
Abstract
Aims/hypothesis
Hepatic steatosis is characterised by excessive triacylglycerol accumulation and is strongly associated with insulin resistance. An inability to efficiently mobilise liver triacylglycerol may be a key event mediating hepatic steatosis. Adipose triacylglycerol lipase (ATGL) is a key triacylglycerol lipase in the liver and we hypothesised that liver-specific overproduction of ATGL would reduce steatosis and enhance insulin action in obese rodents.
Methods
Studies of fatty acid metabolism were conducted in primary hepatocytes isolated from wild-type and Atgl (also known as Pnpla2)−/− mice. An ATGL adenovirus was utilised to overproduce ATGL in the livers of obese insulin-resistant C57Bl/6 mice (Ad-ATGL). Blood chemistry, hepatic lipid content and insulin sensitivity were assessed in mice.
Results
Triacylglycerol content was increased in Atgl −/− hepatocytes and was associated with increased fatty acid uptake and impaired fatty acid oxidation. ATGL adenovirus administration in obese mice increased the production of hepatic ATGL protein and reduced triacylglycerol, diacylglycerol and ceramide content in the liver. Overproduction of ATGL improved insulin signal transduction in the liver but did not affect fasting glycaemia or insulinaemia. Inflammatory signalling was not suppressed by ATGL overproduction. While ATGL overproduction increased plasma non-esterified fatty acids, neither lipid deposition nor insulin-stimulated glucose uptake were affected in skeletal muscle.
Conclusions/interpretation
Liver ATGL overproduction decreases hepatic steatosis and mildly enhances liver insulin sensitivity. These effects are not sufficient to improve fasting glycaemia or insulinaemia in rodent obesity.
Similar content being viewed by others


Introduction
Obesity is accompanied by the accumulation of triacylglycerol (TAG) and other lipids in non-adipose tissues, including the skeletal muscle, pancreas and liver [1, 2]. Hepatic steatosis is the accumulation of TAG in the liver and is a major element of non-alcoholic fatty liver disease (NAFLD). NAFLD affects ~20% of the adult population and is found in over two-thirds of obese people [3]. Hepatic steatosis is strongly associated with insulin resistance, which is characterised by an inability of insulin to suppress hepatic glucose production (HGP) and stimulate glucose uptake into peripheral tissues such as skeletal muscle [4]. Elevated HGP due to the defective suppression of gluconeogenesis and impaired glucose uptake are primary defects contributing to fasting hyperglycaemia in type 2 diabetes [5, 6]. The majority of studies in humans and animals have shown that insulin-resistant states can be accompanied by hepatic steatosis. For example, rodents fed a high-fat diet, ob/ob mice and lipodystrophic mice all develop hepatic steatosis and insulin resistance [7–9]. However, recent data suggest that hepatic TAG accumulation per se is not sufficient to cause insulin resistance [10–12]; instead, it is the inflammation that often accompanies hepatic steatosis that is likely to be the actual cause of the insulin resistance.
The liver plays a key role in systemic lipid homeostasis through its ability to synthesise and store fatty acids as TAG, and to secrete TAG in the form of VLDL. Hepatic fatty acids are derived from several sources, including adipose tissue lipolysis, chylomicron-TAG lipolysis and de novo lipogenesis, and can be stored as TAG in lipid droplets located within the cytosol [13]. Hepatic lipases mobilise the fatty acids from these TAGs for oxidation, or for re-esterification to TAG at the endoplasmic reticulum where the TAG is packaged into an apolipoprotein-B-containing VLDL precursor for secretion [13–15]. The regulation of these key processes of hepatic TAG homeostasis remains poorly defined.
The metabolic events leading to the development of hepatic steatosis are unresolved. While decreases in fatty acid oxidation [16] or increases in fatty acid uptake [17] play important roles, another possibility is that TAG accumulates as a consequence of an inability to mobilise the stored TAG in the presence of increased fatty acid import and biosynthesis [14]. The first step in the mobilisation of TAG is catalysed by several lipases, which include adipose triacylglycerol lipase (ATGL), hormone sensitive lipase (HSL) and triacylglycerol hydrolase (TGH). ATGL is an essential TAG lipase in many cell types [18] and Atgl (also known as Pnpla2) −/− mice store vast quantities of TAG in the major metabolic tissues, including the liver [19]. Post-translational regulation of ATGL TAG lipase activity is mediated by ATGL association with comparative gene identification (CGI)-58 [20], which enhances ATGL activity and its association with lipid droplets [21]. While hepatic ATGL content is low, it is proposed to account for a third of TAG lipase activity [22] and Atgl overexpression (>200-fold) in a hepatoma cell line or the livers of ob/ob mice reduces TAG levels [22]. Similarly, CGI-58 enhances the use of stored TAG in hepatoma cells by facilitating the packaging of TAG into lipoprotein particles [23, 24].
