
Abstract
Aims/hypothesis
The aim of this study was to examine whether the cytosolic NADPH/NADP+ ratio of beta cells serves as an amplifying signal in fuel-induced insulin secretion and whether such a function is mediated by cytosolic α-ketoglutarate.
Methods
Pancreatic islets and islet cells were isolated from albino mice by collagenase digestion. Insulin secretion of incubated or perifused islets was measured by ELISA. The NADPH and NADP+ content of incubated islets was determined by enzymatic cycling. The cytosolic Ca2+ concentration ([Ca2+]c) in islets was measured by microfluorimetry and the activity of ATP-sensitive K+ channels in islet cells by patch-clamping.
Results
Both 30 mmol/l glucose and 10 mmol/l α-ketoisocaproate stimulated insulin secretion and elevated the NADPH/NADP+ ratio of islets preincubated in the absence of fuel. The increase in the NADPH/NADP+ ratio was abolished in the presence of 2.7 μmol/l glipizide (closing all ATP-sensitive K+ channels). However, α-ketoisocaproate, but not glucose, still stimulated insulin secretion. That glipizide did not inhibit α-ketoisocaproate-induced insulin secretion was not the result of elevated [Ca2+]c, as glucose caused a more marked [Ca2+]c increase. Insulin release triggered by glipizide alone was moderately amplified by dimethyl α-ketoglutarate (which is cleaved to produce cytosolic α-ketoglutarate), but there was no indication of a signal function of cytosolic α-ketoglutarate.
Conclusions/interpretation
The results strongly suggest that the NADPH/NADP+ ratio in the beta cell cytosol does not serve as an amplifying signal in fuel-induced insulin release. The study supports the view that amplification results from the intramitochondrial production of citrate by citrate synthase and from the associated export of citrate into the cytosol.
Similar content being viewed by others
Introduction
Stimulation of insulin secretion by glucose and by some other fuels results from the combination of triggering and amplifying signals produced in the fuel metabolism of pancreatic beta cells [1]. There is strong evidence that an increase in cytosolic ATP and the associated decrease in cytosolic ADP trigger insulin release by closing the ATP-sensitive K+ channels (KATP channels) in the beta cell plasma membrane [1–3]. The ensuing membrane depolarisation opens voltage-dependent Ca2+ channels, thereby raising the cytosolic Ca2+ concentration ([Ca2+]c) and initiating the exocytosis of insulin [2]. However, about two thirds of glucose-induced insulin secretion are probably due to the amplifying pathway [4]. Two experimental approaches have been employed to demonstrate amplification: all KATP channels of beta cells are closed by sulfonylurea (e.g. glipizide) [5] or all KATP channels are held open by diazoxide while the membrane is depolarised by high K+ [6]. The insulin secretion stimulated under these conditions is still enhanced by glucose and other insulin-releasing fuels [1]. Despite considerable efforts, it has remained unclear so far which signals mediate the metabolic amplification. Current signal candidates are an increase in the cytosolic NADPH/NADP+ ratio [7, 8], an increase in cytosolic α-ketoglutarate [9], the export of citrate cycle intermediates to the cytosol [10–13], the accumulation of cytosolic acyl-CoAs [10, 11, 13–16] and an increase in mitochondrial succinyl-CoA [11, 17]. Metabolic amplification does not appear to result from an increase in the cytosolic ATP/ADP ratio [12].
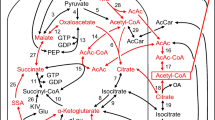
In the beta cell cytosol, two sources yield NADPH [7, 8, 11, 13, 18, 19]. Cytosolic malic enzyme transforms malate + NADP+ into pyruvate + CO2 + NADPH and cytosolic isocitrate dehydrogenase transforms isocitrate + NADP+ into α-ketoglutarate + CO2 + NADPH (Fig. 1). In the cytosol of mouse beta cells, isocitrate is the only important source of NADPH, since the activity of cytosolic malic enzyme is very low [20]. The substrates malate and isocitrate are exported from the mitochondria to the cytosol (Fig. 1) when the activated fuel metabolism provides citrate cycle intermediates [11]. The NADPH/NADP+ ratio in insulin-secreting cells exposed to increasing glucose concentrations rises in parallel with insulin release [7, 8]. However, these findings do not prove a causal link between the changes in NADPH/NADP+ ratio and insulin release. Intracellular application of NADPH via a micropipette was shown to stimulate the exocytosis of insulin granules in whole-cell capacitance measurements [7]. But these findings might not reflect the situation in intact beta cells.

