV-2. Similarly to other coronaviruses, SARS-CoV-2 contains four essential structural proteins: the Spike (S) glycoprotein, the Envelope (E) protein, the Membrane (M) protein, and the Nucleocapsid (N) protein, which binds the single-stranded positive-sense RNA genome
Tracing the origin of this novel virus has been the subject of intense research. Zhou et al. analyzed full-length genome sequences from five patients at the early stage of the outbreak and reported that SARS-CoV-2 shares 79.6% sequence identity to SARS-CoV-1 and is very closely related to the intermediate horseshoe bat (Rhinolophus affinis) RaTG13 coronavirus, with 96.2% genomic similarity [13]. This close phylogenetic relationship suggests SARS-CoV-2 may have originated in bats. The Rhinolophus genus of horseshoe bats is now recognized as a natural reservoir of SARS-related coronaviruses, some of which have crossed the species barrier into humans. Whereas palm civets, raccoon dogs, and dromedary camels have been recognized as the intermediate hosts for zoonotic transmission of SARS and MERS respectively, the SARS-CoV-2 intermediate host remains the subject of debate [11, 13]. Zhang et al. reported genomic evidence of the occurrence of a coronavirus similar to SARS-CoV-2 in dead Malayan pangolins (Pangolin-CoV). Based on whole-genome level comparisons, the Pangolin-CoV is the second closest related coronavirus to SARS-CoV-2 with 91.02% genomic similarity. Interestingly, five key amino acid residues involved in the tight interaction between RBD and ACE2 are completely consistent between Pangolin-CoV and SARS-CoV-2, whereas only four of them are present in RaTG13. Neither Pangolin-CoV nor RaTG13-CoV contain the furin cleavage site present in SARS-CoV-2 [14]. This higher sequence identity at a crucial segment of the genome involved in host infection between Pangolin-CoV and SARS-CoV-2 suggests a more complex origin for the latter.
Angiotensin-Converting Enzyme 2: a Functional Receptor for SARS-CoV-2
SARS-CoV-2, following proteolytic cleavage of its S protein by a serine protease, binds to the transmembrane ACE2, a homolog of ACE [7]. ACE2 is a counter-regulatory enzyme to the Renin-Angiotensin-Aldosterone System (RAAS) widely distributed throughout the body. It degrades angiotensin II to angiotensin-(1–7) and angiotensin I to angiotensin-(1–9), attenuating their effects on vasoconstriction and sodium retention [15]. Following the 2002 SARS epidemic, several authors studied the expression of ACE2 in human tissues. ACE2 receptor is expressed by type I and II pneumocytes in the epithelial alveolar lining, and to a variable extent on a variety of cells including arterial and venous endothelial cells, including those in cerebral capillaries, arterial smooth muscle cells, perivascular pericytes, neurons and microglia, macrophages, and cardiomyocytes. [16, 17] Renal proximal and distal tubular cells and enterocytes of the small intestine also express ACE2 [18, 19]. Through the use of human small intestinal organoids, Lamers et al. recently demonstrated that intestinal epithelium supports SARS-CoV-2 replication [20]. This may explain the gastrointestinal symptoms seen in a subset of patients with COVID-19 [21] and the detection of viral RNA and even infectious virions in stool samples and rectal swabs after nasopharyngeal testing has turned negative [21, 22]. Remarkably, SARS-CoV-2 binds ACE2 with higher affinity (at least ten times) than SARS-CoV-1 S glycoprotein [7]. These findings could explain, at least partially, the higher transmissibility and infectivity of this emergent pathogen compared to the phylogenetically related SARS-CoV-1. One critical property of ACE2 is its hypoxia-inducibility regulated by hypoxia-inducible factor-1 [23]. Although low levels of SARS-CoV-2 are detected in post-mortem samples of the kidney, liver, heart and brain [24], in hypoxic patients, such as those with severe COVID-19, an enhanced ACE2 expression on endothelial cells could increase the binding of SARS-CoV-2 in remote organs such as the CNS.
Vascular Injury and Microvascular Dysfunction
Several histological studies in COVID-19 patients reveal marked vascular pathological changes including vascular wall thickening, lumen occlusion, microthrombi, vascular hyperplasia and congestion, focal hemorrhage, edematous changes, and accumulation of inflammatory cells with infiltration of monocytes and lymphocytes within and around blood vessels [25, 26]. The pathophysiological mechanisms of the damage associated with COVID-19 are partially understood, but several experimental and clinical observations suggest that COVID-19 pathology is, at least in part, linked to a vascular injury (Fig. 9). These include (i) the expression of ACE2 receptor on vascular cells including endothelial cells and pericytes, [18, 27] (ii) the demonstration of SARS-CoV-2-induced vascular cell infection and apoptosis, (iii) the presence of a frequent angiocentric mixed inflammatory infiltrate, (iv) the prominence of interstitial exudates likely to be linked to increased vascular permeability, and (v) the presence of numerous microvascular microthrombi.

