
Abstract
Aims/hypothesis
To study the secondary consequences of impaired suppression of endogenous glucose production (EGP) we have created a transgenic rat overexpressing the gluconeogenic enzyme phosphoenolpyruvate carboxykinase (PEPCK) in the kidney. The aim of this study was to determine whether peripheral insulin resistance develops in these transgenic rats.
Methods
Whole body rate of glucose disappearance (Rd) and endogenous glucose production were measured basally and during a euglycaemic/hyperinsulinaemic clamp in phosphoenolpyruvate carboxykinase transgenic and control rats using [6-3H]-glucose. Glucose uptake into individual tissues was measured in vivo using 2-[1-14C]-deoxyglucose.
Results
Phosphoenolpyruvate carboxykinase transgenic rats were heavier and had increased gonadal and infrarenal fat pad weights. Under basal conditions, endogenous glucose production was similar in phosphoenolpyruvate carboxykinase transgenic and control rats (37.4±1.1 vs 34.6±2.6 µmol/kg/min). Moderate hyperinsulinaemia (810 pmol/l) completely suppressed EGP in control rats (−0.6±5.5 µmol/kg/min, p<0.05) while there was no suppression in phosphoenolpyruvate carboxykinase rats (45.2±7.9 µmol/kg/min). Basal Rd was comparable between PEPCK transgenic and control rats (37.4±1.1 vs 34.6±2.6 µmol/kg/min) but under insulin-stimulated conditions the increase in Rd was greater in control compared to phosphoenolpyruvate carboxykinase transgenic rats indicative of insulin resistance (73.4±11.2 vs 112.0±8.0 µmol/kg/min, p<0.05). Basal glucose uptake was reduced in white and brown adipose tissue, heart and soleus while insulin-stimulated transport was reduced in white and brown adipose tissue, white quadriceps, white gastrocnemius and soleus in phosphoenolpyruvate carboxykinase transgenic compared to control rats. The impairment in both white and brown adipose tissue glucose uptake in phosphoenolpyruvate carboxykinase transgenic rats was associated with a decrease in GLUT4 protein content. In contrast, muscle GLUT4 protein, triglyceride and long-chain acylCoA levels were comparable between PEPCK transgenic and control rats.
Conclusions/interpretation
A primary defect in suppression of EGP caused adipose tissue and muscle insulin resistance.
Similar content being viewed by others


Insulin resistance is a characteristic feature of Type 2 diabetes. Both reduced insulin-stimulated glucose uptake and metabolism in peripheral tissues (skeletal muscle and fat) and impaired suppression of endogenous glucose production (EGP) by insulin have been documented [1, 2].
Multiple mechanisms for the insulin resistance have been proposed including both primary genetic [3, 4] and secondary causes such as nutrient excess [5]. The current increase in the prevalence of Type 2 diabetes in many parts of the world [6] is linked to an increase in obesity in the population [7, 8]. While there could be many causes of insulin resistance, fat-induced defects in insulin action are probably the most important.
Excess fat has been shown to cause peripheral insulin resistance via multiple mechanisms which include among others, excess supply of non-esterified fatty acids [9], increased intracellular accumulation of muscle triglycerides and long chain acyl CoAs (LCACoAs) [9], excessive production of tumor necrosis factor-α [10], reduction in the concentration of adiponectin [11] and increased c-Jun amino-terminal kinase (JNK) activity [12].
Diets high in fat have also been shown to cause impaired insulin suppression of endogenous glucose production [13, 14, 15]. This has been associated with a NEFA-induced increase in gluconeogenesis [15, 16, 17, 18]. Recently, we have confirmed that the dietary fat-induced increase in gluconeogenesis is associated with increased levels of fructose-1,6-bisphosphatase, a regulatory enzyme in the glucose synthesising pathway [15, 19]. Interestingly, it has been suggested that fat-induced hepatic insulin resistance precedes muscle insulin resistance [13] raising the possibility that hepatic insulin resistance can contribute to muscle and fat insulin resistance observed under states of increased dietary fat intake.
The secondary consequences of impaired suppression of gluconeogenesis have not been formally tested. An increase in glucose production could enhance glucose entry into muscle and fat cells causing 'glucose toxicity', a phenomenon that has been attributed to a number of processes including increased flux through the hexosamine biosynthesis pathway (HBP) [5, 20, 21] and the intracellular accumulation of triglycerides [22].