Interaction of the citrate cycle with the metabolism of glucose, α-ketoisocaproate, monomethyl succinate and dimethyl α-ketoglutarate in pancreatic beta cells. The plasma membrane and the inner mitochondrial membrane are shown. For clarity, not all compounds participating in the enzyme reactions and transport processes are indicated. The shuttle systems translocating the reducing equivalents of glycolytically formed NADH into the mitochondrion are also not shown. The broken lines indicate that the activities of cytosolic and mitochondrial malic enzyme are very low in mouse beta cells. α-Ketoglutarate is transformed into succinate by sequential reactions catalysed by α-ketoglutarate dehydrogenase and succinyl-CoA synthase, respectively, and perhaps also by GABA transaminase followed by succinate semialdehyde dehydrogenase [50]. α-Ketoisocaproate is transaminated both in the cytosol and the mitochondrion, generating leucine and α-ketoglutarate. Intramitochondrially generated leucine allosterically activates the glutamate dehydrogenase. For further explanations and references, see the text. GABA, γ-aminobutyric acid; Glu, glutamate; KIC, α-ketoisocaproate; Leu, leucine; SSA, succinate semialdehyde. The numbers indicate the following enzymes: 1, cytosolic malic enzyme; 2, mitochondrial malic enzyme; 3, pyruvate carboxylase; 4, pyruvate dehydrogenase; 5, citrate synthase; 6, mitochondrial aconitase; 7, NAD+-dependent isocitrate dehydrogenase; 8, NADP+-dependent isocitrate dehydrogenase; 9, α-ketoglutarate dehydrogenase; 10, succinyl-CoA synthase; 11, malate dehydrogenase; 12, glutamate dehydrogenase; 13, intramitochondrial aminotransferase; 14, GABA aminotransferase; 15, succinate semialdehyde dehydrogenase; 16, cytosolic aminotransferase; 17, esterase; 18, cytosolic isocitrate dehydrogenase; 19, cytosolic aconitase; 20, ATP-dependent citrate lyase
The signal function of citrate cycle intermediates is difficult to verify by extracellular application because these compounds do not cross the beta cell plasma membrane. The membrane-permeable ester dimethyl α-ketoglutarate is converted into α-ketoglutarate in the beta cell (Fig. 1) and elicits insulin release [9, 21]. But it is unclear how α-ketoglutarate is involved in dimethyl α-ketoglutarate-induced insulin secretion and whether its mechanism is compatible with a role as an amplifying signal.
The specific aims of the present study were to examine whether the cytosolic NADPH/NADP+ ratio in beta cells functions as an amplifying signal in fuel-induced insulin secretion and to investigate the role of cytosolic α-ketoglutarate in insulin secretion stimulated by dimethyl α-ketoglutarate.
Methods
Materials and media
Sigma/Fluka (Taufkirchen, Germany) provided α-ketoisocaproate (4-methyl-2-oxopentanoate, sodium salt), α-ketoglutarate (disodium salt dihydrate), dimethyl α-ketoglutarate (α-ketoglutaric acid dimethyl ester), l-glutamine, glucose 6-phosphate (disodium salt hydrate), 6-phosphogluconate (trisodium salt), 6-phosphogluconate dehydrogenase (from yeast) and imidazole. NADP+ (disodium salt), NADPH (tetrasodium salt), glucose-6-phosphate dehydrogenase (from yeast) and glutamate dehydrogenase (from beef liver) were from Roche (Mannheim, Germany). EDTA and l-cysteine were from Merck (Darmstadt, Germany). Fura-PE3/AM (acetoxymethyl ester), cell culture medium RPMI 1640 and fetal calf serum were supplied by Molecular Probes (Leiden, the Netherlands), Gibco BRL (Gaithersburg, MD, USA) and Biochrom (Berlin, Germany), respectively. All other chemicals were obtained from sources described elsewhere [12, 22, 23]. The media for isolation, perifusion and incubation of pancreatic islets consisted of basal medium (HEPES-buffered Krebs–Ringer bicarbonate medium) containing 2 mg/ml BSA [22]. Other additions to the media are detailed below.
Isolation and culture of pancreatic islets and islet cells
Albino mice (NMRI) were purchased and bred as described previously [12]. The study was conducted in accordance with the Principles of Laboratory Care. Pancreatic islets from mice of both sexes (9–13 weeks old, fed an unrestricted diet) were isolated by a collagenase digestion technique (in the presence of 5 mmol/l glucose) and hand-picked under a stereomicroscope. Islets were dissociated into single cells by incubation for 10 min in a Ca2+-free medium (135 mmol/l NaCl, 4.8 mmol/l KCl, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO4, 25 mmol/l HEPES, 0.5 mmol/l EGTA, 3 mmol/l glucose, 1% BSA, pH 7.4), subsequent vortex-mixing for 1 min, centrifugation for 1 min at 200 g and suspension in culture medium. Isolated islets were cultured in the presence of 5 mmol/l glucose (for microfluorimetric measurements) and islet cells in the presence of 10 mmol/l glucose (for patch-clamp experiments) as described previously [23, 24].
Measurement of insulin secretion
Insulin secretion from perifused freshly isolated islets was measured as described previously [12]. For static incubations, 50 freshly isolated islets were preincubated in 1 ml of control medium (either in the absence of secretagogue or in the presence of 2.7 μmol/l glipizide) for 60 min at 37°C. Batches of 15 preincubated islets were incubated in 200 μl of control medium or medium plus test compound and the insulin concentration in the media was determined after 40 min at 37°C.