Diagram summarizing the three proposed pathophysiological processes that underlie the neurovascular injury associated with COVID-19. Three main pathophysiological processes including infection, inflammation, and thrombosis are responsible for the neurovascular injury and could separately or together cause the pathology seen in COVID-19 patients
Indeed, several observations have been made confirming the microvascular injury and dysfunction including direct endothelial and pericytic viral infection. For example, the ultrastructural detection of viral particles in endothelial cells and associated endothelial cell apoptosis observed across vascular beds of different organs of patients with COVID-19 [28] is powerful evidence that the virus can infect endothelial cells and pericytes. Some literature, however, has suggested that these particles may have been misinterpreted as coronavirus and instead represent the rough endoplasmic reticulum [29]. SARS-CoV-2 can directly infect engineered human blood vessel organoids in vitro [30], and the occurrence of apoptosis underscores the vascular damage and the SARS-CoV-2-induced endotheliitis. This could explain, at least in part, the systemic impairment of microcirculatory function in vascular beds of organs including the lung, kidney, myocardium, and brain seen in patients with COVID-19 [28].
Several additional mechanisms have been proposed to explain the vascular injury seen in COVID-19 patients. Frequently cited is an exaggerated and uncontrolled inflammatory response resulting in a “cytokine storm” associated with COVID-19. Immune cells release high levels of proinflammatory cytokines including interleukin (IL)-6, IL-7, IL-8, IL-10, IL-17, IL-22, IL-1β, and tumor necrosis factor-α [31] resulting in overactivation of T cells, Th17 skewing, and increased cytotoxicity of CD8 T cells [25]. In addition, immune complexes, associating an immunoglobulin and a viral antigen, can lead to vasculitis activating complement and inflammatory cells [32]. Indeed, the fact that histopathologic alterations are reduced in MERS-CoV inoculated but immunosuppressed rhesus macaques emphasizes the role of the deleterious immune response in coronavirus-induced diseases [33].
Further highlighting the important pathophysiological role of vascular injury are reports of COVID-19 manifestations in children and adolescents likely resulting from endothelial damage and microthrombi, initially reported as “acute acro-ischemia” of the extremities. These skin lesions usually present as multiple, often painful, variably sized (5–15 mm in diameter), erythematous reddish-purple irregular round lesions of the plantar and/or dorsal surfaces of the toes and occasionally the hands with intense itching and burning. These sometimes become bullous with dark crusts and can be associated with fever, muscle pain, and headache but usually regress within days [34]. More recently, another serious manifestation of vascular involvement is the multisystem inflammatory syndrome in children (MIS-C) also seen in children and young adults [35]. This syndrome shares features with Kawasaki disease (Fig. 10). Both acro-ischemia and MIS-C are linked to vascular inflammation and injury, formation of microthrombi, and release of cytokines produced in association with an excessive immune response to SARS-CoV-2 [34].

Multisystem inflammatory syndrome in children (MIS-C). a Superficial and deep scant perivascular inflammation (arrows) with microthrombi in the papillary dermal capillaries (arrowheads) (features of thrombotic microangiopathy). b The perivascular inflammatory infiltrate (arrows) consists mostly of lymphocytes and some histiocytes (hematoxylin and eosin; original magnification a × 100; b × 400) (courtesy of Drs Michael Occidental and George Jour)
SARS-CoV-2 induces vascular pathology because of the persistent dysregulated inflammatory response marked by the overproduction of proinflammatory cytokines causing vascular injury, cell death, and increased vascular permeability (capillary leak syndrome). A recent publication reports on the analysis of SARS-CoV-2-infected lung tissue and reveals the upregulation of several angiogenic factors [36] including some that are known to increase vascular permeability [37]. Moreover, SARS-CoV-2 binding to the ACE2 receptor impairs ACE2 activity [8], indirectly activating the kallikrein–bradykinin pathway and increasing vascular permeability [38].