To determine the consequences of chronically impaired suppression of gluconeogenesis we have produced transgenic rats that overexpress the regulated enzyme of gluconeogenesis, phosphoenolpyruvate carboxykinase (PEPCK) predominantly in the kidney under the control of a non-insulin responsive promoter [23]. These rats developed a number of features associated with the metabolic syndrome including obesity, mild hyperglycaemia, hyperinsulinaemia, and dyslipidaemia [24]. The aim of this study was to investigate the consequences of impaired EGP regulation on peripheral glucose uptake. We show that an impaired suppression of EGP leads to insulin resistance in muscle and adipose tissue.
Methods
Animals
PEPCK transgenic rats expressing the PEPCK gene predominantly in the kidney under the control of the metallothionein promoter were produced as previously described on the PVG/c background strain. We have previously shown that there is no PEPCK expression in fat, heart, muscle, lung and brain of transgenic rats [23]. The rats were housed in the University of Melbourne, Department of Medicine Animal Research Facility at the Royal Melbourne Hospital. Control rats (PVG/c) were purchased from the Animal Resource Centre (Perth, WA, Australia) at 11 to 12 weeks of age and then housed in the University of Melbourne, Department of Medicine animal facility for 3 weeks. Lighting was artificial and timer controlled with a 12-h light/dark cycle. Room temperature was kept constant at 22°C. Rats were fed a standard laboratory chow diet consisting of 74% carbohydrate, 6% fat and 20% protein, by weight (Barastoc Pty, Pakenham, Victoria, Australia). All procedures were approved by the Royal Melbourne Hospital Research Foundation Animal Ethics Committee, approval number AEC 2000.002.
Experimental procedure
After an overnight fast, polyethylene catheters (Crichley Electrical, NSW, Australia) were inserted into the right jugular vein and left carotid artery. The venous catheter was used for infusion and the arterial catheter for blood sampling. A tracheostomy was carried out to prevent upper airway obstruction during the experiment. Anaesthesia was monitored regularly and maintained throughout the experiment by intravenous administration of pentobarbitone sodium (Nembutal, Rhone Merieux, QLD, Australia). Body temperature was monitored with a rectal temperature probe and maintained at 37°C with a heating lamp.
Glucose turnover
A primed (2-min, 100 µBq·min−1) continuous infusion (5.5 µBq·min−1) of [6-3H]-glucose was administered during basal and euglycaemic-hyperinsulinaemic clamp experiments to measure whole body glucose turnover [25]. During the hyperinsulinaemic clamp experiments, following an initial priming dose, insulin was infused at a constant rate at either 2 mU·kg−1·min in transgenic rats or 4 mU·kg−1·min in control rats to produce similar plasma insulin concentrations in the two groups of rats. In a separate group of PEPCK transgenic rats, insulin was infused at the higher 4 mU·kg−1·min. Blood glucose concentration was maintained by the infusion of a 5% glucose solution. Blood samples were collected during steady state conditions at 90, 100 and 110 min. Under steady-state conditions the rate of glucose appearance equals the rate of glucose disappearance. The rate of glucose disappearance (Rd) was calculated by dividing the infusion rate of [6-3H]-glucose (dpm·min−1) by the plasma [6-3H]-glucose specific activity. The rate of endogenous glucose production (EGP) was measured as the difference between the calculated Rd and the rate of infused glucose.
Measurement of individual tissue glucose uptake
A modification of the labelled 2-[1-14C]-deoxyglucose technique [26] was used to measure glucose uptake into individual tissues [25]. Following the collection of blood samples for measurement of whole body glucose turnover, a bolus of 2-[1-14C]-deoxyglucose (370 µBq) was injected intravenously and blood samples were collected at 2, 5, 10, 15, 30 and 45 min. Immediately following the collection of the last blood sample at 45 min, the rats were killed by a lethal overdose of pentobarbitone sodium and tissues including heart, diaphragm, quadriceps (red and white), soleus, gastrocnemius (red and white), brown adipose and white adipose tissue were rapidly removed, frozen in liquid nitrogen and stored at −70°C for subsequent analysis. The time course of 2-[1-14C]-deoxyglucose disappearance from plasma and the degree of phosphorylated 2-deoxyglucose accumulation in individual tissues were determined [25]. Glucose uptake into individual tissues was calculated as previously described [26].
Analysis of Plasma Glucose, Insulin and NEFA
The concentration of glucose was measured by the glucose oxidase assay using an automated glucose analyser (YSI 2300 Stat Plus, Yellow Springs Instruments, Yellow Springs, Ohio, USA). Plasma insulin concentrations were measured using a rat insulin radioimmunoassay kit (Linco Research, St. Charles, Mo., USA). Fasting NEFA concentrations were measured in plasma samples that were collected in sequestrene tubes before the infusion of heparin using a colorimetric assay kit (Wako Pure Chemical Industries, Va., USA).