NADPH and NADP+ measurement
NADPH and NADP+ contents were measured by enzymatic cycling as described by Passonneau and Lowry [25] with minor modifications. Batches of 15 freshly isolated islets were preincubated in 200 μl of control medium (either in the absence of secretagogue or in the presence of 2.7 μmol/l glipizide) for 60 min at 37°C (control period). Then 180 μl of the medium was replaced with control medium or with medium plus test compound and the islets were incubated for 40 min at 37°C. Incubations were stopped by removing 180 μl of medium and placing the incubation tube (0.5 ml polypropylene tube; Sarstedt, Nümbrecht, Germany) in liquid N2. After thawing the tube in an ice bath, 40 μl of ice-chilled NaOH (60 mmol/l + 7.5 mmol/l cysteine) was added. The stoppered tube was immediately sonicated as described previously [26] and centrifuged for 1 min at 20,000 g (4°C). Aliquots (15 μl) of the supernatant were added to 7.5 μl of NaOH (40 mmol/l + 5 mmol/l cysteine; for NADPH determination) and to 7.5 μl of HCl (200 mmol/l; for NADP+ determination). The samples were heated for 10 min at 60°C (to destroy NADP+ and NADPH, respectively) and kept at −80°C (usually for 1–3 days). Addition of 100 μl cycling reagent (100 mmol/l Tris, 80 mmol/l HCl, 7.5 mmol/l α-ketoglutarate, 2 mmol/l glucose 6-phosphate, 0.1 mmol/l ADP, 25 mmol/l ammonium acetate, 0.1% BSA, 6 U/ml glucose-6-phosphate dehydrogenase, 10 U/ml glutamate dehydrogenase, pH 7.4) to each sample started the cycling reaction, which lasted for 1 h at 37°C. The reaction was stopped by heating for 3 min at 97°C and centrifugation for 10 min at 20,000 g (4°C). For the fluorimetrical indicator reaction, 90 μl supernatant was mixed with 900 μl indicator reagent [25] without 6-phosphogluconate dehydrogenase. An aliquot (900 μl) of the mixture was transferred to a quartz cuvette in a filter fluorimeter (Eppendorf 1101 M with attachment 1030; Eppendorf, Hamburg, Germany), the fluorescence (excitation at 366 nm, emission at 400–3,000 nm) was followed with a chart recorder and the indicator reaction was started by addition of 6-phosphogluconate dehydrogenase. Recording was continued until the reaction was finished. Appropriate blanks, NADPH standards and NADP+ standards were run in parallel through the entire procedure, including the extraction steps. For each single incubation, the NADPH/NADP+ ratio was calculated from the measured NADPH and NADP+ content.
Microfluorimetric measurement of the cytosolic Ca2+ concentration
Pancreatic islets were cultured on collagen-coated glass cover slips in Petri dishes and were used from day 2 to day 4 after isolation. Fura-PE3/AM was used as Ca2+ indicator to minimise dye leakage during the experiments. The loading concentration was 2 μmol/l in basal medium containing 2 mg/ml BSA and 5 mmol/l glucose (45 min at 37°C). As described previously [24], the islets were then perifused at 0.2 ml/min and at 35°C using basal medium (additions as detailed under Results, no BSA) and the epifluorescence was recorded (six islet subregions evaluated per experiment).
Patch-clamp experiments
Using single islet cells cultured for 24–72 h, KATP channel currents in inside-out patches were measured at room temperature (20–22°C) with an experimental design (including calculations) exactly as described previously [23]. No effort was made to determine the proportion of beta cells in our islet cell preparation (intact mouse islets contain >80% beta cells [27]), because the properties of alpha cell KATP channels are closely similar to those of beta cell KATP channels [28].
Statistical analysis
Results are presented as means ± SEM. Differences between groups were analysed using the two-tailed t test for paired observations. Statistical significance was assumed at p < 0.05.
Results
A recent observation led us to conceive the present experiments: in mouse islets exposed to 2.7 μmol/l glipizide (blocking all KATP channels, [12]), 10 mmol/l α-ketoisocaproate (a fuel secretagogue) amplified insulin secretion, but 30 mmol/l glucose did not, even though both 10 mmol/l α-ketoisocaproate and 30 mmol/l glucose produced similar changes in the islet contents of ATP and ADP [12]. For methodical reasons, the ATP and ADP measurements and the intended NADPH and NADP+ measurements had to be performed with statically incubated islets, whereas insulin secretion was measured in perifused islets. Thus, we had to ascertain that the above observation on secretagogues is also valid for the conditions of our measurements of metabolic intermediates in islets. After preincubating mouse islets for 60 min in the absence of any fuel or secretagogue, 40 min incubations in the presence of 30 mmol/l glucose or 10 mmol/l α-ketoisocaproate stimulated insulin secretion by 3.8- or 4.7-fold, respectively (Fig. 2). A small proportion of the secretory response to α-ketoisocaproate was probably due to direct KATP channel inhibition [23]. After preincubating mouse islets for 60 min in the absence of fuel but in the presence of 2.7 μmol/l glipizide, 40 min incubations in the presence of 2.7 μmol/l glipizide plus 30 mmol/l glucose did not stimulate insulin secretion (Fig. 2), whereas 40 min incubations in the presence of 2.7 μmol/l glipizide plus 10 mmol/l α-ketoisocaproate stimulated insulin secretion by 9.4-fold (Fig. 2). So the secretory responses of the incubated islets clearly reflect those of the perifused islets (Fig. 2) [12].

Effects of glucose and α-ketoisocaproate on insulin secretion by incubated mouse pancreatic islets. The experimental series consisted of three (i) or two (ii , iii) simultaneous incubations (40 min) in the absence (i) or presence (ii, iii) of 2.7 μmol/l glipizide. The incubation media contained no fuel secretagogue (white bars), 30 mmol/l glucose (grey bars) or 10 mmol/l α-ketoisocaproate (black bars). During the preincubations (60 min in the absence of fuel secretagogue), the media in ii and iii contained 2.7 μmol/l glipizide. The insulin concentration in the media at the end of the incubation period is indicated. Data are the means ± SEM of results from 14 (i), 10 (ii) or 9 (iii) separate experiments. *p < 0.001 vs corresponding control
To assess the signal function of the islet NADPH/NADP+ ratio in fuel-induced amplification of insulin secretion, the contents of NADPH and NADP+ were measured in mouse islets. After preincubating islets for 60 min in the absence of any fuel or secretagogue, 40-min incubations in the presence of 30 mmol/l glucose or 10 mmol/l α-ketoisocaproate significantly increased the NADPH/NADP+ ratio in the islets (Table 1). This increase was exclusively due to an increase of the NADPH content, the content of NADP+ remained unchanged. Unexpectedly, the increase in the NADPH content and the NADPH/NADP+ ratio by 30 mmol/l glucose or 10 mmol/l α-ketoisocaproate was abolished when the preincubation and incubation media contained 2.7 μmol/l glipizide (Table 1). Again, there was no fuel-induced change in the NADP+ content and consequently no change in the NADPH/NADP+ ratio. Interestingly, the NADPH content after control incubation, i.e. the continued absence of fuels, was the same in the absence and presence of glipizide. Thus glipizide specifically abolished the fuel-induced increase in NADPH.