Coagulation Abnormalities as a Feature of COVID-19
Another critical pathophysiological process seen in COVID-19 is the increased incidence of thrombosis. Early in the pandemic, coagulation abnormalities were recognized as a prominent feature in COVID-19 patients that were noted to have a predisposition for developing thrombotic complications [39,40,41]. During quiescence, the endothelium and the adjacent pericytic cells maintain vascular integrity and function. For example, the normal endothelium expresses several molecules that prevent platelet aggregation and initiation of coagulation, providing a resting-state “anticoagulant” surface. These include serine protease inhibitors anchored to glycosaminoglycans (GAGs) such as the tissue factor pathway inhibitor (TFPI), which prevents the activation of Factor X by the TF-FVIIa complex; and antithrombin (AT), which inhibits the activated forms of several factors of the contact activation (formerly intrinsic) pathway, predominantly Factor X and thrombin. The endothelium also expresses thrombomodulin (TM) and endothelial protein C receptor (EPCR), which support the generation of activated protein C by thrombin, and secretes tissue-type plasminogen activator (tPA), which promotes fibrinolysis [42]. Abundant experimental evidence suggests that viral induced endothelial damage results in the disruption of this natural antithrombotic state controlled by several molecules and that endothelial activation in response to inflammatory cytokines and other signals released during cell death induces a prothrombotic transformation. Several events contribute to this prothrombotic state: (i) production of GAGs is downregulated, and anticoagulant proteins like TFPI, AT, TM, and EPCR are cleaved from the endothelial cell surface [42]; (ii) increased production of tissue factor (TF) with thrombin generation and subsequent fibrinogen transformation into fibrin; (iii) IL-1β and TNF-α activation of endothelial cells leads to P-selectin expression that causes platelet recruitment and aggregation [38]; (iv) activation of platelets by coagulation proteases such as thrombin; and (v) release of preformed granules containing von Willebrand factor (vWF), by the activated endothelium further promoting platelet aggregation [42] and vascular endothelial growth factor (VEGF) upregulation [43] that leads to TF expression by endothelial cells [38] and increased vascular permeability [37].
It is well known that microbial penetrance into the organism initiates thrombogenesis and coagulation pathways with fibrin deposition and platelet aggregation. This dense network contributes to host defense by preventing spread of infectious microorganisms in a manner similar to neutrophil extracellular traps (NETs) and by providing anchoring sites for leukocytes, antimicrobial proteins, and lytic enzymes [44]. The interplay between hemostasis and innate immune responses inhibits the spread of pathogens and provides an activation site for innate immune cells through the formation of “immunothrombi.” [45] This process is inherently beneficial at the local level, but uncontrolled stimulation of the release of cytokines can lead to dysregulation of the coagulation system, widespread microvascular thrombosis, ischemic events, and subsequent depletion of hemostatic factors with concomitant potential for bleeding [46]. For example, thrombotic microvascular injury, accompanied by extensive deposition of alternative and lecithin-based complement components in the microvasculature, has been reported in lung and skin samples from patients with severe COVID-19 [32]. Increased markers of endothelial inflammation have been reported in intensive care unit patients with COVID-19, including increased vWF activity, circulating vWF antigen, and Factor VIII [47]. This could explain the post-mortem findings in some COVID-19 patients where microvascular platelet-rich thrombotic depositions are found in the lung [32, 48] (Fig. 11), liver (Fig. 12), and other organs [32]. Of note, although thrombi formation is a common feature seen in diffuse alveolar damage (DAD) regardless of the etiology [49], severe COVID-19 likely causes a separate, true thrombotic vasculopathy. Evidence to support this hypothesis lies in the fact that the amount of platelet-rich thrombi found in the lungs is greater than usually seen in DAD [50] and that they can be present even in the absence of prominent DAD pathology [32]. Furthermore, DAD alone would certainly not explain the microvascular thromboses seen in distant organs in some cases [32].

Alveolar and vascular pathology in lung. a Large-vessel thrombus (arrows) in lung with DAD picture with hyaline membranes (arrowheads). b DAD with hyaline membranes (arrowheads) and thrombosis seen in medium- and small-sized vessels (arrows) highlighted with CD61 immunoreactive platelets (c). d Mononuclear mixed perivascular inflammatory infiltrate composed of CD163-immunopositive macrophages (e) and CD3-immunopositive T lymphocytes (f). (a, b, and d hematoxylin and eosin and c CD61, e CD163, f CD3 immunostains; original magnification a × 40, b–f × 100)

Venous thrombosis within portal space. The thrombus is seen in the portal vein with highlighted CD61 immunoreactive platelets. The hepatic artery (HA) is patent and the bile duct (BD) is unaffected. (a hematoxylin and eosin and b and c CD61 immunostains, original magnification a × 40, b and c × 100) (courtesy of Drs Neil Theise and Christina Hajdu)
Importantly, the high incidence of venous and arterial thromboembolism in patients with COVID-19 despite anticoagulation therapy with low molecular weight heparin (LMWH) [40, 47] could indicate that additional procoagulant mechanisms are at play, specifically those that bypass Factor Xa, the molecular target of LMWH. One of these potential pathways lies in fibrinogen-like protein 2 (FGL2), a prothrombinase that has been implicated in thrombotic processes related to the innate arm of immunity, such as in acute vascular xenograft rejection [51]. During the past SARS epidemic, some studies showed that SARS-CoV-1-induced transcription of FGL2 [52] an observation that was debated [53]. Nevertheless, the emerging evidence that COVID-19 has important vascular and thrombotic consequences has prompted clinical trials to evaluate different anticoagulant/antithrombotic therapies.