Kidney and liver PEPCK mRNA expression
Total RNA was extracted from kidney and liver using the guanidinium thiocyanate procedure as previously described [23]. RNA was then reverse transcribed using random priming with an AMV reverse transcriptase kit (Promega, Annandale, NSW, Australia). Relative expression of the PEPCK transcript were measured by quantitative Real-Time PCR using SYBR Green (Applied Biosystems, Scoresby, Victoria, Australia) to detect products formed from PEPCK specific primers (PEPCK forward primer 5′-AGTTGAATGTGTGGGTGATGACA-3′; reverse primer 5′-AAAACCGTTTTCTGGGTTGATG-3′), which was compared against the product amplified by β-actin specific primers (β-actin forward primer 5′-CGTGAAAAGATGACCCAGATCA-3′; reverse primer 5′-CACAGCCTGGATGGCTACGT-3′).
PEPCK enzyme activity
Kidney and liver was collected in groups of rats that were fasted overnight, but had not been infused with 14C labelled tracer. Tissue samples were homogenised and assayed for PEPCK enzyme activity using the oxaloacetate-H14CO3 exchange method [27].
Tissue glycogen measurement
Glycogen content was measured in red and white gastrocnemius muscle obtained under basal and hyperinsulinaemic clamp conditions. Muscle was freeze dried, dissected free of visible connective tissue and blood and powdered and glycogen concentrations were determined [28].
Muscle and fat GLUT4 protein content
GLUT4 protein content was measured in muscle and brown and white adipose tissue samples from PEPCK transgenic and control rats using a rabbit polyclonal antibody specific for GLUT4 (R1159) [29].
Tissue triglyceride and LCA-CoA measurement
Muscle samples were collected from PEPCK transgenic and control rats that were fasted overnight and administered a lethal overdose of pentobarbitone sodium. Previously described methods were used to measure the content of triglyceride and long-chain acyl-CoA [22, 30] in red and white gastrocnemius.
Statistical analysis
Results are expressed as mean ± SEM. A non-paired, Student's two-tailed t test was used to determine statistical significance between the means of two sample populations. A p value of less than 0.05 was considered statistically significant.
Results
Animal characteristics
PEPCK transgenic and control rats of both sexes were studied at approximately 14 weeks of age. At this age, the body weight of both male and female PEPCK transgenic rats was higher than control rats (Table 1). This increase in body weight was accompanied by higher weights of gonadal and infrarenal fat depots in PEPCK transgenic compared to control rats . As there was no sex difference in any glucose metabolism parameters measured, data from male and female rats were pooled.
Plasma glucose, insulin and NEFA concentrations were measured in PEPCK transgenic and control rats during the glucose turnover studies. Under basal conditions plasma glucose concentrations were comparable between PEPCK transgenic and control rats. Basal insulin concentrations tended to be higher in PEPCK transgenic rats but did not reach statistical significance (p=0.13). Despite increased adipose tissue depots, there was no difference in basal plasma NEFA concentrations between PEPCK transgenic and control rats (Table 2).
During the hyperinsulinaemic clamp, exogenous glucose was infused to prevent hypoglycaemia. This resulted in higher plasma glucose concentration in the PEPCK transgenic compared to control rats. To match plasma insulin concentrations during the hyperinsulinaemic clamp, PEPCK transgenic rats were infused with 2 mU·kg–1·min–1 and control rats were infused at a rate of 4 mU·kg–1·min–1 insulin. This resulted in comparable plasma insulin concentration between PEPCK transgenic and control rats. This enabled direct comparison of glucose metabolism parameters between the two groups of rats. A separate group of PEPCK transgenic rats were infused with 4 mU·kg–1·min–1 insulin (designated as higher insulin infusion rate in Table 2) and the plasma insulin level achieved was 1260±150 pmol/l (p<0.05 vs 2 mU·kg–1·min–1 PEPCK transgenic group).
Endogenous Glucose Production
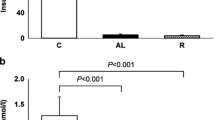
Under basal conditions the rate of EGP was comparable between PEPCK transgenic and control rats. During the hyperinsulinaemic clamp, EGP was completely suppressed in control rats, whereas in PEPCK transgenic rats EGP was not different from basal, despite comparable insulin concentrations. Therefore during the hyperinsulinaemic clamp, EGP was increased in PEPCK transgenic compared to control rats. Even at the higher insulin infusion rate EGP was decreased but not completely suppressed in PEPCK transgenic rats (Fig. 1A).