To elucidate why mouse islets exposed to 2.7 μmol/l glipizide lost glucose-induced, but not α-ketoisocaproate-induced amplification of insulin secretion [12], [Ca2+]c was measured as stimulus–secretion coupling signal. Perifusion of mouse islets for 60 min in the absence of nutrients caused a slow increase in [Ca2+]c which reached a steady state prior to the addition of glucose or α-ketoisocaproate (Fig. 3a). Within 1 min, 30 mmol/l glucose caused a marked transient decrease of [Ca2+]c. After 6 min, the [Ca2+]c level prior to glucose addition was re-established, then after 15 min a clear further increase was noted. The addition of 10 mmol/l α-ketoisocaproate caused a similarly fast decrease of [Ca2+]c and again there was an increase after a 6 min time interval. However, the [Ca2+]c levels remained clearly below the values prior to nutrient addition for the remaining 30 min of perifusion time (Fig. 3a). The addition of 2.7 μmol/l glipizide in the absence of any nutrient caused a strong increase of [Ca2+]c after a time lag of 2 min (Fig. 3b). The elevated [Ca2+]c level remained constant until to the addition of the nutrients. Both 30 mmol/l glucose and 10 mmol/l α-ketoisocaproate caused an immediate decrease of [Ca2+]c, the kinetics of which showed a biphasic pattern. Again, the ensuing return to elevated [Ca2+]c levels was clearly more marked in the presence of glucose than of α-ketoisocaproate (Fig. 3b). As in the previous set of experiments the fura ratio values at the end of the nutrient perifusion were significantly different for glucose and α-ketoisocaproate, whereas no significant difference existed prior to the nutrient exposure.

Effects of glucose and α-ketoisocaproate on [Ca2+]c of perifused mouse islets. a Fura-loaded islets were perifused with medium containing no glucose for 60 min. Thereafter, either 30 mmol/l glucose (black line) or 10 mmol/l α-ketoisocaproate (ared line) was added to the perifusion, which continued for another 40 min. The data are the means±SEM of five (glucose) or six (α-ketoisocaproate) experiments. b Fura-loaded islets were perifused with medium containing no glucose for 60 min. From t = 10 to t = 100, 2.7 μmol/l glipizide was present. From t = 60 onwards, either 30 mmol/l glucose (black line) or 10 mmol/l α-ketoisocaproate (ared line) was added to the perifusion, which continued for another 40 min. The data are the means±SEM of five (glucose) or six (α-ketoisocaproate) experiments
Dimethyl α-ketoglutarate, which is split into α-ketoglutarate by cytosolic esterases (Fig. 1), stimulated insulin release from pancreatic islets in the absence of other secretagogues [9, 21]. Under our conditions, a moderate amplifying component of the secretory response was observed (Fig. 4). After perifusing islets for 60 min in the presence of 2.7 μmol/l glipizide (control period), perifusion of islets with a medium containing 2.7 μmol/l glipizide plus dimethyl α-ketoglutarate (15 mmol/l) elicited a monophasic increase in insulin release peaking at time (t) 62.5 min (Fig. 4). As compared with the secretory rate at t = 58, the secretory rates were higher from t = 62.5 to t = 74 for every experiment. This amplifying effect was weaker than that of 15 mmol/l 2-aminobicyclo[2,2,1]heptane-2-carboxylic acid (BCH) in conjunction with 2 mmol/l glutamine. The sole application of 2 mmol/l glutamine did not enhance glipizide-induced insulin secretion (Fig. 4), a finding similar to that made previously with glibenclamide (known as glyburide in the USA and Canada) [29].

Effects of dimethyl α-ketoglutarate, glutamine and BCH on the kinetics of insulin secretion by mouse pancreatic islets. Islets were perifused from t = 0 to t = 60 with medium containing 2.7 μmol/l glipizide (control period) and from t = 61 to t = 104 (test period) with medium containing 2.7 μmol/l glipizide plus 15 mmol/l dimethyl α-ketoglutarate (white circles) or 2.7 μmol/l glipizide plus 2 mmol/l glutamine (black squares) or 2.7 μmol/l glipizide plus 2 mmol/l glutamine plus 15 mmol/l BCH (black circles). The rate of insulin secretion is expressed as a percentage of the secretion rate at the end of the control period. The data points are the means of six (dimethyl α-ketoglutarate) or three (glutamine, glutamine + BCH) separate experiments (with SEM shown when larger than symbols) and are plotted in the middle of the sampling intervals
As stimulation of insulin secretion requires closure of KATP channels in the beta cell plasma membrane, it is conceivable that α-ketoglutarate derived from dimethyl α-ketoglutarate directly inhibits the KATP channels. To test this possibility, KATP channel currents in inside-out patches of islet cell membranes were recorded using the experimental design described previously [23]. The continuous current trace in Fig. 5 revealed pronounced rundown of KATP channel activity. Rundown was considered by using the mean current during control periods before and after application of α-ketoglutarate as reference (100%). The KATP channel current in the presence of 5 mmol/l α-ketoglutarate plus 1 mmol/l ADP was 100.2 ± 4.1% (n = 6). Thus, α-ketoglutarate did not inhibit the KATP channels of islet cells.

