
Abstract
Aims
Although ACE inhibitors slow progression of diabetic renal disease, the mortality and morbidity is still high. As other hormonal factors are involved, inhibition of vasopeptidases could further reduce progression. We studied dual inhibition of angiotensin converting enzyme and neutral endopeptidase in a model of progressive diabetic renal injury. The major endpoints were reductions in systemic blood pressure, albuminuria and renal structural injury.
Methods
Diabetic spontaneously hypertensive rats were treated with the ACE inhibitor perindopril (mg·kg−1·day−1) or the vasopeptidase inhibitor omapatrilat at doses of 10 (oma10) and 40 (oma40) mg·kg−1·day−1 for 32 weeks. In vivo ACE and NEP inhibition was quantitated by in vitro autoradiography. Renal structural injury was assessed by measurement of the glomerulosclerotic (GS) index and tubulointerstitial area (TI). The expression of transforming growth factor β, β-inducible gene-h3 and nephrin were also quantitated.
Results
Despite a similar reduction in blood pressure by perindopril and oma10, greater attenuation of albuminuria was afforded by oma10, with a complete amelioration observed with oma40. Oma40 lead to a 33% reduction in renal NEP binding and this was associated with less albuminuria and prevention of GS, TI area and overexpression of TGFβ and βig-h3. Diabetes-associated reduction in nephrin expression was restored by both drugs.
Conclusion/Interpretation
These findings suggest that other vasoactive mechanisms in addition to angiotensin II are important in the prevention of diabetic nephropathy, and that vasopeptidase inhibition might confer an advantage over blockade of the RAS alone in the treatment of diabetic renal disease.
Similar content being viewed by others


Despite advances in antihypertensive therapy for diabetic patients, hypertension continues to remain a major risk factor for the progression of diabetes related microvascular complications and in particular diabetic nephropathy [1, 2, 3]. Although antihypertensive therapy per se has been shown to retard diabetic nephropathy, as assessed by both functional and structural parameters, there is increasing evidence that agents that interrupt the renin-angiotensin system (RAS) clearly seem to be superior to other antihypertensive agents on albuminuria and renal structural damage, for similar reductions in systemic blood pressure. Other studies have shown the renoprotective effects of RAS inhibition in experimental diabetes [4], and these effects have been confirmed using both angiotensin converting enzyme inhibitors (ACEi) and angiotensin II receptor antagonists in models of hypertensive and normotensive diabetic nephropathy [5, 6, 7, 8]. However, blockade of the RAS alone, both experimentally and clinically, only retards but does not prevent or reverse diabetic nephropathy [6, 9]. Furthermore, it has been postulated that the effects of blockade of the RAS could be limited by the salt and water retention often found in experimental and clinical diabetes [10, 11].
Recently single compounds have been developed which can inhibit not only angiotensin converting enzyme (ACE) but also another zinc dependent metallopeptidase, neutral endopeptidase (NEP). Known as vasopeptidase inhibitors (VPIs), these agents provide a potential therapeutic option to confer superior renoprotection in a state such as diabetes, particularly in the context of associated systemic hypertension. VPIs simultaneously inhibit ACE and NEP, thereby acting to decrease circulating levels of vasoconstrictor effector molecules such as angiotensin II (AII) as well as increasing the levels of vasodilatory agents such as atrial natriuretic peptide and bradykinin (BK) [12, 13]. Previous research using the first of the VPIs, omapatrilat, has examined the beneficial effects on ameliorating developing clinical and experimental hypertension [14, 15, 16]. In addition, an increasing number of studies have explored these agents in models of non-diabetic renal disease [13, 17, 18]. In a study comparing both selective ACE and NEP inhibitors and another less potent dual ACE/NEP inhibitor mixanpril, albuminuria was partially prevented in a 4 week study in diabetic SHR [19].
This study investigated the effects of the ACE/NEP inhibitor omapatrilat in a long term model of experimental diabetic renal disease. It compared the ACE inhibitor perindopril and two doses of omapatrilat, including a low dose of this vasopeptidase inhibitor to achieve similar blood pressure reduction to the ACE inhibitor, as confirmed by 24-h blood pressure monitoring using radiotelemetry in conscious diabetic animals. Furthermore, detailed assessment of renal structural injury and investigation of molecular mechanisms suggested to play a potential role in diabetes associated albuminuria and renal extra cellular matrix accumulation were also explored. Specifically, the recently described slit-pore protein nephrin, which has been postulated to modulate glomerular albumin permeability, was assessed [20]. In addition, expression of TGFβ, a prosclerotic cytokine implicated in diabetes-associated extracellular matrix accumulation [21] and the TGFβ dependent matrix protein, βig-h3 [22] were evaluated.
Materials and methods
Animal model
Eight-week-old male spontaneously hypertensive rats (SHR 200–250 g), housed in the Biological Research Laboratory at the Austin and Repatriation Medical Centre, were used in this study. The research project was approved by the Animal Ethics Committee of the Austin and Repatriation Medical Centre. Diabetes was induced by tail vein injection of streptozotocin (Boehringer-Mannheim, Mannhein, Germany) at a dose of 45 mg/kg in citrate buffer after a 16-h fast [23]. Long-acting insulin (Protophane, Novo Nordisk Pharmaceuticals Pty., Denmark) at a dose of 4 U/day was given to all diabetic animals by subcutaneous injection to avoid ketonuria and promote weight gain.