(A) Endogenous glucose production, (B) kidney PEPCK mRNA and (C) liver PEPCK mRNA under basal and hyperinsulinaemic clamp conditions in PEPCK transgenic (open and grey bars) and control rats (closed bars). Values are presented as means ± SEM. * p<0.05 vs control, # p<0.05 vs basal
PEPCK mRNA and enzyme activity
PEPCK mRNA was measured in liver and kidney samples obtained from control and PEPCK transgenic rats under basal conditions and following insulin infusion. Kidney PEPCK mRNA was higher in PEPCK transgenic compared to control rats under basal and during insulin infusion conditions (Fig. 1B). This was consistent with the enzyme assay which showed that kidney PEPCK activity was 16% higher in PEPCK transgenic compared to control rats (p<0.05, Fig. 2). Insulin infusion at the higher rate did suppress kidney PEPCK mRNA towards control values in PEPCK transgenic rats (Fig. 1B). In contrast, liver PEPCK mRNA was not different basally and suppressed to similar levels following insulin infusion in both PEPCK transgenic and control rats (Fig. 1C). This suggests that the transgene was not expressed in liver of PEPCK transgenic rats. Furthermore, liver PEPCK enzyme activity was the same in PEPCK transgenic and control rats (Fig. 2).

Liver and kidney PEPCK enzyme activity in PEPCK transgenic (open bars) and control rats (closed bars). Values are presented as means ± SEM. * p<0.05 vs control
Rate of glucose disappearance
During basal conditions there was no difference in the rate of glucose disappearance (Rd) between control and PEPCK transgenic rats (Fig. 3A). During the hyperinsulinaemic clamp, the Rd in PEPCK rats was lower than that in the control rats, indicative of whole body insulin resistance. At the higher plasma insulin concentration, Rd was not different between PEPCK transgenic and control rats.

(A) Rate of glucose disappearance and (B) metabolic clearance rate under basal and hyperinsulinaemic clamp conditions in PEPCK transgenic (open and grey bars) and control rats (closed bars). Values are presented as means ± SEM. * p<0.05 vs control
As stated earlier, the plasma glucose concentration in the hyperinsulinaemic clamp was higher in the PEPCK transgenic compared to control rats. To account for the mass action effect of glucose on the Rd, metabolic clearance rate (MCR) was calculated (Fig. 3B). Basal MCR was the same in PEPCK transgenic and control rats. However under hyperinsulinaemic clamp conditions MCR was lower in PEPCK transgenic compared to control rats. MCR was increased with the higher insulin infusion rate in PEPCK transgenic rats such that it was comparable to that achieved at a lower plasma insulin concentration in control rats.
Tissue glucose uptake
To determine the site(s) of insulin resistance in PEPCK transgenic rats, glucose uptake into individual tissues was measured using 2-[1-14C]-deoxyglucose (Table 3, Fig. 4). Compared to basal values, glucose uptake into all tissues was increased in PEPCK transgenic and control animals in response to the hyperinsulinaemic clamp (p<0.05). The only exception was white adipose tissue of PEPCK transgenic rats, which was completely unresponsive to the stimulatory effects of insulin.

Rate of glucose uptake in (A) brown adipose tissue and (B) white adipose tissue under basal and hyperinsulinaemic clamp conditions in PEPCK transgenic (open and grey bars) and control rats (closed bars). Values are presented as means ± SEM. * p<0.05 vs control
Under basal conditions the rate of glucose uptake into heart, diaphragm, red quadriceps, soleus, white and brown adipose tissue was lower in PEPCK transgenic than control rats, while uptake into the other tissues (white quadriceps, red and white gastrocnemius) was comparable between the two groups).
During the hyperinsulinaemic clamp, the rate of glucose uptake into white quadriceps and gastrocnemius, soleus and white and brown adipose tissue was lower in PEPCK transgenic compared to control rats while uptake into other tissues (heart, diaphragm, red quadriceps and gastrocnemius) was comparable between the two groups of rats. Insulin resistance persisted in white gastrocnemius, soleus and white adipose tissue even at the higher insulin infusion rate in PEPCK transgenic compared to control rats (Table 3, Fig. 4).
Muscle glycogen content
Glycogen content was measured in red gastrocnemius (which showed comparable glucose uptake between PEPCK transgenic and control rats) and white gastrocnemius (which showed reduced glucose uptake in PEPCK transgenic compared to control rats) under basal and hyperinsulinaemic clamp conditions (Table 4). Glycogen content of both red and white gastrocnemius muscle was the same in control and PEPCK transgenic rats under basal conditions. Insulin infusion caused an increase in muscle glycogen content in both PEPCK transgenic and control rats but there was no difference in insulin-stimulated glycogen accumulation between the two groups.