Effect of α-ketoglutarate on KATP channel current in an inside-out patch of a mouse islet cell. Free Mg2+ (0.7 mmol/l) was always present in the solutions applied at the cytoplasmic membrane side. Segments of channel activities from the continuous trace are shown below the trace on an expanded time scale. Scale bar (bottom right): horizontal corresponds to 1 min for the continuous trace and to 1 s for the expanded segments; vertical corresponds to 20 pA for the continuous trace and to 10 pA for the expanded segments. Application of intracellular solution containing 1 mmol/l ATP, 1 mmol/l ADP or 1 mmol/l ADP plus 5 mmol/l α-ketoglutarate (KG) was by the bath and at the times/durations indicated (bold horizontal bars). The sodium concentrations in the solutions containing only ATP or ADP were made equal to the sodium concentration in the solution containing α-ketoglutarate by adding NaCl. In this experiment, α-ketoglutarate (5 mmol/l) inhibited the KATP channel current by only 2.4% of the mean KATP channel current during the control periods before and after the test period with α-ketoglutarate
Discussion
The findings in this study argue against a role of cytosolic NADPH as an amplifying signal in fuel-induced insulin secretion. The data leading to this conclusion were obtained by use of a high (2.7 μmol/l) glipizide concentration as a tool blocking all beta cell KATP channels. It has previously been shown in mouse islets perifused with 2.7 μmol/l glipizide (no fuel during the pre-stimulatory period) that 10 mmol/l α-ketoisocaproate amplified insulin secretion, whereas 30 mmol/l glucose did not [12]. A similar difference was also observed, when insulin secretion was determined in islets incubated under conditions corresponding to those during our measurements of metabolic intermediates (Fig. 2). The present results support the view that this difference was not due to a lower [Ca2+]c in the presence of 30 mmol/l glucose (Fig. 3).
In the absence of fuels, whole-islet and mitochondrial NADPH/NADP+ ratios were similar in islets and isolated islet cells, respectively [30, 31]. Both ratios rose in parallel in the presence of glucose. Glucose also raised the cytosolic NADPH/NADP+ ratio [32, 33]. These findings allow conclusions to be drawn from the whole-islet NADPH/NADP+ ratio about the mitochondrial and the cytosolic NADPH/NADP+ ratios. The prevention by glipizide of glucose- and α-ketoisocaproate-induced elevation of the islet NADPH/NADP+ ratio (Table 1) argues against a signal function of the cytosolic NADPH/NADP+ ratio in fuel-induced amplification of insulin secretion. This is concluded because glipizide failed to suppress α-ketoisocaproate-induced insulin release (Fig. 2) [12]. It is very unlikely that the resistance to glipizide resulted from an as yet undetected direct amplifying effect of α-ketoisocaproate as such, since inhibition of α-keto acid transamination abolished amplification by α-ketoisocaproate [23]. Moreover, amplification of glipizide-induced insulin secretion was also seen with BCH, which is not structurally related to α-ketoisocaproate [12]. The prevention by glipizide of an increase in the islet NADPH/NADP+ ratio also argues against formation of isocitrate by reversal of the mitochondrial NADP+-dependent isocitrate dehydrogenase reaction (Fig. 1) [12], since reversal of this reaction requires NADPH levels to be elevated by the mitochondrial electrochemical proton gradient [34].
The glucose- and α-ketoisocaproate-induced increase in the islet NADPH/NADP+ ratio resulted from an elevation of the NADPH content, whereas the NADP+ content remained unchanged (Table 1). These observations are assumed to reflect a dual action of the fuels. First, the fuels stimulate NAD regeneration from precursors and conversion of some of the NAD into NADP [35]. These reactions require ATP. Second, a more reduced state of the NADPH–NADP+ couple is brought about. The expected decrease of the NADP+ content seems to be hidden by the synthesis of NADP+.
In isolated pancreatic islets and insulin-secreting cells, glucose and α-ketoisocaproate were consistently found to raise the NADPH/NADP+ ratio (Table 1) [7, 8, 13, 19, 30, 36, 37]. How could the presence of 2.7 μmol/l glipizide abolish this effect? The reason is probably the depolarisation of the inner mitochondrial membrane potential by the high glipizide concentration. The KATP channels in the beta cell plasma membrane are half-maximally inhibited by 7 μmol/l tolbutamide or 4 nmol/l glipizide (the therapeutic free plasma concentrations are 6–25 μmol/l and 4–40 nmol/l, respectively; 6 or 2% of the total plasma concentration of tolbutamide or glipizide, respectively, are free) [22]. At much higher concentrations, sulfonylureas uncouple oxidative phosphorylation [38–40]. In mouse beta cells perifused in the absence of exogenous fuels, 500 μmol/l tolbutamide generated a small depolarisation of the inner mitochondrial membrane potential [41]. The depolarisation amounted to only 10% of the depolarisation induced by the mitochondrial protonophoretic uncoupler carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP, 1 μmol/l) and seemed to be associated with a decrease in the cytosolic ATP concentration. The depolarisation was believed to result either from direct inhibition of the mitochondrial KATP channels or from uncoupling of oxidative phosphorylation. In mouse islets incubated in the absence of exogenous fuels, tolbutamide (about 400 μmol/l free concentration) slightly stimulated oxygen consumption [26, 42]. Decrease in ATP content induced by high sulfonylurea concentrations was observed in mouse and rat islets incubated in the absence of exogenous fuels or in the presence of low glucose concentrations [43–46]. Mild mitochondrial depolarisation is the likely reason why glipizide caused glucose and α-ketoisocaproate to induce only a small increase in the islet ATP/ADP ratio [12]. The contrasting effects of glipizide on the secretory responses to glucose on the one hand and to α-ketoisocaproate on the other hand (Fig. 2) [12] probably indicate that reactions involved in the generation of the amplification signal by glucose need a higher ATP/ADP ratio than reactions following formation of the signal [47]. Another likely consequence of a low ATP/ADP ratio is an inhibition of ATP-dependent regeneration of NADP (see above). In conjunction with this effect, mitochondrial depolarisation and the associated increase in consumption of reducing equivalents (NADH and NADPH) in the respiratory chain can explain why glipizide prevented the fuel-induced rise in the islet content of NADPH.