Study groups
The animals were allocated into five groups and treated for 32 weeks: Diabetic SHR with no treatment, diabetic SHR treated with the ACE inhibitor, perindopril (Per) (Servier Laboratories, Neuilly, France) at a dose of 1.25 mg·kg−1·day−1 administered in drinking water, diabetic SHR treated with the dual vasopeptidase inhibitor, omapatrilat (Bristol-Myers Squibb, Princeton, New Jersey, USA) at a dose of 10 mg·kg−1day−1 (oma10) and diabetic SHR treated with omapatrilat at a higher dose of 40 mg·kg−1·day−1 (oma40). In addition, non-diabetic SHRs served as controls and were studied concurrently.
Radiotelemetry studies
Radiotelemetric blood pressure monitoring was used to assess 24-h blood pressure control [24]. Male SHR, aged eight weeks, weighing between 180 to 220 g were synchronized to 12:12 h of light (07.00–19.00) and dark (19.00–07.00) for 5 days. In anaesthetised rats, the abdominal aorta was exposed and clamped inferior to the renal artery bifurcation. The sensor's catheter was inserted 1 cm into the clamped area, glued into place with Histoacryl (Braun, Mengulsen, Switzerland) and the radio frequency transmitter sutured to the abdominal wall. After allowing a minimum of 5 days for full recovery, the rats were assigned to treatment groups as previously mentioned. Each rat was housed individually and exposed to standard food chow and water ad libitum, with the plastic cage (30 cm×40 cm×13 cm) placed on top of a receiver (RPC-1) that was connected to a BCM-100 consolidation matrix. This receiver then transmitted parameter and waveform information to the Dataquest Gold Acquisition system (Data Sciences International, St. Paul, Minn., USA). Telemetric systolic BP (tSBP) was measured for a 10 s duration at 30 min intervals over 10 weeks.
Physiological and metabolic parameters
Animals were placed in metabolic cages every 4 weeks (Iffa Credo, L'Arbesele, France) for collection of urine over 24 h for measurement of albumin concentration by radioimmunoassay as previously described [25]. Systolic blood pressure (SBP) was assessed by tail cuff plethsymography in conscious, preheated rats [26] every 4 weeks. HbA1c was measured by a high performance liquid chromatography method (Bio-Rad, Richmond Laboratories, Va., USA) [10]. Plasma renin activity (PRA) was measured by radioimmunoassay [27, 28].
NEP/ACE inhibition
NEP inhibition: For NEP in vitro autoradiography, the p-nitrophenolic ester of RB104, a specific NEP inhibitor ligand (0.51 µg in 5.1 µl) was iodinated according to a minor modification of a previously published method using 1 mCi of 125I (Amersham Radiochemicals, Amersham, UK) and chloramine T (25 µg in 50 µl) [29]. Autoradiography was done as previously described [15, 19]. Quantification of binding density was determined by computerised densitometry and the optical density of the autoradiographs was converted to dpm·mm−2 with the use of radioactive standards. ACE inhibition: ACE inhibition was assessed using a previously established method [19, 30, 31] including the radioligand, 125I MK-351A, a derivative of the ACE inhibitor lisinopril. Specific binding densities were calculated as the difference between total and nonspecific binding densities and quantitation carried out as the same for NEP binding.
Quantitation of nephrin
Immunohistochemistry was done as previously described according to a modified method using a specific antibody to mAb 5-1-6 antigen which is identical to rat nephrin [32, 33]. This antibody was raised in the rabbit against a synthetic peptide sequence derived from the cytoplasmic region of the nephrin protein [32]. These experiments were done using 16 micron frozen kidney sections, as previously reported [34]. Quantitation of nephrin immunostaining was carried out by calculation of the proportion of area occupied by the brown staining within each glomerulus [35]. An observer who was masked to the study group of origin assessed 20 glomeruli per slide and eight slides per treatment group.
Localisation of ACE
Immunohistochemistry for the ACE protein was carried out on 16 µm paraffin sections as previously described [36, 37]. Formalin-fixed paraffin sections were dewaxed, hydrated and incubated using a specific primary antibody raised against rat ACE (Chemicon, Tenecula, Calif., USA) at a dilution of 1/400. The secondary biotin conjugated rabbit anti-mouse antibody (DAKO, Copenhagen, Denmark) was used at 1/250 dilution. Negative control sections omitted the primary antibody.
Histological assessment of kidney injury
At week 32 animals were killed and organs harvested. The right kidney was removed, halved and snap frozen in liquid nitrogen for autoradiography studies. The left kidney was fixed in either 4% paraformaldehyde or neutral buffered formalin for histological studies. The degree of glomerulosclerosis was evaluated by a semiquantitative method as described previously [38]. In brief, kidney sections were stained with haematoxylin and eosin and observed under light microscope in a masked fashion at a magnification of ×400 using the Analysis Imaging System (AIS, Imaging Research, St. Catherines, Ontario, Canada). In each kidney 40 glomeruli were graded according to the severity of the glomerular damage: 0, normal; 1, slight glomerular damage, the mesangial matrix and /or hyalinosis with focal adhesion, involving less than 25% of the glomerulus; 2, sclerosis of 25 to 50%; 3, sclerosis of 50 to 75%; 4, sclerosis of more than 75% of the glomerulus. The indices for glomerulosclerosis were calculated using the following formula:
where nx=number of glomeruli in each grade of glomerulosclerosis. The coefficient of variation in this method was 1.7%.