Clinically, COVID-19 coagulation abnormalities include marked elevation of d-dimer and fibrinogen levels, normal or modestly decreased platelet count, and concurrent large-vessel and microvascular venous and arterial thrombosis, an unusual pattern when compared to coagulopathies seen in other severe inflammatory disorders such as DIC, cytokine release syndrome, macrophage activation syndrome, or catastrophic anti-phospholipid antibody syndrome [54]. Abnormal coagulation tests at the time of hospitalization including longer prothrombin time (PT) and significantly elevated d-dimer and fibrin degradation products (FDPs) are associated with poor prognosis and correlate with increased mortality risk [55], suggesting a potential role for d-dimer as a tool for risk stratification [56]. The prothrombotic state can lead to both venous and arterial thromboembolism [41, 57], even in the absence of overt DIC [47, 58] and despite prophylactic or therapeutic anticoagulation [40, 47]. Similar to other viral infections, some authors have detected the presence of circulating lupus anticoagulant (anti-phospholipid antibodies) in COVID-19 patients which may contribute to the prothrombotic state [47, 59]. Of note, one report describes the association between acro-ischemia and coagulation disorders [34].
COVID-19-Associated Neurological Injury
COVID-19-related neurological clinical manifestations are increasingly recognized in the current pandemic and include headache, nausea, vomiting, and confusion [4, 60]. Cerebrovascular events including ischemic and hemorrhagic strokes and cerebral venous thrombosis are also emerging as important complications [4, 60, 61]. Common especially in elderly COVID-19 patients [3, 60], their prevalence increases threefold in severe cases [62]. However, It is also possible that the COVID-19 infection in neurological patients is purely coincidental and not directly implicated in the pathogenesis of the neurological complication. Also worrisome are the reports of younger COVID-19 patients presenting with large-vessel stroke [63, 64]. Both venous and arterial thromboembolic events are likely to be, at least in part, responsible for the neurological complications seen in COVID-19 patients. Moreover, vascular inflammation and cytokines released during the acute infection can elicit activation of cells within pre-existing atherosclerotic lesions, augmenting thromboembolic risk and ischemic syndromes [65]. Other neurological complications involving the central and peripheral nervous system include epileptic seizures [66, 67], anosmia [68], ageusia, meningoencephalitis, necrotizing hemorrhagic encephalitis [68, 69], acute disseminated encephalomyelitis-like pathology [70], and severe cases of Guillain-Barré syndrome [71]. Of great relevance is the difficulty in clinical diagnosis of a neurological event in patients sedated for prolonged mechanical ventilation.
Neuroimaging has demonstrated that acute ischemic and hemorrhagic stroke with an overall incidence of 1–2% in all hospitalized COVID-19 patients is the most common abnormality and is a strong prognostic marker of poor outcome [72]. Importantly, many of these hospitalized patients are treated with anticoagulation due to high thrombotic risk. In one recent publication, approximately 80% of all hemorrhagic stroke patients were shown to be on therapeutic or prophylactic anticoagulation [61]. Leukoencephalopathy and cerebral microhemorrhages are also seen in critically ill COVID-19 patients and are likely associated with thrombotic microangiopathy as well as prolonged hypoxemia [73]. Of note, MRI evaluation is more sensitive than CT scan yield and should be preferred when possible.
Self-reported anosmia and dysgeusia by up to a third of COVID-19 patients [74] are common features of most upper respiratory infections. ACE2 expression has been found in olfactory epithelial support cells, olfactory stem cells, and nasal respiratory epithelium, providing a possible explanation for these symptoms as well as a SARS-CoV-2 entry route into the brain. Indeed, studies using transgenic mice expressing human ACE2 showed that SARS-CoV-1 has the potential to infect the CNS, entering through the olfactory bulb and rapidly disseminating via transneural spread to connected areas of the brain [75, 76]. In addition, at least two other pathogenic pathways have also been proposed to explain the CNS entry and neurotropic properties of this virus: (i) hematogenous spread of SARS-CoV-2 and binding of the virus to ACE2 receptors present on cerebral endothelial cells and (ii) crossing of SARS-CoV-2-infected activated monocytes through a permeable blood-brain barrier. These pathways have been observed in experimental animal models of SARS and MERS, both of which are known to occasionally cause clinically relevant CNS infections, further supporting the neuroinvasive capacity of coronaviruses [6, 77]. These animal models address the possibility of neuroinfection by SARS-CoV-2 and importantly provide a mechanism for its entry into the CNS. Some have also proposed that SARS-CoV-2 may reach the brainstem through a neural route from lung chemoreceptors, severely affecting the cardiorespiratory center and suggesting that the acute respiratory failure in some COVID-19 patients may have a central origin [78]. Further stressing the neurotropism of SARS-CoV-2, it has been shown that SARS-CoV-2 binds the ACE2 receptor present on endothelial cells and neurons with a higher affinity compared to SARS-CoV-1 [79].