Muscle triglyceride and long-chain acyl-CoA content
To further investigate other possible causes of reduced insulin muscle sensitivity, triglyceride and long-chain acyl-CoA content was measured in red and white gastrocnemius from PEPCK transgenic and control rats under basal conditions (Table 5). The triglyceride content of both white and red gastrocnemius was lower in PEPCK transgenic compared to control rats. Long-chain acyl-CoA content of both gastrocnemius muscles was the same between transgenic and control rats.
Muscle and adipose tissue GLUT4 protein content
Total GLUT4 protein content was determined in red and white gastrocnemius and in white and brown adipose tissue from PEPCK transgenic and control animals (Fig. 5). There was no difference in total GLUT4 content in red or white gastrocnemius muscle between PEPCK transgenic and control animals. In contrast, in the more severely insulin-resistant adipose tissue from PEPCK transgenic rats, total GLUT4 was reduced by 59% in white fat (p<0.001) and by 30% in brown adipose tissue compared to control rats (p<0.001).

GLUT4 protein content in (A) white adipose tissue (WAT), (B) brown adipose tissue (BAT), (C) red gastrocnemius (RG) and (D) white gastrocnemius (WG) in PEPCK transgenic (open bars) and control rats (closed bars). Values are presented as means ± SEM. * p<0.001 vs control
Discussion
Insulin resistance is an early defect in the development of Type 2 diabetes. The increase in the incidence of insulin resistance and Type 2 diabetes in many societies is thought to be due to changes in diet and lifestyle [6]. A diet high in fat has been shown to reduce insulin-stimulated glucose uptake and to impair suppression of EGP [13, 14]. Furthermore, it has been suggested that the defect in the suppression of EGP by insulin precedes the defect in insulin action in peripheral tissues [13]. While a high-fat diet might cause peripheral insulin resistance by direct effects on muscle and adipose tissue, the effect of high fat to stimulate gluconeogenesis [15, 16, 17, 18] could have more consequences than simply impaired suppression of endogenous glucose output, as chronic impaired suppression of glucose production leads to obesity and peripheral insulin resistance.
PEPCK has been proposed to be an important regulatory enzyme of gluconeogenesis [32]. In support of this we have previously shown that over-expression of PEPCK in a rat hepatoma cell line led to increased glucose production [33]. Furthermore transgenic mice overexpressing liver PEPCK showed increased glucose production, hyperglycaemia, hyperinsulinaemia and glucose intolerance [34, 35]. The transgenic rats used in this study over-express PEPCK predominantly in the kidney under the control of a non-insulin responsive promoter [23] and resulted in impaired suppression of EGP during an hyperinsulinaemic clamp. Thus the same concentration of plasma insulin that resulted in complete suppression in control rats was unable to suppress EGP from basal values in the PEPCK transgenic rats. In fact, even at a higher insulin infusion rate, EGP was not completely suppressed in the transgenic rats, which is indicative of the importance of PEPCK to the regulation of gluconeogenesis. Like liver PEPCK over-expressing mice [35], renal PEPCK mRNA was more than twice as high in the transgenic compared to wild-type rats. However PEPCK enzyme activity measured in the basal state was increased only by 16%. It can be concluded therefore that even a modest increase of renal PEPCK activity can have major consequences if no suppression occurs when insulin concentrations are increased post-prandially. Enzyme activity was not measured in the liver over-expressing mice [35].
Interestingly moderate increase of insulin suppressed liver, but not kidney, PEPCK mRNA in both the PEPCK transgenic and control rats. As has been previously suggested [32], this implies that kidney PEPCK is not as insulin responsive as liver PEPCK and that increased renal glucose production can be a substantial contributor to glucose turnover [36, 37]. It has been shown that in patients undergoing liver transplantation endogenous glucose production falls about 50% after removal of the liver, representing renal glucose production [38]. Indeed it has been suggested that if gluconeogenesis accounts for approximately 50% of endogenous glucose production in the postabsorptive state [39], that the kidney accounts for approximately 40% of overall gluconeogenesis [40]. Early studies in animals showed an increase in the rate of glucose infusion required to maintain euglycaemia when the liver and kidneys were removed as compared to when only the liver was excised [41, 42]. Furthermore experimental diabetes in animals results in increased renal gluconeogenesis and glucose production in vitro [43, 44]. Recently increased renal glucose production has been described in patients with Type 2 diabetes which could contribute to the ensuing hyperglycaemic state [45].