The evaluation of tubulointerstitial area (TIA) was assessed using a point-counting technique and carried out in the renal cortex and corticomedullary junction for each animal following routine established methods [39]. In each field, 100 points were counted on a 1 cm2 eyepiece graticule with 10 equidistant grid lines. A total of 12 high power fields (×400) per section were counted for each animal in all groups in the corticomedullary field. Each high power field was 0.076 mm2 with 0.91 mm2 being the total area counted per slide.
% Fractional area (FA) was calculated as below:
where %FA corresponds to the percent of tubulointerstitial area in the total area counted of the section.
Gene expression of transforming growth factor β1 (TGFβ1) and β-inducible gene-h3 (βig-h3)
The site specific expression of both TGF-β and βig-h3 mRNA was assessed by in situ hybridization, and done as previously reported [40, 41]. Briefly, four micron kidney sections were hybridised following digestion with PronaseE at 37°C. The hybridization buffer containing 2×104 cpm/µl 35S labelled riboprobe, 0.72 mg/ml yeast RNA, 50% deionized formamide, 100 mmol/l DTT, 10% dextran sulfate, 0.3 M NaCl, 10 mmol/l Na2HPO4, 10 mmol/l pH 7.5 and 5 mmol/l EDTA pH 8.0, 0.02% bovine serum albumin (BSA) 0.02% Ficoll 400 and 0.02% PVP was added to each section and incubated at 60°C overnight. Following stringent washing with 50% formamide, 2×SSC at 55°C the slides were air-dried and exposed to BioMax MR film (Kodak Company, Rochester, N.Y., USA) for 3 to 5 days. Slides were coated with desiccant for a period of 2 to 4 weeks according to the autoradiography results. The slides were developed using Kodak D19 developer for 4 min, 1% acetic acid for 1 min and Illford Hypan fixative for 4 min followed by rinsing in distilled water for at least 15 min. The sections were then fixed in 4% paraformaldehyde and treated with a progressive haematoxylin/eosin stain. Background hybridization was controlled for by the inclusion of a sense riboprobe in a method previously described [22, 40]. Quantitation of the data was done on the renal cortex and corticomedullary junction from the autoradiography film for each probe. The probe binding density was analysed by computerised densitometry. Prior to each analysis session the analysis software was densitometrically calibrated to the Linotype-Hell scanner using an AGFA transmissive greyscale step wedge. The densitometric values for the AGFA step wedge were acquired using an optically calibrated densitometer. Using a densitometrically calibrated look-up table, greyscale values were converted to optical density values.
Statistics
All data are shown as means ± SEM unless otherwise specified. Since the albuminuria data were not normally distributed, these data were logarithmically transformed before being subjected to statistical analysis and are shown as geometric mean ×/÷ tolerance factors (geometric SEM). Data were initially analysed by analysis of variance (ANOVA) with post hoc comparisons between group means carried out by Fisher's least significant difference method [42]. Plasma renin activity results were analysed using a Mann-Whitney U unpaired t test. A p value of less than 0.05 was statistically significant.
Results
Metabolic parameters, blood pressure and kidney weight
HbA1c was increased in all diabetic groups with no difference among the various drug regimens (Table 1). Diabetes was associated with reduced body weight gain. None of the drug therapies influenced body weight. There was an increase in kidney weight/body weight ratio (KW/BW) between the control group and the diabetic untreated group (control, 3.3±0.1 vs diabetic, untreated 4.8±0.1 mg/kg; p<0.001), which was significantly reduced by both doses of omapatrilat (oma10, 4.3±0.1; oma40 4.3±0.1 mg/kg; p<0.05 vs diabetic), but not perindopril (4.6±0.1 mg/kg). Left ventricular weight was not affected by the induction of diabetes, however this parameter was reduced by each treatment group to a similar degree (Table 1).
Biochemical assays
There was no difference in measurements of plasma renin activity (PRA) between the control and untreated diabetic groups. Perindopril was associated with an increase in PRA. The administration of omapatrilat was associated with a marked increase in PRA in a dose-dependent manner (Table 1).
Systolic blood pressure (assessment by tail cuff method)
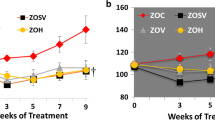
Diabetes was associated with a reduced SBP compared to controls over the 32-week study period (ANOVA; F=24.4, p<0.001; average wk 8–32 control 206±2 vs untreated diabetic, 185±2 mmHg, p<0.001) (Fig. 1A). Perindopril significantly reduced SBP compared to the untreated diabetic group (ANOVA; F=109, p<0.001; average wk 8–32 Per 152±2 mmHg p<0.001 vs diabetic) as did the oma10 group (ANOVA; F=132, p<0.001; average wk 8–32 oma10 148±2 mmHg, p<0.001 vs diabetic). There was no difference in blood pressure between the perindopril and oma10 treated groups. The oma40 treatment significantly reduced systolic blood pressure when compared to the oma10 treatment (ANOVA; F=26.4, p<0.001, average wk 8–32 133±2 mmHg; p<0.001 vs oma10).