Some clinical observations suggest a regulatory role of ATGL TAG lipase activity in steatosis and insulin resistance. ATGL and CGI-58 protein abundance is reduced in the livers of insulin-resistant patients with NAFLD [25] and genetic variations within the ATGL gene are associated with type 2 diabetes and plasma TAG [26]. Together, these findings suggest that enhancing ATGL-mediated lipolysis in the liver may be a novel treatment for NAFLD and hepatic insulin resistance. We tested the hypothesis that ATGL is an important control point for TAG mobilisation in hepatocytes, and that overproduction of ATGL would reduce steatosis and enhance insulin action in the setting of obesity.
Methods
Animals
Male C57Bl/6 and ob/ob mice aged 10 weeks were purchased from Monash Animal Services (Clayton, VIC, Australia). Atgl −/− mice were generated by targeted homologous recombination as described by Haemmerle et al. [19]. Mice were maintained on a 12 h light/dark cycle and had free access to regular chow and water. Male C57Bl/6 mice were fed a high-fat diet (59% energy from fat; Specialty Feeds, Glen Forrest, WA, Australia) for 24–28 weeks prior to experiments. Body composition was determined by dual-energy X-ray absorptiometry (Lunar PIXImus2 mouse densitometer; GE Healthcare, Piscataway, NJ, USA). The Monash University School of Biomedical Sciences Animal Ethics Committee approved all procedures and the principles of laboratory animal care were followed.
Primary hepatocyte isolation
Atgl −/− mice (n = 6) and sex-matched wild-type (Wt) littermates (n = 6) aged 6–8 weeks were killed and hepatocytes isolated [27]. Hepatocytes were maintained in Medium 199 (Gibco, Invitrogen, Mulgrave, VIC, Australia) containing 20 ng/ml dexamethasone, 10 ng/ml epidermal growth factor, 10% (vol./vol.) fetal bovine serum and 1% glutamine and penicillin. Experiments were performed within 3 days of isolation.
Assessment of hepatocyte fatty acid metabolism and insulin sensitivity
Hepatocytes were incubated for 6 h with Medium 199 containing 2% fatty-acid-free bovine serum albumin, [1-14C]oleic acid (18.5 MBq/ml; GE Healthcare, Piscataway, NJ, USA) and 0.5 mmol/l unlabelled oleate to determine exogenous fatty acid oxidation. To determine endogenous fatty acid oxidation, hepatocytes were ‘pulsed’ for 24 h with the fatty acid medium described above to load endogenous lipid pools with [14C]oleate. The hepatocytes were washed with PBS then incubated for 6 h in fresh unlabelled fatty acid medium. Fatty acid oxidation was determined by measuring 14CO2 in the culture media and acid soluble metabolite production. Fatty acid incorporation into TAG was assessed by a Folch extraction of cellular lipids followed by thin layer chromatography [28]. Fatty acid uptake was calculated as the sum of [14C]oleate oxidation and 14C incorporation into all lipids. Hepatic insulin sensitivity was assessed by measuring hepatic glucose output and the suppression of Pepck and G6Pase (also known as G6pc) mRNA expression with insulin. Hepatocytes were incubated without or with increasing concentrations of insulin for 4 h before lysing in Qiazol lysis reagent (Qiagen, Doncaster, VIC, Australia).
Adenoviral vectors and administration
The recombinant adenoviral vectors were Ad-ATGL expressing wild-type Atgl (1.7 × 1011 pfu/ml) and Ad-GFP (2.9 × 1011 pfu/ml). See Electronic supplementary material (ESM) for cloning and adenovirus production. Adenovirus was injected into the jugular vein of anaesthetised mice (2.2% [vol./vol.] isoflurane in O2) at a final concentration of 1.5 × 109 pfu/g.
Insulin and pyruvate tolerance tests
Insulin tolerance tests were conducted 12 days after adenovirus administration. Mice were fasted for 4 h and insulin (1.0 U/kg body mass, Actrapid; Novo Nordisk, Baulkham Hills NSW, Australia) or pyruvate (0.35 or 2 mg/kg) were injected into the intraperitoneal cavity. Blood glucose levels were measured from a tail cut (Accu-Chek; Roche, Mannheim, Germany).