Impaired suppression of EGP led to peripheral insulin resistance with Rd being reduced in our PEPCK transgenic compared to control rats. This is in contrast to transgenic liver PEPCK overexpressing mice which did not show a decrease in Rd compared to wild-type [35]. This might be due to species differences (rat vs mouse) or to a difference in the tissue in which PEPCK is over-expressed (liver vs kidney). To determine the site(s) responsible for insulin resistance we measured glucose uptake in individual tissues using radioactively labelled 2-deoxyglucose. We found that adipose tissue was the most resistant tissue. Skeletal muscle sensitivity was differentially affected in the transgenic rats. Thus insulin-stimulated glucose uptake was lower in white but not red quadriceps and gastrocnemius muscles. While the metabolic differences between red, which is predominantly oxidative, and white skeletal muscle, which is predominantly glycolytic, have been well characterised [46], the soleus, a pure red muscle did demonstrate insulin resistance. The difference in the responsiveness of different muscles remains unclear.
The decrease in white and brown adipose tissue glucose uptake was associated with reduced GLUT4 protein content in PEPCK transgenic compared to control rats. GLUT4 is predominantly present in muscle and adipose tissue and is responsible for glucose transport during insulin stimulation [47]. In contrast, muscle GLUT4 protein content was not different between the two groups of rats, despite white gastrocnemius muscle being insulin resistant in the PEPCK transgenic rats. This tissue-specific reduction of GLUT4 is similar to that reported in patients with Type 2 diabetes with a decrease in adipose tissue but normal skeletal muscle GLUT4 content [48, 49, 50]. Tissue-specific regulation of GLUT4 has also been described in animals fed a high fat diet, which resulted in decreased GLUT4 mRNA expression in white adipose tissue but not skeletal muscle [51, 52]. Further analysis has suggested that the defect in muscle glucose uptake in patients with Type 2 diabetes could be associated with impairments in trafficking and/or translocation of GLUT4 to the plasma membrane [53, 54, 55]. In agreement, high fat-induced skeletal muscle insulin resistance in animals has been associated with defects in the translocation and intrinsic activity of GLUT4. Therefore in our PEPCK transgenic rats insulin resistance in white and brown adipose tissue is associated with decreased GLUT4 protein content. However insulin resistance in skeletal muscle of the PEPCK transgenic rats was not associated with a reduction in the amount of GLUT4 but could be due to defects in trafficking and/or translocation of this glucose transporter.
Muscle triglyceride and LCACoA levels, which have been associated with insulin resistance [9, 22], were also not different between PEPCK transgenic and control rats. Furthermore, given that muscle insulin resistance was selective and relatively mild (compared to the adipose tissue), we suggest that it was the decrease in adipose tissue glucose uptake that was primarily responsible for the reduction in the rate of whole body glucose disposal and consequently insulin resistance in our PEPCK transgenic rats. In support of this, mice with a specific deletion in adipose tissue GLUT4 also developed glucose intolerance and whole body insulin resistance [56].
In conclusion, we have shown that a defect in the suppression of endogenous glucose production can secondarily lead to peripheral insulin resistance. Although there is no evidence that increased PEPCK activity is a mechanism for impaired suppression of endogenous glucose production in human diabetes, it has been shown that high fat availability stimulates gluconeogenesis [18, 57, 58]. Thus it is possible that fat can cause peripheral insulin resistance not only by direct effects on muscle and adipose tissue but secondarily by stimulation of gluconeogenesis.