Serial measurements of systolic blood pressure are shown for weeks 4–32 of the study period for control rats (white squares), untreated diabetic rats (black squares), diabetic + perindopril (white circles), diabetic + omapatrilat (10 mg/kg/day) (white triangles) and diabetic + omapatrilat (40 mg/kg/day) (black triangles). Diabetic vs control, F=24.4, p<0.001; diabetic vs perindopril F=109, p<0.001; diabetic vs oma10, F=134, p<0.001; diabetic vs oma40 F=300, p<0.001; oma10 vs om 40, F=26.6, p<0.001). ANOVA with repeated measures. B Data for systolic blood pressure data obtained by radiotelemetry are shown as means ± SEM. *p<0.001 vs control; †p<0.001 vs diabetic. C, control; D, diabetic; Per, perindopril; Oma10, omapatrilat 10 mg/kg/day; Oma40, omapatrilat 40 mg/kg/day
Systolic blood pressure (assessment by telemetry)
Diabetes was associated with a significant decrease in SBP compared to the controls over the 10-week period (control 183±4 vs diabetic untreated 143±6 mmHg; p<0.001), (Fig. 1B). Drug therapy with both Per and oma10 significantly decreased SBP to a similar degree (Per 119±4 mmHg, oma10 118±3 mmHg). There was a further reduction in blood pressure with oma40 treatment (111±3 mmHg), however this did not reach statistical significance (p<0.10).
Inhibition of NEP and ACE
There was no difference in NEP observed between the control and untreated diabetic group, nor with the oma10 group. There was significant inhibition of NEP in the oma40 treated group compared to the diabetic group (untreated diabetic, 1768±44 vs oma40 1362±94 dpm/mm2, p<0.005). Renal ACE binding was reduced in diabetic rats (control 1569±139 vs untreated diabetic, 616±124 dpm/mm2, p<0.01). Both oma10 and oma40 groups completely inhibited the degree of ACE binding (oma10 50.3±23.2, oma40 24.2±4.5 dpm/mm2; p<0.001). There was no difference in renal ACE inhibition between the two omapatrilat treatment groups. Ex vivo inhibition of ACE by perindopril could not accurately be assessed because of disassociation of the drug from the enzyme during the preparation and rinsing of the tissue.
In view of the previous report of ACE redistribution to the glomerulus in diabetic rats [43], ACE immunostaining was assessed in all five groups. Only in untreated diabetic rats was there evidence of glomerular ACE immunostaining. In all treated diabetic groups, there was minimal ACE immunostaining in glomeruli with the majority of staining in renal tubules (Fig. 2).

Immunohistochemical staining for renal ACE in cortical glomeruli and tubules from control (A), untreated diabetic (B), diabetic + Per (C), diabetic + Oma10 (D) and diabetic + Oma40 (E). Original magnification ×200
Albuminuria
Diabetes was associated with a progressive rise in albuminuria compared with non-diabetic SHR during weeks 16 to 32 of the study (ANOVA; F=84.9, p<0.001; week 32 diabetes 145.8×/÷1.1 mg/24 h vs control 48.9×/÷1.2 mg/24 h, p<0.001) (Fig. 3). Perindopril retarded the increase in albuminuria compared to the untreated diabetic group over the treatment period (ANOVA; F=17.0 p<0.001; week 32 diabetes + perindopril 97.1×/÷1.1 mg/24 h, vs diabetic p<0.001), as did the oma10 group (ANOVA; F=38.6, p<0.001). There was a difference in the ability of the lower dose of omapatrilat to prevent the increase in albuminuria compared to perindopril (F=11.8, p<0.005). The oma40 group completely prevented the rise in albuminuria to below that of the control group (ANOVA; F=55.3, p<0.001; week 32 diabetic + oma40). Furthermore, oma40 was more effective than oma10 in retarding the development of albuminuria in diabetic SHR (F=5.9, p<0.05).

Serial monthly measurements of albumin excretion rate are shown for weeks 16–32 of the study period for control rats (white squares), untreated diabetic rats (black squares), diabetic + perindopril (white circles), diabetic + omapatrilat 10 mg·kg−1·day (white triangles) and diabetic + omapatrilat 40 mg/kg/day (black triangles). Geometric means and tolerance factors are shown. Control vs diabetic F=84.9, p<0.001; diabetic vs perindopril F=19.1, p<0.001; diabetic vs oma 10 F=38.6, p<0.001; oma 10 vs oma 40 F=5.9, p<0.05). ANOVA with repeated measures
Nephrin expression
Diabetes was associated with a significant decrease in nephrin protein expression compared to control SHR (control 40.0±1.9 vs untreated diabetic 18.6±1.8%; p<0.001) (Figs. 4, 5). Perindopril and oma10 treatment partially prevented the loss in glomerular nephrin expression (Per 37.6±4.0%, Oma10 32.8±3.8%; p<0.001 vs untreated diabetic). Oma40 treatment resulted in nephrin protein values similar to that observed in non-diabetic controls (47.6±1.5%; p<.001 vs untreated diabetic; p<0.01 vs Oma10).

Immunohistochemical staining for nephrin in glomeruli from control (A), untreated diabetic (B), diabetic + Per (C), diabetic + Oma 10 (D) and diabetic + Oma 40 (E). Original magnification ×200

Quantitation for the immunohistochemical staining for nephrin protein expression. Data are shown as mean ± SEM. *p<0.01 vs control; †p<0.01 vs diabetic; #p<0.01 vs Oma10
Glomerulosclerotic index (GSI) and tubulointerstitial area (TIA)
Diabetes was associated with an increase in the glomerulosclerotic index (GSI) compared to the control rats (control 2.0±0.1 vs diabetic untreated 3.4±0.8; p<0.001) (Figs. 6, 7A). The perindopril and oma10 groups significantly prevented the increase in GSI (Per 2.2±0.2, oma10 2.2±0.1; p<0.001 vs untreated diabetic). The high dose omapatrilat group completely prevented the increase in GSI to levels below that seen in the control group (oma40 1.6±0.1; p<0.05 vs control SHR).