Evidence has been presented that α-ketoisocaproate, some related α-keto acid anions and the nonmetabolisable leucine analogue BCH amplify insulin secretion by enhancing the intramitochondrial formation of α-ketoglutarate and thereby stimulating the citrate cycle in beta cells [12, 23, 48]. The present study supports the view that formation of citrate from α-ketoglutarate via reversal of the NADP+-dependent isocitrate dehydrogenase reaction does not take place in the presence of 2.7 μmol/l glipizide (see above). Therefore, citrate production induced by α-ketoglutarate represents citrate synthesis from oxaloacetate and acetyl-CoA by the citrate synthase (Fig. 1). Acetyl-CoA comes from degradation of the fuel secretagogue (e.g. α-ketoisocaproate, Fig. 1) or from endogenous sources (e.g. endogenous fatty acids in the case of BCH) [49]. It has been proposed that succinyl-CoA generated from succinate or α-ketoglutarate (Fig. 1) is important for insulin secretion due to its reaction with mitochondrial acetoacetate to form succinate and acetoacetyl-CoA, a precursor of acetyl-CoA (reaction not shown in Fig. 1) [17]. However, it is unclear whether this reaction is significant for BCH-induced amplification of insulin secretion, since low acetoacetate levels are expected in the absence of exogenous fuels or presence of glutamine (Fig. 4) [12].
Not only NADPH, but also α-ketoglutarate is provided by isocitrate exported to the cytosol (Fig. 1). Direct stimulation of insulin secretion by α-ketoglutarate has been proposed to explain the insulin-releasing effect of dimethyl α-ketoglutarate [9]. This membrane-permeable ester is split via monomethyl α-ketoglutarate (two isomers) into α-ketoglutarate (Fig. 1) [21]. But as initiation of insulin release requires closure of KATP channels in the beta cell plasma membrane, the lack of direct KATP channel inhibition by α-ketoglutarate (Fig. 5) suggests channel inhibition by activation of mitochondrial energy metabolism and the associated changes in the cytosolic ATP and ADP concentrations. The amplification of insulin secretion by dimethyl α-ketoglutarate cannot result from mitochondrial uptake of α-ketoglutarate, which occurs in exchange for malate and causes no net increase in citrate cycle intermediates of mouse beta cells, as suggested by findings for monomethyl succinate (Fig. 1) [12, 19, 20]. It is unlikely that glutamate formed by transamination of cytosolic α-ketoglutarate mediates the amplifying effect of dimethyl α-ketoglutarate. The islet content of glutamate, but not the amplification of insulin secretion, is much more enhanced by 2 mmol/l glutamine than by 10–20 mmol/l dimethyl α-ketoglutarate (Fig. 4) [21, 29]. Whatever is the precise mechanism of insulin release by dimethyl α-ketoglutarate, our present observations do not support a signal function of cytosolic α-ketoglutarate.
α-Ketoglutarate-induced citrate production causes a net increase in citrate cycle intermediates and enhanced export of these intermediates from the beta cell mitochondria into the cytosol [11]. In the cytosol, citrate, but not the other intermediates, apparently mediates amplification of fuel-induced insulin secretion. First, the cytosolic NADPH/NADP+ ratio and the cytosolic α-ketoglutarate level probably do not serve as amplification signals (this study). Therefore mitochondrial export of isocitrate and α-ketoglutarate does not appear to amplify insulin release. Second, the very weak amplification of insulin secretion by monomethyl succinate in mouse islets [12] argues against a signal function of cytosolic succinate, fumarate and malate (Fig. 1). Fumarate is formed from succinate and malate and is exported to the cytosol in exchange for succinate (not shown in Fig. 1). Third, succinyl-CoA and oxaloacetate cannot cross the inner mitochondrial membrane. Cytosolic citrate is converted into acetyl-CoA and oxaloacetate by the ATP-dependent citrate lyase (Fig. 1). It has been proposed that compounds enhancing insulin secretion exist among acyl-CoAs and lipids, which are synthesised from acetyl-CoA, using NADPH [10, 11, 14–16]. The lack of a signal function of the cytosolic NADPH/NADP+ ratio does not mean that the supply of NADPH is insufficient for these biosynthetic reactions.
In conclusion, the present study strongly suggests that the NADPH/NADP+ ratio in the beta cell cytosol does not serve as an amplifying signal in fuel-induced insulin release. The findings support the view that amplification results from intramitochondrial production of citrate by the citrate synthase. Citrate exported to the cytosol probably mediates an increase in insulin secretion by promoting the generation of acyl-CoAs and lipids, some of which are putative amplifying signals.