The pathophysiological processes responsible for the neurological impact of COVID-19 are incompletely understood. Variable direct and indirect mechanisms have been proposed. Overall, five potentially linked mechanisms have emerged as important pathophysiological processes causing the neurological damage: (i) direct viral infection of vascular, neuronal, microglial, and astrocytic cells and injury; (ii) direct viral infection of circulating monocytes and macrophages; (iii) excessive host immune-mediated release of proinflammatory cytokines, including IL-6, TNF-α, IL-1β, and IL12 [79] and associated systemic inflammatory response syndrome causing CNS dysfunction; (iv) microvascular damage and dysfunction with intravascular coagulation and microthrombi; and (v) increased frequency of embolic events. Thus, the varying degree of cellular injury, host immune-mediated responses, and thromboembolic events may explain the heterogeneity in the neurological clinical manifestations. The relative contribution of the direct viral infection of cellular elements of the CNS and of the secondary processes including immune exaggerated inflammatory-mediated injury and thromboembolic events remains to be determined.
Conclusions
Although it is clear that SARS-CoV-2 primarily affects the respiratory system, vascular and neurological injury are important components of COVID-19. Major routes of CNS infection include intranasal olfactory route and hematogenous spread with vascular cell infection, subsequent inflammation, associated cytokines release, and permeable blood-brain barrier. We have reviewed clinical and neuroradiological findings in five COVID-19 patients who suffered severe neurological disease and illustrated the pathological findings in a 7-year-old boy with COVID-19-induced encephalopathy whose brain tissue sample showed angiocentric mixed mononuclear inflammatory infiltrate. We have emphasized the extent of the neurovascular injury and reviewed neurological complications associated with COVID-19. We summarized the key concepts linked to the pathogenesis of the neurovascular complications of COVID-19. Direct viral and immune-mediated injury result in widespread microvascular dysfunction including vasoconstriction, inflammation, a procoagulant state, ischemic and hemorrhagic damage, and edematous changes. Our central thesis is that three major pathophysiological processes, vascular injury, inflammation, and thrombosis, sometimes associated with embolic complications, resulting from SARS-CoV-2 infection can explain the broad spectrum of pathology seen in COVID-19 patients. Individual susceptibility factors that underlie the variable presentation and severity of neurovascular disease, as well as the occurrence of serious complications such as stroke and encephalitis seen in individual COVID-19 patients, remain unclear. Although we have emphasized the neurovascular complications, it is now apparent that COVID-19 can also affect the vasculature in several organs, and in severe cases, this disease could be best characterized as a multisystemic vascular disease [80]. Further investigations to test our proposed mechanistic models and help develop novel preventive and therapeutic strategies to improve patient outcome are needed.
References
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–9. https://doi.org/10.1038/s41586-020-2008-3.
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–74. https://doi.org/10.1016/S0140-6736(20)30251-8.
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19–associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features. Radiology. 2020;296:246–8. https://doi.org/10.1148/radiol.2020201222.
Mao L, Jin H, Wang M, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China [published online ahead of print, 2020 Apr 10]. JAMA Neurol. 2020;e201127. https://doi.org/10.1001/jamaneurol.2020.1127.
McCray PB Jr, Pewe L, Wohlford-Lenane C, et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. 2007;81(2):813–21. https://doi.org/10.1128/JVI.02012-06.
Desforges M, Le Coupanec A, Brison E, Meessen-Pinard M, Talbot PJ. Neuroinvasive and neurotropic human respiratory coronaviruses: potential neurovirulent agents in humans. Adv Exp Med Biol. 2014;807:75–96. https://doi.org/10.1007/978-81-322-1777-0_6.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–3. https://doi.org/10.1126/science.abb2507.
Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. 1992;360(6402):358–61. https://doi.org/10.1038/360358a0.
Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11(1):1620. Published 2020 Mar 27. https://doi.org/10.1038/s41467-020-15562-9.
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. https://doi.org/10.1016/j.cell.2020.02.058.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742. https://doi.org/10.1016/j.antiviral.2020.104742.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3. https://doi.org/10.1038/s41586-020-2012-7.
Zhang T, Wu Q, Zhang Z. Probable Pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020;30(7):1346–1351.e2. https://doi.org/10.1016/j.cub.2020.03.022.
Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with COVID-19. N Engl J Med. 2020;382(17):1653–9. https://doi.org/10.1056/NEJMsr2005760.
Peña Silva RA, Chu Y, Miller JD, et al. Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke. 2012;43(12):3358–63. https://doi.org/10.1161/STROKEAHA.112.667063.
Yamagata R, Nemoto W, Nakagawasai O, Takahashi K, Tan-No K. Downregulation of spinal angiotensin converting enzyme 2 is involved in neuropathic pain associated with type 2 diabetes mellitus in mice. Biochem Pharmacol. 2020;174:113825. https://doi.org/10.1016/j.bcp.2020.113825.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7. https://doi.org/10.1002/path.1570.
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R373–81. https://doi.org/10.1152/ajpregu.00292.2006.
Lamers MM, Beumer J, van der Vaart J, et al. SARS-CoV-2 productively infects human gut enterocytes [published online ahead of print, 2020 May 1]. Science. 2020;eabc1669. https://doi.org/10.1126/science.abc1669.
Cheung KS, Hung IF, Chan PP, et al. Gastrointestinal manifestations of SARS-CoV-2 infection and virus load in fecal samples from the Hong Kong Cohort and Systematic Review and Meta-analysis [published online ahead of print, 2020 Apr 3]. Gastroenterology2020;S0016–5085(20)30448–0. https://doi.org/10.1053/j.gastro.2020.03.065.
Wang W, Xu Y, Gao R, et al. Detection of SARS-CoV-2 in different types of clinical specimens [published online ahead of print, 2020 Mar 11]. JAMA. 2020;e203786. https://doi.org/10.1001/jama.2020.3786.
Zhang R, Wu Y, Zhao M, Liu C, Zhou L, Shen S, et al. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2009;297(4):L631–40. https://doi.org/10.1152/ajplung.90415.2008.
Puelles VG, Lütgehetmann M, Lindenmeyer MT, et al. Multiorgan and renal tropism of SARS-CoV-2 [published online ahead of print, 2020 May 13]. N Engl J Med. 2020;NEJMc2011400. https://doi.org/10.1056/NEJMc2011400.
Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome [published correction appears in Lancet Respir Med. 2020 Feb 25;:]. Lancet Respir Med. 2020;8(4):420–2. https://doi.org/10.1016/S2213-2600(20)30076-X.
Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China [published online ahead of print, 2020 Mar 12]. Clin Infect Dis. 2020;ciaa248. https://doi.org/10.1093/cid/ciaa248.
Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020;116(6):1097–100. https://doi.org/10.1093/cvr/cvaa078.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8. https://doi.org/10.1016/S0140-6736(20)30937-5.
Goldsmith CS, Miller SE, Martines RB, Bullock HA, Zaki SR. Electron microscopy of SARS-CoV-2: a challenging task. Lancet. 2020;395(10238):e99. https://doi.org/10.1016/S0140-6736(20)31188-0.
Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2 [published online ahead of print, 2020 Apr 17]. Cell. 2020;S0092–8674(20)30399–8. https://doi.org/10.1016/j.cell.2020.04.004
Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention [published online ahead of print, 2020 Apr 28]. Nat Rev Immunol. 2020;1–12. https://doi.org/10.1038/s41577-020-0311-8.
Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases [published online ahead of print, 2020 Apr 15]. Transl Res. 2020;S1931–5244(20)30070–0. https://doi.org/10.1016/j.trsl.2020.04.007
Prescott J, Falzarano D, de Wit E, Hardcastle K, Feldmann F, Haddock E, et al. Pathogenicity and viral shedding of MERS-CoV in immunocompromised rhesus macaques. Front Immunol. 2018;9:205.
Wollina U, Karadağ AS, Rowland-Payne C, Chiriac A, Lotti T. Cutaneous signs in COVID-19 patients: a review [published online ahead of print, 2020 May 10]. Dermatol Ther. 2020; https://doi.org/10.1111/dth.13549.
Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395(10237):1607–8. https://doi.org/10.1016/S0140-6736(20)31094-1.
Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19 [published online ahead of print, 2020 May 21]. N Engl J Med. 2020; https://doi.org/10.1056/NEJMoa2015432.
Dvorak HF. Discovery of vascular permeability factor (VPF). Exp Cell Res. 2006;312(5):522–6. https://doi.org/10.1016/j.yexcr.2005.11.026.
Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–15.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–13. https://doi.org/10.1016/S0140-6736(20)30211-7.
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–7. https://doi.org/10.1016/j.thromres.2020.04.013.
Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18:1421–4. https://doi.org/10.1111/jth.14830.
Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83(3):536–45. https://doi.org/10.1189/jlb.0607373.