Haemotoxylin and eosin staining of glomeruli from control (A), untreated diabetic (B), diabetic + Per (C), diabetic + Oma10 (D) and diabetic + Oma40 (E). Original magnification ×200

Glomerulosclerotic indices (A) and tubulointerstitial area (B) in rats from control, diabetic untreated and diabetic treated with perindopril or omapatrilat groups. *p<0.01 vs control; †p<0.01 vs diabetic untreated; #p<0.01 vs omapatrilat 10 mg·kg−1·day−1. C, control; D, diabetic; Per, perindopril; Oma10, omapatrilat 10 mg·kg−1·day−1; Oma40, omapatrilat 40 mg·kg−1·day−1
An increase in TIA was observed in the untreated diabetic group compared to the control group (control 7.2±0.6 vs diabetic untreated 17.1±1.0%, p<0.001). TIA was reduced by both the perindopril (8.6±0.8%) and oma10 treatment (9.6±0.6%) to similar levels (P<0.001). This parameter was further decreased by oma40 treatment (oma10 vs oma40 6.7±0.3%, p<0.001) (Fig. 7B).
Gene expression of Transforming Growth Factor β (TGFβ1) and β-inducible gene h3 (βig-h3) by in situ hybridisation
Diabetes was associated with an increase in renal TGFβ1 gene expression when compared to the control group (control 215±13 vs diabetic untreated 305±17 dpm/mm2 (p<0.05) (Fig. 8, 9A). There was a reduction in gene expression observed in the oma40 group (oma40 178±15dpm/mm2 vs diabetic untreated, p<0.001. Neither Per or oma10 treatment had any effect on TGFβ1 gene expression (Fig. 9A).

X-ray film of TGFβ1 in situ hybridisation of kidney from control (A), untreated diabetic (B), diabetic + Per (C), diabetic + Oma10 (D) and diabetic + Oma40 (E). Original magnification ×4

Quantitation of gene expression as assessed by in situ hybridisation for TGFβ1 (A) and βig-h3 (B). Data are shown as means ± SEM. *p<0.001 vs control; †p<0.001 vs diabetic. C, control; D, diabetic; Per, perindopril; Oma10, omapatrilat 10 mg·kg−1·day−1; Oma40, omapatrilat 40 mg·kg−1·day−1
Diabetes was also associated with an increase in gene expression of the TGFβ-dependent matrix protein, βig-H3 when compared to the control group (control 88±16 vs diabetic 178±21 dpm/mm2, p<0.001) (Figs. 8, 9B). This parameter was also reduced by oma40 treatment (77±14 dpm/mm2, p<0.001 vs diabetic) but not by perindopril or oma10 group therapy (Per, 209±25; Oma10, 158±16 dpm/mm2) (Fig. 9B).
Discussion
This study shows that a low dose of omapatrilat, despite similar effects on blood pressure to perindopril, was more powerful at retarding the rise in albuminuria in this experimental model. Furthermore, high dose omapatrilat (40 mg·kg−1·day−1) achieved not only a greater reduction in blood pressure but was also associated with reduced nephropathy as assessed by both functional and structural parameters. These observations are consistent with our previous work showing that ACE inhibition in this model attenuates but does not prevent the development of nephropathy [44].
Blood pressure is a major determinant of the rate of progression of diabetic complications, including nephropathy. Therefore, it is critical to accurately determine the blood pressure response to the various treatment groups. This was facilitated by the use of radiotelemetry to carefully monitor blood pressure over the entire 24-h period in conscious animals. The doses of perindopril and omapatrilat (low dose, 10 mg·kg−1·day−1 used in this study achieved similar reductions in blood pressure and complemented the findings from the tail-cuff measurements carried out every 4 weeks over the 8-month study period. Despite similar blood pressure reduction between perindopril and low dose omapatrilat, VPI therapy was associated with greater retardation in the rate of development of albuminuria in these diabetic SHR. This discordance in proteinuria, despite similar blood pressure control between low-dose omapatrilat and perindopril has also been observed in a study of the subtotal nephrectomy model of progressive renal injury [18]. In that study, despite omapatrilat and enalapril achieving similar blood pressure, omapatrilat treatment was associated with less proteinuria in association with a delay in the development of chronic renal impairment [18].
The underlying mechanisms responsible for the superiority of omapatrilat over ACE inhibition on albuminuria despite similar reductions in blood pressure remain to be determined. One possibility is that omapatrilat more effectively blocks the intrarenal RAS by being a more powerful inhibitor of renal ACE. Such a possibility is suggested by the reduction in renal ACE activity with omapatrilat being associated with a greater increase in the plasma renin activity than seen with perindopril treatment. In some of the previous studies comparing ACE/NEP inhibition to ACE inhibition, there was a significant difference in blood pressure between the two groups and therefore it was not possible to determine blood pressure-independent effects of agents such as omapatrilat [13, 17].
In the present study, low dose omapatrilat was more effective in retarding albuminuria than perindopril yet both agents were similar in their benefits on renal structural parameters. The results of this study extend previous findings which indicate that one cannot assume that functional and structural markers of renal injury will respond in an identical manner. For example, administration of a neutralizing antibody to TGFβ has been reported to influence renal structural injury yet have no effect on albuminuria [21].