Abbreviations
- EGP:
-
endogenous glucose production
- Rd :
-
rate of glucose disappearance
- LCACoA:
-
long chain acyl CoA
- TG:
-
triglycerides
- MCR:
-
metabolic clearance rate
References
DeFronzo RA, Bonadonna RC, Ferrannini E (1992) Pathogenesis of NIDDM. A balanced overview. Diabetes Care 15:318–368
Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176
Groop L (2000) Genetics of the metabolic syndrome. Br J Nutr 83 [Suppl 1]:S39–S48
Stern MP (2000) Strategies and prospects for finding insulin resistance genes. J Clin Invest 106:323–327
Proietto J, Filippis A, Nakhla C, Clark S (1999) Nutrient-induced insulin resistance. Mol Cell Endocrinol 151:143–149
Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414:782–787
Haffner SM (2000) Obesity and the metabolic syndrome: the San Antonio Heart Study. Br J Nutr 83 [Suppl 1]:S67–S70
McCarty MF (2000) Toward practical prevention of type 2 diabetes. Med Hypotheses 54:786–793
Kraegen EW, Cooney GJ, Ye JM, Thompson AL, Furler SM (2001) The role of lipids in the pathogenesis of muscle insulin resistance and beta cell failure in type II diabetes and obesity. Exp Clin Endocrinol Diabetes 109:S189–S201
Hotamisligil GS (2000) Molecular mechanisms of insulin resistance and the role of the adipocyte. Int J Obes Relat Metab Disord 24 [Suppl 4]:S23–S27
Weyer C, Funahashi T, Tanaka S et al. (2001) Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86:1930–1935
Hirosumi J, Tuncman G, Chang L et al. (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Kraegen EW, Clark PW, Jenkins AB, Daley EA, Chisholm DJ, Storlien LH (1991) Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes 40:1397–1403
Oakes ND, Cooney GJ, Camilleri S, Chisholm DJ, Kraegen EW (1997) Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes 46:1768–1774
Song S, Andrikopoulos S, Filippis C, Thorburn AW, Khan D, Proietto J (2001) Mechanism of fat-induced hepatic gluconeogenesis: effect of metformin. Am J Physiol Endocrinol Metab 281:E275–E282
Williamson JR, Kreisberg RA, Felts PW (1966) Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc Natl Acad Sci USA 56:247–254
Blumenthal SA (1983) Stimulation of gluconeogenesis by palmitic acid in rat hepatocytes: evidence that this effect can be dissociated from the provision of reducing equivalents. Metabolism 32:971–976
Roden M, Stingl H, Chandramouli V et al. (2000) Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 49:701–707
Andrikopoulos S, Proietto J (1995) The biochemical basis of increased hepatic glucose production in a mouse model of type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 38:1389–1396
Filippis A, Clark S, Proietto J (1997) Increased flux through the hexosamine biosynthesis pathway inhibits glucose transport acutely by activation of protein kinase C. Biochem J 324:981–985
Filippis C, Filippis A, Clark S, Proietto J (2002) Activation of PI 3-kinase by the hexosamine biosynthesis pathway. Mol Cell Endocrinol 194:29
Laybutt DR, Schmitz-Peiffer C, Saha AK, Ruderman NB, Biden TJ, Kraegen EW (1999) Muscle lipid accumulation and protein kinase C activation in the insulin-resistant chronically glucose-infused rat. Am J Physiol 277:E1070–E1076
Rosella G, Zajac JD, Baker L et al. (1995) Impaired glucose tolerance and increased weight gain in transgenic rats overexpressing a non-insulin-responsive phosphoenolpyruvate carboxykinase gene. Mol Endocrinol 9:1396–1404
Thorburn AW, Baldwin ME, Rosella G et al. (1999) Features of syndrome X develop in transgenic rats expressing a non-insulin responsive phosphoenolpyruvate carboxykinase gene. Diabetologia 42:419–426
Nolan CJ, Proietto J (1994) The feto-placental glucose steal phenomenon is a major cause of maternal metabolic adaptation during late pregnancy in the rat. Diabetologia 37:976–984
Kraegen EW, James DE, Jenkins AB, Chisholm DJ (1985) Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol 248:E353–E362
Andrikopoulos S, Rosella G, Gaskin E et al. (1993) Impaired regulation of hepatic fructose-1,6-bisphosphatase in the New Zealand obese mouse model of NIDDM. Diabetes 42:1731–1736
Passonneau JV, Lauderdale VR (1974) A comparison of three methods of glycogen measurement in tissues. Anal Biochem 60:405–412
Kelada AS, Macaulay SL, Proietto J (1992) Cyclic AMP acutely stimulates translocation of the major insulin-regulatable glucose transporter GLUT4. J Biol Chem 267:7021–7025
Antinozzi PA, Segall L, Prentki M, McGarry JD, Newgard CB (1998) Molecular or pharmacologic perturbation of the link between glucose and lipid metabolism is without effect on glucose-stimulated insulin secretion. A re-evaluation of the long-chain acyl-CoA hypothesis. J Biol Chem 273:16146–16154
Proietto J, Harewood M, Aitken P, Nankervis A, Caruso G, Alford F (1982) Validation of a practical in vivo insulin dose-response curve in man. Metabolism 31:354–361