Abbreviations
- BCH:
-
2-aminobicyclo[2,2,1]heptane-2-carboxylic acid
- [Ca2+]c :
-
cytosolic Ca2+ concentration
- KATP channel:
-
ATP-sensitive K+ channel
- t :
-
time (min)
References
Henquin J-C (2000) Perspectives in diabetes. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760
Ashcroft FM, Rorsman P (1989) Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 54:87–143
Tarasov AI, Girard CA, Ashcroft FM (2006) ATP sensitivity of the ATP-sensitive K+ channel in intact and permeabilized pancreatic beta-cells. Diabetes 55:2446–2454
Henquin JC, Ravier MA, Nenquin M, Jonas JC, Gilon P (2003) Hierarchy of the β-cell signals controlling insulin secretion. Eur J Clin Invest 33:742–750
Panten U, Schwanstecher M, Wallasch A, Lenzen S (1988) Glucose both inhibits and stimulates insulin secretion from isolated pancreatic islets exposed to maximally effective concentrations of sulfonylureas. Naunyn-Schmiedeberg’s Arch Pharmacol 338:459–462
Gembal M, Gilon P, Henquin J-C (1992) Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse β-cells. J Clin Invest 89:1288–1295
Ivarsson R, Quintens R, Dejonghe S et al (2005) Redox control of exocytosis. Regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54:2132–2142
Ronnebaum SM, Ilkayeva O, Burgess S et al (2006) A cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem 281:30593–30602
Rabaglia ME, Gray-Keller MP, Frey BL, Shortreed MR, Smith LM, Attie AD (2005) α-Ketoisocaproate-induced hypersecretion of insulin by islets from diabetes-susceptible mice. Am J Physiol Endocrinol Metab 289:E218–E224
Flamez D, Berger V, Kruhøffer M, Orntoft T, Pipeleers D, Schuit FC (2002) Critical role for cataplerosis via citrate in glucose-regulated insulin release. Diabetes 51:2018–2024
MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, Kendrick MA (2005) Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am J Physiol Endocrinol Metab 288:E1–E15
Urban KA, Panten U (2005) Selective loss of glucose-induced amplification of insulin secretion in mouse pancreatic islets pretreated with sulfonylurea in the absence of fuels. Diabetologia 48:2563–2566
Joseph JW, Jensen MV, Ilkayeva O et al (2006) The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J Biol Chem 281:35624–35632
Yaney GC, Corkey BE (2003) Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 46:1297–1312
Roduit R, Nolan C, Alarcon C et al (2004) A role for the malonyl-CoA/long-chain acyl-CoA pathway of lipid signaling in the regulation of insulin secretion in response to both fuel and nonfuel stimuli. Diabetes 53:1007–1019
Martens GA, Vervoort A, Van de Casteele M et al (2007) Specificity in beta cell expression of l-3-hydroxyacyl-CoA dehydrogenase, short-chain (HADHSC) and potential role in down-regulating insulin release. J Biol Chem 282:21134–21144
MacDonald MJ (2007) Synergistic potent insulin release by combinations of weak secretagogues in pancreatic islets and INS-1 cells. J Biol Chem 282:6043–6052
Pongratz R, Kibbey RG, Shulman GI, Cline GW (2007) Cytosolic and mitochondrial malic enzyme isoforms differentially control insulin secretion. J Biol Chem 282:200–207
Heart E, Yaney G, Corkey RF et al (2007) Ca2+, NAD(P)H and membrane potential changes in pancreatic β-cells by methyl succinate: comparison with glucose. Biochem J 403:197–205
MacDonald MJ (2002) Differences between mouse and rat pancreatic islets: succinate responsiveness, malic enzyme and anaplerosis. Am J Physiol Endocrinol Metab 283:E302–E310
Fernández-Pascual S, Mukala-Nsengu-Tshibangu A, Martín del Río R, Tamarit-Rodríguez J (2004) Conversion into GABA (γ-aminobutyric acid) may reduce the capacity of l-glutamine as an insulin secretagogue. Biochem J 379:721–729
Panten U, Burgfeldt J, Goerke F et al (1989) Control of insulin secretion by sulfonylureas, meglitinide and diazoxide in relation to their binding to the sulfonylurea receptor in pancreatic islets. Biochem Pharmacol 38:1217–1229
Heissig H, Urban KA, Hastedt K, Zünkler BJ, Panten U (2005) Mechanism of the insulin-releasing action of a-ketoisocaproate and related α-keto acid anions. Mol Pharmacol 68:1097–1105
Bleck C, Wienbergen A, Rustenbeck I (2005) Essential role of the imidazoline moiety in the insulinotropic effect but not the KATP channel-blocking effect of imidazolines; a comparison of the effects of efaroxan and its imidazole analogue, KU14R. Diabetologia 48:2567–2575
Passonneau JV, Lowry OH (1993) Enzymatic analysis. A practical guide. Humana Press, Totowa, pp 85–110
Panten U, Zünkler BJ, Scheit S, Kirchhoff K, Lenzen S (1986) Regulation of energy metabolism in pancreatic islets by glucose and tolbutamide. Diabetologia 29:648–654
Gepts W, Christophe J, Mayer J (1960) Pancreatic islets in mice with the obese-hyperglycemic syndrome. Lack of effect of carbutamide. Diabetes 9:63–69
Boquist K, Olson HL, Høy M et al (1999) Characterisation of sulphonylurea and ATP-regulated K+ channels in rat pancreatic A-cells. Pflügers Arch 438:428–436