The importance of the local compared with systemic RAS continues to be a major area of investigation. We have shown two different models of renal disease that local activation on the tubular RAS is a major determinant of progressive renal injury [45, 46]. The importance of ACE per se in the diabetic kidney also needs to be reconsidered. Although renal ACE is reduced in the diabetic kidney, another study clearly showed up-regulation of ACE in diabetic glomeruli [43], a phenomenon we have now confirmed in the kidneys from diabetic SHR. It is postulated that a drug with greater inhibition of ACE in the kidney could confer superior effects on renal functional parameters such as albuminuria.
A major finding of this study was the superior renoprotection afforded by high dose omapatrilat. This is unlikely to be related to intrarenal ACE inhibition since low and high doses both inhibited renal ACE binding to a similar degree. However the higher dose of omapatrilat also inhibited the enzyme neutral endopeptidase (NEP) in addition to ACE. This reduction in NEP activity could explain the superior blood pressure reduction since NEP is involved in the degradation of vasodilators such as bradykinin and natriuretic peptides. The observed reduction in systemic blood pressure afforded by the high dose VPI, if translated to the intrarenal microcirculation, could lead to further reduction in intraglomerular pressure (PG). Furthermore, based on the postulated in vivo actions of NEP inhibition with associated potentiation of vasodilatory substances such as NP, it is postulated that omapatrilat, for a given reduction in systemic blood pressure, could be more effective at decreasing PG than other hypertensive agents. It is important to note that NEP inhibition alone has not been shown to confer renoprotection and the only context where such an approach has been shown to have beneficial effects on the kidney has been in the setting of concomitant inhibition of vasoconstrictors as reported with dual ACE/NEP and ECE/NEP inhibition [47].
In our study, expression of the slit pore protein nephrin was documented to be reduced in the diabetic kidney. This confirms our previous report that the angiotensin II (AII) antagonist, irbesartan can prevent renal nephrin depletion in diabetes associated with attenuation of albuminuria [33]. We have also shown that interruption of the RAS with an ACE inhibitor also has this effect, suggesting that AII-dependent mechanisms participate in modulation of renal nephrin. It remains to be seen if this is a direct effect of blocking the RAS, or related to the ability of these drugs to attenuate diabetes associated renal injury. Interestingly the high dose of omapatrilat was associated with a higher level of renal nephrin content that with the other treatment and this was associated with greater effects on urinary albumin excretion. These findings extend the important associations that have now been reported linking albuminuria, antihypertensive treatment and nephrin expression not only in diabetes [33, 34] but also in non-diabetic models of renal disease [41, 48].
Both the ACE inhibitor and omapatrilat not only retarded the development of albuminuria and glomerulosclerosis but also had effects on reducing tubulointerstitial injury. The tubulointerstitium is now considered a major site of diabetes-related injury and is an important determinant of the rate of decline in renal function in diabetic nephropathy [49]. High dose omapatrilat was the most effective treatment in reducing both the glomerular and tubulointerstitial injury. Furthermore, this drug at the high dose prevented overexpression of TGFβ1 in the diabetic kidney. In the context of the prosclerotic effects of this growth factor and that inhibition of TGFβ1 using a neutralizing antibody leads to decreased renal extracellular matrix (ECM) accumulation in diabetes [21], it is possible that reduction in TGFβ1 partly explains the superior effects of high dose omapatrilat on GSI and TI injury. High dose omapatrilat not only reduced TGFβ1 expression but also decreased TGFβ1 bioactivity in a similar manner, as measured by the expression of the TGFβ1 dependent matrix protein βig-h3. This novel matrix protein has been previously shown to reflect the activity of TGFβ1 in both the human and experimental context [40, 50], although the function of βig-h3 in renal physiology and pathogenic states remains unclear [22]. The lack of effect of perindopril or low-dose omapatrilat on TGFβ1 expression in these diabetic SHR, yet improvement in renal structural injury, emphasises that TGFβ1 alone cannot explain diabetes associated renal structural injury. The effects on albuminuria are unlikely to be linked to TGFβ1 expression as infusion of TGFβ1 antibody has been shown not to affect this parameter [21].
In recent international guidelines for the treatment of diabetes and hypertension, aggressive blood pressure targets have been defined which often cannot be achieved with single agents, including ACE inhibition [51]. Our study has identified that high dose omapatrilat which has multiple effects on vasoactive hormones, has the potential to not only decrease blood pressure more than other monotherapies but could also confer superior effects on the kidney for a given decrease in systemic blood pressure. Although one cannot extrapolate directly from the experimental to the clinical context, it seems that VPIs such as omapatrilat could have an important role not only in decreasing blood pressure in diabetes, but also in conferring superior renoprotection over agents that act solely to interrupt the RAS.