Webb NJ, Bottomley MJ, Watson CJ, Brenchley PE. Vascular endothelial growth factor (VEGF) is released from platelets during blood clotting: implications for measurement of circulating VEGF levels in clinical disease. Clin Sci (Lond). 1998;94(4):395–404. https://doi.org/10.1042/cs0940395.
Franco AT, Corken A, Ware J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood. 2015;126(5):582–8. https://doi.org/10.1182/blood-2014-08-531582.
Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis [published online ahead of print, 2020 Apr 26]. Clin Chim Acta. 2020;507:167–73. https://doi.org/10.1016/j.cca.2020.04.027.
Kitchens CS. Thrombocytopenia and thrombosis in disseminated intravascular coagulation (DIC). Hematol Am Soc Hematol Educ Program. 2009;2009:240–6. https://doi.org/10.1182/asheducation-2009.1.240.
Helms J, Tacquard C, Severac F, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study [published online ahead of print, 2020 May 4]. Intensive Care Med. 2020:1–10. https://doi.org/10.1007/s00134-020-06062-x.
Fox SE, Atmakbekov A, Harbert JL, Li G, Brown JQ, Vander Heide RS. Pulmonary and cardiac pathologyin COVID-19: the first autopsy series from New Orleans. MedRxiv 2020; in press/available on-line.
Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID-19 autopsies, Oklahoma, USA. Am J Clin Pathol. 2020;153(6):725–33. https://doi.org/10.1093/ajcp/aqaa062.
Rapkiewicz AV, Mai X, Carsons SE, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020;24:100434. Published 2020 Jun 25. https://doi.org/10.1016/j.eclinm.2020.100434.
Ghanekar A, Mendicino M, Liu H, He W, Liu M, Zhong R, et al. Endothelial induction of fgl2 contributes to thrombosis during acute vascular xenograft rejection. J Immunol. 2004;172(9):5693–701. https://doi.org/10.4049/jimmunol.172.9.5693.
Han M, Yan W, Huang Y, Yao H, Wang Z, Xi D, et al. The nucleocapsid protein of SARS-CoV induces transcription of hfgl2 prothrombinase gene dependent on C/EBP alpha. J Biochem. 2008;144(1):51–62. https://doi.org/10.1093/jb/mvn042.
Siu KL, Chan CP, Chan C, Zheng BJ, Jin DY. Severe acute respiratory syndrome coronavirus nucleocapsid protein does not modulate transcription of the human FGL2 gene. J Gen Virol. 2009;90(Pt 9):2107–13. https://doi.org/10.1099/vir.0.009209-0.
Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19 [published online ahead of print, 2020 May 11]. Lancet Haematol. 2020;7(6):e438–40. https://doi.org/10.1016/S2352-3026(20)30145-9.
Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–7. https://doi.org/10.1111/jth.14768.
Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Middeldorp S, Coppens M, van Haaps TF, et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19 [published online ahead of print, 2020 May 5]. J Thromb Haemost. 2020; https://doi.org/10.1111/jth.14888.
Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy [published online ahead of print, 2020 Apr 23]. Thromb Res. 2020;191:9–14. https://doi.org/10.1016/j.thromres.2020.04.024.
Bowles L, Platton S, Yartey N, et al. Lupus anticoagulant and abnormal coagulation tests in patients with COVID-19 [published online ahead of print, 2020 May 5]. N Engl J Med. 2020; https://doi.org/10.1056/NEJMc2013656.
Li Y, Wang M, Zhou Y, Chang J, Xian Y, Mao L, Hong C, Chen S, Wang Y, et al. Acute cerebrovascular disease following COVID-19: a single center, retrospective, observational study. The Lancet. 2020;In Press, https://doi.org/10.2139/ssrn.3550025.
Dogra S, Jain R, Cao M, Bilaloglu S, Zagzag D, Hochman S, et al. Hemorrhagic stroke and anticoagulation in COVID-19. J Stroke Cerebrovasc Dis. 2020;29(8):104984. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.104984.
Li Z, Liu T, Yang N, et al. Neurological manifestations of patients with COVID-19: potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain [published online ahead of print, 2020 May 4]. Front Med. 2020;1–9. https://doi.org/10.1007/s11684-020-0786-5.
Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-vessel stroke as a presenting feature of COVID-19 in the young. N Engl J Med. 2020;382(20):e60. https://doi.org/10.1056/NEJMc2009787.
González-Pinto T, Luna-Rodríguez A, Moreno-Estébanez A, Agirre-Beitia G, Rodríguez-Antigüedad A, Ruiz-Lopez M. Emergency room neurology in times of COVID-19: malignant ischemic stroke and SARS-COV2 infection [published online ahead of print, 2020 Apr 30]. Eur J Neurol. 2020; https://doi.org/10.1111/ene.14286.
Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116(10):1666–87. https://doi.org/10.1093/cvr/cvaa106.
Kuroda N. Epilepsy and COVID-19: associations and important considerations [published online ahead of print, 2020 Apr 22]. Epilepsy Behav. 2020;108:107122. https://doi.org/10.1016/j.yebeh.2020.10712.
Lu L, Xiong W, Liu D, et al. New onset acute symptomatic seizure and risk factors in coronavirus disease 2019: a retrospective multicenter study [published online ahead of print, 2020 Apr 18]. Epilepsia. 2020; https://doi.org/10.1111/epi.16524.
Lechien JR, Chiesa-Estomba CM, De Siati DR, et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study [published online ahead of print, 2020 Apr 6]. Eur Arch Otorhinolaryngol. 2020;1–11. https://doi.org/10.1007/s00405-020-05965-1.
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. 2020;94:55–8. https://doi.org/10.1016/j.ijid.2020.03.062.
Reichard RR, Kashani KB, Boire NA, Constantopoulos E, Guo Y, Lucchinetti CF. Neuropathology of COVID-19: a spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology [published online ahead of print, 2020 May 24]. Acta Neuropathol. 2020;1–6. https://doi.org/10.1007/s00401-020-02166-2.
Toscano G, Palmerini F, Ravaglia S, et al. Guillain-Barré syndrome associated with SARS-CoV-2 [published online ahead of print, 2020 Apr 17]. N Engl J Med. 2020;NEJMc2009191. https://doi.org/10.1056/NEJMc2009191.
Jain R, Young M, Dogra S, et al. COVID-19 related neuroimaging findings: a signal of thromboembolic complications and a strong prognostic marker of poor patient outcome [published online ahead of print, 2020 May 19]. J Neurol Sci. 2020;116923. https://doi.org/10.1016/j.jns.2020.116923.
Radmanesh A, Derman A, Lui Y, et al. COVID-19 associated diffuse leukoencephalopathy and microhemorrhages. Radiology. 2020; in press;297:E223–7.
Giacomelli A, Pezzati L, Conti F, et al. Self-reported olfactory and taste disorders in SARS-CoV-2 patients: a cross-sectional study [published online ahead of print, 2020 Mar 26]. Clin Infect Dis. 2020;ciaa330. https://doi.org/10.1093/cid/ciaa330.
Brann DH, Tsukahara T, Weinreb C, Logan DW, Datta SR. Non-neural expression of SARS-CoV-2 entry genes in the olfactory epithelium suggests mechanisms underlying anosmia in COVID-19 patients. Preprint at bioRxiv. https://doi.org/10.1101/2020.03.25.009084.
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. 2008;82(15):7264–75. https://doi.org/10.1128/JVI.00737-08.
Li Y, Fu L, Gonzales DM, Lavi E. Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J Virol. 2004;78(7):3398–406. https://doi.org/10.1128/jvi.78.7.3398-3406.2004.
Li YC, Bai WZ, Hashikawa T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients [published online ahead of print, 2020 Feb 27]. J Med Virol. 2020; https://doi.org/10.1002/jmv.25728.
Natoli S, Oliveira V, Calabresi P, Maia LF, Pisani A. Does SARS-Cov-2 invade the brain? Translational lessons from animal models [published online ahead of print, 2020 Apr 25]. Eur J Neurol. 2020; https://doi.org/10.1111/ene.14277.
Spuntarelli V, Luciani M, Bentivegna Eet al. COVID-19: is it just a lung disease? A case-based review [published online ahead of print, 2020 Jul 28]. SN Compr Clin Med. 2020;1–6. https://doi.org/10.1007/s42399-020-00418-6.
Acknowledgments
We would like to acknowledge the NYU Family Connect Program as well as the thousands of front-line healthcare workers as an inspiration for this project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
R. Jain is a consultant for Cancer Panels Inc., has received royalties from Thieme Inc., and is on the advisory board for Nuevozen Inc. D. Placantonakis has received consultant fees from Tocagen, Synaptive Medical, Monteris, and Robeaute. D. Placantonakis, D. Zagzag, and NYU Grossman School of Medicine have filed a patent application titled “Method for treating high-grade gliomas by administering GPR133 inhibitors” on the use of GPR133 as a treatment target in glioma. C. Muh holds a patent on a cerebrospinal fluid shunt. The other authors report no conflicts.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on COVID-19
Rights and permissions
About this article

Cite this article
Gutierrez Amezcua, J., Jain, R., Kleinman, G. et al. COVID-19-Induced Neurovascular Injury: a Case Series with Emphasis on Pathophysiological Mechanisms. SN Compr. Clin. Med. 2, 2109–2125 (2020). https://doi.org/10.1007/s42399-020-00598-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-020-00598-1