Glucose uptake in isolated skeletal muscle
Mice were fasted for 4 h and anaesthetised with an i.p. injection (60 mg/kg) of pentobarbital sodium. The soleus muscles were excised and glucose uptake was assessed for 10 min using 2-deoxy-d-[2,6-3H]glucose (1 mmol/l, 18.5 MBq/ml) in the presence or absence of 10 nmol/l insulin.
Plasma biochemistry
Plasma insulin was determined using a rat/mouse insulin ELISA (Millipore, Billerica, MA, USA). Plasma NEFA (Wako Chemicals, Wako, VA, USA) and TAG (Triacylglycerol E kit; Wako Chemicals) were determined by enzymatic colorimetric assays. Fasting plasma levels of alanine aminotransferase (ALT) was determined using ALT assay kits (Thermo Electron, Melbourne, VIC, Australia). TAG secretion was performed as described by Reid et al. [22].
Quantitative real-time PCR
RNA was extracted from whole liver or hepatocytes using Qiazol and 1 μg mRNA was reverse transcribed (iScript cDNA Synthesis Kit, BioRad Laboratories, Hercules, CA, USA). Quantitative real-time PCR was performed on a realplex Mastercycler (Eppendorf, North Ryde, NSW, Australia) using the TaqMan Universal PCR Master Mix and TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA; ESM Table 1). The relative quantification was calculated using the ΔΔCt method, normalising values to Ad-GFP (in vivo studies) or Wt (in vitro studies).
Western blotting
Tissues were rapidly dissected and frozen in liquid N2. Tissues were homogenised in ice cold RIPA lysis buffer, centrifuged and the supernatant fraction was resolved by SDS-PAGE and immunoblotted. See the ESM for detailed methods.
Tissue lipid analysis
Livers were sectioned and stained for Oil Red O. For TAG quantification, lipids were extracted by a Folch extraction, the lipid reconstituted in 100% ethanol and TAG content determined using the TG-GPO-PAP reagent (Roche). Diacylglycerol and ceramide content was determined by an [32P]ATP-linked enzymatic method as described by Watt et al. [29].
Statistics
Values are expressed as means ± SEM. Differences were tested by unpaired two-tailed t tests or two-way analysis of variance with Bonferroni post hoc analysis (GraphPad Prism version 5.02). Statistical significance was set a priori at p < 0.05.
Results
ATGL is decreased in the livers of obese mice
Liver TAG was elevated in high-fat-fed and ob/ob mice (Fig. 1a). High-fat-fed mice exhibited a marked reduction in liver Atgl (Fig. 1b), but there were no changes in the expression of other important TAG lipases, Hsl (also known as Lipe; Fig. 1c) or Tgh (also known as Ces3; Fig. 1d). Similar expression patterns were noted in the livers of ob/ob mice compared with lean littermates (Fig. 1e–g). TAG content was increased in the liver of Atgl –/– mice, indicative of an important role for ATGL in modulating liver TAG content (Fig. 1h).

ATGL is dysregulated in obese insulin-resistant rodents. a Liver triacylglycerol content in C57Bl/6 mice fed a chow or high-fat diet (HFD) and ob/ob mice. *p < 0.05, n = 4 per group. (b) Atgl, (c) Hsl and (d) Tgh mRNA expression in lean mice fed a chow diet and age-matched littermates made obese by feeding a high-fat diet. *p < 0.05, n = 4–10 per group. (e) Atgl, (f) Hsl and (g) Tgh mRNA expression in obese ob/ob mice and lean littermates. *p < 0.05, n = 5 per group. h Liver triacylglycerol content in Atgl −/− mice and wild-type age-matched littermates. *p < 0.05, n = 3 per group
Atgl −/− hepatocytes exhibit defects in fatty acid metabolism and are steatotic
Isolated hepatocytes from Atgl −/− mice exhibited marked TAG accumulation (Fig. 2a). Diacylglycerol was increased (1.00 ± 0.05 vs 1.97 ± 0.12 arbitrary units, p < 0.05) and ceramide decreased (1.00 ± 0.04 vs 0.77 ± 0.06 arbitrary units, p < 0.05) in Atgl –/– hepatocytes. Fatty acid uptake was increased in Atgl –/– hepatocytes (Fig. 2b) but fatty acid oxidation was decreased (Fig. 2c). The reduction in fatty acid oxidation was not due to defects in the contents of key mitochondrial enzymes or regulators of mitochondrial transcription, which were, surprisingly, increased (Fig. 2d). Activating phosphorylation of 5′-AMP-activated protein kinase on T172 of the α-subunit was not different (Fig. 2e). Instead, the decreases in fatty acid oxidation were associated with increased fatty acid incorporation into TAG (Fig. 2f). This was not explained by increased levels of transcripts for proteins involved in fatty acid uptake, storage or TAG lipolysis (Fig. 2g). We investigated whether fatty acids stored within TAG could be efficiently catabolised in the absence of ATGL. TAG-derived fatty acid oxidation was reduced by 85% in Atgl −/− hepatocytes (Fig. 2h). TAG secretion was not affected by Atgl ablation (Fig. 2i). This finding indicates that ATGL is a rate-limiting TAG lipase in hepatocytes that is important for providing substrate for fatty acid oxidation, but not TAG synthesis or export [22].