Abbreviations
- ACEi:
-
angiotensin converting enzyme inhibitor
- AII:
-
angiotensin II
- βig-h3:
-
β-inducible gene-h3
- GSI:
-
glomerulosclerotic index
- NEP:
-
neutral endopeptidase
- Oma:
-
omapatrilat
- Per:
-
perindopril
- PRA:
-
plasma renin activity
- RAS:
-
renin-angiotensin system
- SBP:
-
systolic blood pressure
- TIA:
-
tubulointerstitial area
- VPI:
-
vasopeptidase inhibitor
References
Adler AI, Stratton IM, Neil HA et al. (2000) Association of systolic blood pressure with macrovascular and microvascular complications of type 2 diabetes (UKPDS 36): Prospective observational study. BMJ 321:412-419
Thomas W, Shen Y, Molitch ME, Steffes MW (2001) Rise in albuminuria and blood pressure in patients who progressed to diabetic nephropathy in the Diabetes Control and Complications Trial. J Am Soc Nephrol 12:333–340
Jandeleit-Dahm K, Cooper ME (2002) Hypertension and diabetes. Curr Opin Nephrol Hypertens 11:221–228
Zatz R, Dunn BR, Meyer TW, Brenner B (1986) Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest 77:1925–1930
Cooper ME, Allen TJ, Macmillan PA, Clarke BE, Jerums G, Doyle AE (1989) Enalapril retards glomerular basement membrane thickening and albuminuria in the diabetic rat. Diabetologia 32:326–338
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD (1993) The effect of angiotensin converting enzyme inhibition on diabetic nephropathy. N Engl J Med 329:1456–1462
Remuzzi A, Perico N, Amuchastegui CS et al. (1993) Short- and long-term effect of angiotensin II receptor blockade in rats with experimental diabetes. J Am Soc Nephrol 4:40–49
Allen TJ, Cao ZM, Youssef S, Hulthen UL, Cooper ME (1997) Role of angiotensin II and bradykinin in experimental diabetic nephropathy—functional and structural studies. Diabetes 46:1612–1618
Anderson S, Rennke HG, Garcia DL, Brenner BM (1989) Short and long term effects of antihypertensive therapy in the diabetic rat. Kidney Int 36:526–536
Allen TJ, Cooper ME, O'Brien RC, Bach LA, Jackson B, Jerums G (1990) Glomerular filtration rate in streptozocin-induced diabetic rats. Role of exchangeable sodium, vasoactive hormones, and insulin therapy. Diabetes 39:1182–1190
Houlihan CA, Allen TJ, Baxter AL et al. (2002) A low-sodium diet potentiates the effects of losartan in type 2 diabetes. Diabetes Care 25:663–671
Fournie-Zaluski MC, Gonzalez W, Turcaud S, Pham I, Roques BP, Michel JB (1994) Dual inhibition of angiotensin-converting enzyme and neutral endopeptidase by the orally active inhibitor mixanpril: a potential therapeutic approach in hypertension. Proc Natl Acad Sci USA 91:4072–4076
Cao Z, Burrell LM, Tikkanen I, Bonnet F, Cooper ME, Gilbert RE (2001) Vasopeptidase inhibition attenuates the progression of renal injury in subtotal nephrectomized rats. Kidney Int 60:715–721
French JF, Anderson BA, Downs TR, Dage RC (1995) Dual inhibition of angiotensin-converting enzyme and neutral endopeptidase in rats with hypertension. J Cardiovasc Pharmacol 26:107–113
Burrell LM, Droogh J, Man in't Veld O, Rockell MD, Farina NK, Johnston CI (2000) Antihypertensive and antihypertrophic effects of omapatrilat in SHR. Am J Hypertens 13:1110–1116
Intengan HD, Schiffrin EL (2000) Vasopeptidase inhibition has potent effects on blood pressure and resistance arteries in stroke-prone spontaneously hypertensive rats. Hypertension 35:1221–1225
Cohen DS, Mathis JE, Dotson RA, Graybill SR, Wosu NJ (1998) Protective effects of Cgs 30440, a combined angiotensin-converting enzyme inhibitor and neutral endopeptidase inhibitor, in a model of chronic renal failure. J Cardiovas Pharmacol 32:87–95
Taal MW, Nenov VD, Wong W et al. (2001) Vasopeptidase inhibition affords greater renoprotection than angiotensin-converting enzyme inhibition alone. J Am Soc Nephrol 12:2051–2059
Tikkanen T, Tikkanen I, Rockell MD et al. (1998) Dual inhibition of neutral endopeptidase and angiotensin-converting enzyme in rats with hypertension and diabetes mellitus. Hypertension 32:778–785
Ruotsalainen V, Ljungberg P, Wartiovaara J et al. (1999) Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci USA 96:7962–7967
Ziyadeh FN, Hoffman BB, Han DC et al. (2000) Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA 97:8015–8020
Gilbert RE, Wilkinson-Berka JL, Johnson DW et al. (1998) Renal expression of transforming growth factor-beta inducible gene-h3 (beta ig-h3) in normal and diabetic rats. Kidney Int 54:1052–1062
Cooper ME, Allen TJ, O'Brien RC et al. (1988) Effects of genetic hypertension on diabetic nephropathy in the rat- functional and structural characteristics. J Hypertens 6:1009–1016
Brockway BP, Mills PA, Azar SH (1991) A new method for continuous chronic measurement and recording of blood pressure, heart rate and activity in the rat via radio-telemetry. Clin Exp Hypertens A 13:885–895
Soulis T, Cooper ME, Vranes D, Bucala R, Jerums G (1996) Effects of aminoguanidine in preventing experimental diabetic nephropathy are related to the duration of treatment. Kidney Int 50:627–634
Bunag RD (1973) Validation in awake rats of a tail-cuff method for measuring systolic pressure. J Appl Physiol 34:279–282