Bertrand G, Ishiyama N, Nenquin M, Ravier MA, Henquin J-C (2002) The elevation of glutamate content and the amplification of insulin secretion in glucose-stimulated pancreatic islets are not causally related. J Biol Chem 277:32883–32891
Malaisse WJ, Hutton JC, Kawazu S (1978) The stimulus-secretion coupling of glucose-induced insulin release. Metabolic effects of menadione in isolated islets. Eur J Biochem 87:121–130
Ramirez R, Rasschaert J, Sener A, Malaisse WJ (1996) The coupling of metabolic to secretory events in pancreatic islets. Glucose-induced changes in mitochondrial redox state. Biochim Biophys Acta 1273:263–267
Ashcroft SJH, Christie MR (1979) Effects of glucose on the cytosolic ratio of reduced/oxidized nicotinamide-adenine dinucleotide phosphate in rat islets of Langerhans. Biochem J 184:697–700
Sener A, Malaisse-Lagae F, Dufrane SP, Malaisse WJ (1984) The coupling of metabolic to secretory events in pancreatic islets. The cytosolic redox state. Biochem J 220:433–440
Sazanov LA, Jackson JB (1994) Proton-translocating transhydrogenase and NAD- and NADP-linked isocitrate dehydrogenases operate in a substrate cycle which contributes to fine regulation of the tricarboxylic acid cycle activity in mitochondria. FEBS Lett 344:109–116
Pollak N, Dölle C, Ziegler M (2007) The power to reduce: pyridine nucleotides–small molecules with a multitude of functions. Biochem J 402:205–218
Trus M, Warner H, Matschinsky F (1980) Effects of glucose on insulin release and on intermediary metabolism of isolated perifused pancreatic islets from fed and fasted rats. Diabetes 29:1–14
Hutton JC, Sener A, Malaisse WJ (1979) The stimulus-secretion coupling of 4-methyl-2-oxopentanoate-induced insulin release. Biochem J 184:303–311
De Beer L, De Schepper PJ (1967) Metabolic effects of hypoglycemic sulfonylureas. II. In vitro effect of sulfonylureas on cell-free protein synthesis and energy metabolism in rat tissues. Biochem Pharmacol 16:2355–2367
Mannaerts G, De Beer L, Ceuterick-Goethals AM, De Schepper PJ (1974) Metabolic effects of hypoglycemic sulfonylureas. IV. Interference of sulfonylureas with mitochondrial oxidative phosphorylation. Biochem Pharmacol 23:239–250
White CW, Rashed HM, Patel TB (1988) Sulfonylureas inhibit metabolic flux through rat liver pyruvate carboxylase reaction. J Pharmacol Exp Ther 246:971–974
Smith PA, Proks P, Moorhouse A (1999) Direct effects of tolbutamide on the mitochondrial function, intracellular Ca2+ and exocytosis in pancreatic β-cells. Pflügers Arch 437:577–588
Stork H, Schmidt FH, Westman S, Hellerström C (1969) Action of some hypoglycaemic sulphonylureas on the oxygen consumption of isolated pancreatic islets in mice. Diabetologia 5:279–283
Hellman B, Idahl L-Å, Danielsson Å (1969) Adenosine triphosphate level of mammalian pancreatic B cells after stimulation with glucose and hypoglycemic sulfonylureas. Diabetes 18:509–516
Ashcroft SJH, Weerasinghe LCC, Randle PJ (1973) Interrelationship of islet metabolism, adenosine triphosphate content and insulin release. Biochem J 132:223–231
Kawazu S, Sener A, Couturier E, Malaisse WJ (1980) Metabolic, cationic and secretory effects of hypoglycaemic sulfonylureas in pancreatic islets. Naunyn-Schmiedeberg’s Arch Pharmacol 312:277–283
Elmi A, Idahl L-A, Sehlin J (2000) Relationships between the Na+/K+ pump and ATP and ADP content in mouse pancreatic islets: effects of meglitinide and glibenclamide. Br J Pharmacol 131:1700–1706
Detimary P, Gilon P, Nenquin M, Henquin J-C (1994) Two sites of glucose control of insulin release with distinct dependence on the energy state in pancreatic B-cells. Biochem J 297:455–461
Liu Y-J, Cheng H, Drought H, MacDonald MJ, Sharp GWG, Straub SG (2003) Activation of the KATP channel-independent signaling pathway by the nonhydrolyzable analog of leucine, BCH. Am J Physiol Endocrinol Metab 285:E380–E389
Malaisse-Lagae F, Sener A, Garcia-Morales P, Valverde I, Malaisse WJ (1982) The stimulus-secretion coupling of amino acid-induced insulin release. Influence of a nonmetabolized analog of leucine on the metabolism of glutamine in pancreatic islets. J Biol Chem 257:3754–3758
Hernández-Fisac I, Fernández-Pascual S, Ortsäter H et al (2006) Oxo-4 methylpentanoic acid directs the metabolism of GABA into the Krebs cycle in rat pancreatic islets. Biochem J 400:81–89
Acknowledgements
We thank G. Henze-Wittenberg, V. Lier-Glaubitz and I. Thomsen for excellent technical assistance.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Panten, U., Rustenbeck, I. Fuel-induced amplification of insulin secretion in mouse pancreatic islets exposed to a high sulfonylurea concentration: role of the NADPH/NADP+ ratio. Diabetologia 51, 101–109 (2008). https://doi.org/10.1007/s00125-007-0849-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0849-z