Hanson RW, Reshef L (1997) Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem 66:581–611
Rosella G, Zajac JD, Kaczmarczyk SJ, Andrikopoulos S, Proietto J (1993) Impaired suppression of gluconeogenesis induced by overexpression of a noninsulin-responsive phosphoenolpyruvate carboxykinase gene. Mol Endocrinol 7:1456–1462
Valera A, Pujol A, Pelegrin M, Bosch F (1994) Transgenic mice overexpressing phosphoenolpyruvate carboxykinase develop non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci USA 91:9151–9154
Sun Y, Liu S, Ferguson S et al. (2002) Phosphoenolpyruvate carboxykinase overexpression selectively attenuates insulin signaling and hepatic insulin sensitivity in transgenic mice. J Biol Chem 277:23301–23307
Stumvoll M, Chintalapudi U, Perriello G, Welle S, Gutierrez O, Gerich J (1995) Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest 96:2528–2533
Stumvoll M, Meyer C, Mitrakou A, Nadkarni V, Gerich JE (1997) Renal glucose production and utilization: new aspects in humans. Diabetologia 40:749–757
Joseph SE, Heaton N, Potter D, Pernet A, Umpleby MA, Amiel SA (2000) Renal glucose production compensates for the liver during the anhepatic phase of liver transplantation. Diabetes 49:450–456
Chandramouli V, Ekberg K, Schumann WC, Kalhan SC, Wahren J, Landau BR (1997) Quantifying gluconeogenesis during fasting. Am J Physiol 273:E1209–E1215
Gerich JE, Meyer C, Woerle HJ, Stumvoll M (2001) Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care 24:382–391
Bergman H, Drury DR (1938) The relationship of kidney function to the glucose utilization fo the extra abdominal tissues. Am J Physiol 124:279–284
Reinecke R (1943) The kidney as a source of glucose in the eviscerated rat. Am J Physiol 140:276–285
Teng C (1954) Studies on carbohydrate metabolism in rat kidney slices. II. Effects of alloxan diabetes and insulin administration on glucose uptake and formation. Arch Biochem Biophys 48:415–423
Landau B (1960) Gluconeogenesis and pyruvate metabolism in rat kidney in vitro. Endocrinology 67:744–751
Meyer C, Stumvoll M, Nadkarni V, Dostou J, Mitrakou A, Gerich J (1998) Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest 102:619–624
Pette D, Staron RS (1990) Cellular and molecular diversities of mammalian skeletal muscle fibers. Rev Physiol Biochem Pharmacol 116:1–76
Olson AL, Pessin JE (1996) Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr 16:235–256
Pedersen O, Bak JF, Andersen PH et al. (1990) Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes 39:865–870
Garvey WT, Maianu L, Hancock JA, Golichowski AM, Baron A (1992) Gene expression of GLUT4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes 41:465–475
Garvey WT, Maianu L, Huecksteadt TP, Birnbaum MJ, Molina JM, Ciaraldi TP (1991) Pretranslational suppression of a glucose transporter protein causes insulin resistance in adipocytes from patients with non-insulin-dependent diabetes mellitus and obesity. J Clin Invest 87:1072–1081
Ikemoto S, Thompson KS, Itakura H, Lane MD, Ezaki O (1995) Expression of an insulin-responsive glucose transporter (GLUT4) minigene in transgenic mice: effect of exercise and role in glucose homeostasis. Proc Natl Acad Sci USA 92:865–869
Han DH, Hansen PA, Host HH, Holloszy JO (1997) Insulin resistance of muscle glucose transport in rats fed a high-fat diet: a reevaluation. Diabetes 46:1761–1767
Kelley DE, Mintun MA, Watkins SC et al. (1996) The effect of non-insulin-dependent diabetes mellitus and obesity on glucose transport and phosphorylation in skeletal muscle. J Clin Invest 97:2705–2713
Zierath JR, He L, Guma A, Odegoard Wahlstrom E, Klip A, Wallberg-Henriksson H (1996) Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia 39:1180–1189
Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD (1998) Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest 101:2377–2386
Abel ED, Peroni O, Kim JK et al. (2001) Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409:729–733
Chen X, Iqbal N, Boden G (1999) The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest 103:365–372
Boden G, Chen X, Capulong E, Mozzoli M (2001) Effects of free fatty acids on gluconeogenesis and autoregulation of glucose production in type 2 diabetes. Diabetes 50:810–816
Acknowledgements
We would like to thank Mr. P. Brazodurro for excellent technical assistance. This work was supported by a grant from the National Health and Medical Research Council of Australia (No 970393), and from a Grant-in-Aid from the Diabetes Australia Research Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lamont, B.J., Andrikopoulos, S., Funkat, A. et al. Peripheral insulin resistance develops in transgenic rats overexpressing phosphoenolpyruvate carboxykinase in the kidney. Diabetologia 46, 1338–1347 (2003). https://doi.org/10.1007/s00125-003-1180-y
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1180-y