Johnston CI, Mendelsohn F, Casley D (1971) Evaluation of renin and angiotensin assays and their clinical application. Med J Aust 1:126–128
Burrell LM, Palmer J, Charlton JA, Thomas T, Baylis PH (1990) A new radioimmunoassay for human alpha atrial natriuretic peptide and its physiological validation. J Immunoassay 11:159–175
Burrell LM, Farina N, Risvanis J, Woollard D, Casely D, Johnston CI (1997) Inhibition of neutral endopeptidase, the degradative enzyme for natriuretic peptides, in rat kidney after oral SCH 42495. Clin Sci (Lond) 93:43–50
Mendelsohn FA (1984) Localization of angiotensin converting enzyme in rat forebrain and other tissues by in vitro autoradiography using 125I-labelled MK351A. Clin Exp Pharmacol Physiol 11:431–435
Farina NK, Johnston CI, Burrell LM (2000) Reversal of cardiac hypertrophy and fibrosis by S21402, a dual inhibitor of neutral endopeptidase and angiotensin converting enzyme in SHRs. J Hypertens 18:749–755
Kawachi H, Koike H, Kurihara H et al. (2000) Cloning of rat nephrin: expression in developing glomeruli and in proteinuric states. Kidney Int 57:1949–1961
Bonnet F, Cooper ME, Kawachi H, Allen TJ, Boner G, Cao Z (2001) Irbesartan normalises the deficiency in glomerular nephrin expression in a model of diabetes and hypertension. Diabetologia 44:874–877
Forbes JM, Bonnet F, Russo LM et al. (2002) Modulation of nephrin in the diabetic kidney: association with systemic hypertension and increasing albuminuria. J Hypertens 20:985–992
Soulis T, Thallas V, Youssef S et al. (1997) Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 40:619–628
Hsu SM, Raine L, Fanger H (1981) Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 29:577–580
Forbes JM, Cooper ME, Thallas V et al. (2002) Reduction of the accumulation of advanced glycation end products by ACE inhibition in experimental diabetic nephropathy. Diabetes 51:3274–3282
Saito T, Sumithran E, Glasgow EF, Atkins RC (1987) The enhancement of aminonucleoside nephrosis by the co-administration of protamine. Kidney Int 32:691–699
Hewitson TD, Darby IA, Bisucci T, Jones CL, Becker GJ (1998) Evolution of tubulointerstitial fibrosis in experimental renal infection and scarring. J Am Soc Nephrol 9:632–642
Rumble JR, Cooper ME, Soulis T et al. (1997) Vascular hypertrophy in experimental diabetes. Role of advanced glycation end products. J Clin Invest 99: 1016–1027
Cao Z, Bonnet F, Candido R et al. (2002) Angiotensin type 2 receptor antagonism confers renal protection in a rat model of progressive renal injury. J Am Soc Nephrol 13:1773–1787
Snedecor GW, Cochran WG (1980) Statistical methods. In: Snedeker GW (ed.) Statistical methods, 7th edn. Iowa State University Press, Ames, Iowa, pp 228–236
Anderson S, Jung FF, Ingelfinger JR (1993) Renal renin-angiotensin system in diabetes: functional, immunohistochemical, and molecular biological correlations. Am J Physiol 265: F477–486
Cooper ME, Rumble JR, Allen TJ, O'Brien RC, Jerums G, Doyle AE (1992) Antihypertensive therapy in a model combining spontaneous hypertension with diabetes. Kidney Int 41:898–903
Gilbert RE, Wu LL, Kelly DJ et al. (1999) Pathological expression of renin and angiotensin II in the renal tubule after subtotal nephrectomy. Implications for the pathogenesis of tubulointerstitial fibrosis. Am J Pathol 155:429–440
Kelly DJ, Skinner SL, Gilbert RE, Cox AJ, Cooper ME, Wilkinson-Berka JL (2000) Effects of endothelin or angiotensin II receptor blockade on diabetes in the transgenic (mRen-2)27 rat. Kidney Int 57:1882–1894
Tikkanen I, Tikkanen T, Cao Z et al. (2002) Combined inhibition of neutral endopeptidase with angiotensin converting enzyme or endothelin converting enzyme in experimental diabetes. J Hypertens 20:707–714
Benigni A, Tomasoni S, Gagliardini E et al. (2001) Blocking angiotensin II synthesis/activity preserves glomerular nephrin in rats with severe nephrosis. J Am Soc Nephrol 12:941–948
Gilbert RE, Cooper ME (1999) The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int 56:1627–1637
O'Brien ER, Bennett KL, Garvin MR et al. (1996) Beta ig-h3, a transforming growth factor-beta-inducible gene, is overexpressed in atherosclerotic and restenotic human vascular lesions. Arterioscler Thromb Vasc Biol 16:576–584
Bakris GL, Williams M, Dworkin L et al. (2000) Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 36:646–661
Acknowledgements
This study was supported by grants from the Juvenile Diabetes Research Foundation International (JDRFI) and the National Health and Medical Research Council of Australia. BJD is a recipient of a Dora Lush PhD scholarship from the National Health and Medical Research Council of Australia.
The authors gratefully thank Ms K. Goldring, Mr G. Langmaid, Ms M. Arnstein, Ms D. Paxton and Dr. L. Jordan for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davis, B.J., Johnston, C.I., Burrell, L.M. et al. Renoprotective effects of vasopeptidase inhibition in an experimental model of diabetic nephropathy. Diabetologia 46, 961–971 (2003). https://doi.org/10.1007/s00125-003-1121-9
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1121-9