Atgl deletion alters hepatic lipid metabolism in vitro. Hepatocytes isolated from Wt and Atgl −/− mice were cultured for 2–3 days prior to functional analysis of fatty acid metabolism using radiolabelled oleate. a Hepatocyte triacylglycerol content (n = 6 per group). (b) Uptake and (c) oxidation of fatty acids (n = 8–12 per group). d The mRNA of oxidative metabolism and mitochondrial biogenesis proteins was quantified by qRT-PCR and normalised against Gapdh (n = 6 per group). Gapdh was not different between groups (Wt: 20.3 ± 0.4 vs Atgl −/−: 19.6 ± 0.6 AU, p = 0.34). e 5′-AMP protein kinase T172 phosphorylation (n = 3–4 per group). f Fatty acid incorporation of media-derived fatty acids into intracellular triacylglycerol (n = 12 per group). g The mRNA content of fatty acid synthesis/storage/triacylglycerol lipase proteins (n = 4–5 per group). h Oxidation of fatty acid derived from intracellular stores (presumably TAG, n = 12 per group). i TAG secretion into culture media over 24 h (n = 4–6 per group). *Different from Wt, p < 0.05
Atgl −/− hepatocytes are not insulin resistant
Despite storing vast quantities of TAG in most tissues, whole-body Atgl −/− mice were originally reported to be insulin sensitive in multiple tissues including the liver [19]; however, more recent studies indicate that the liver of Atgl −/− mice may in fact be insulin resistant [30]. We examined insulin sensitivity in hepatocytes isolated from Atgl −/− and Wt mice, which precludes interference from systemic factors and local inflammatory mediators (e.g. Kupffer cells and mast cells). Insulin-stimulated phosphorylation of Akt at S473 and T308 was not different between genotypes (Fig. 3a, b). Consistent with this notion, the capacity of insulin to suppress phosphoenolpyruvate kinase (Pepck) and glucose-6-phosphatase (G6Pase) mRNA expression was not different (Fig. 3c, d). HGP was lower in Atgl −/− hepatocytes (main effect, p < 0.05) and was decreased by insulin in both genotypes (Fig. 3e). Suppression of HGP by insulin was not different between genotypes (Fig. 3f). Insulin-stimulated Akt phosphorylation was not affected in mice injected with insulin in vivo (ESM Table 2). Together, these findings indicate that despite inducing marked steatosis, Atgl ablation does not influence insulin sensitivity in hepatocytes.

Atgl ablation does not appreciably influence insulin action in primary hepatocytes. Hepatocytes isolated from Wt and Atgl −/− mice were cultured for 2–3 days prior to assays. (a) Akt phosphorylation at S473 and (b) T308 under basal conditions or after acute insulin stimulation (n = 3–4 per condition). In separate experiments, hepatocytes were incubated with insulin (0–10 nmol/l) for 4 h and Pepck (c) and G6Pase (d) mRNA was determined by qRT-PCR (n = 4–12 per condition). e Hepatic glucose output was assessed in Wt (white bars) and Atgl −/− hepatocytes (black bars) under basal and insulin stimulated conditions (n = 6 per group). f Hepatic glucose output reported as % suppression by insulin. Wt (white bars) and Atgl −/− (black bars) (n = 6 per group)
ATGL overproduction in the livers of mice alters hepatic lipid metabolism
We next tested the hypothesis that increasing ATGL production in the liver modulates lipid and glucose metabolism in vivo. Adenoviral ATGL overproduction was restricted to the liver, where it increased Atgl mRNA (Fig. 4a) and ATGL protein content (Fig. 4b). Hsl mRNA expression was decreased (31%, p = 0.02) and Tgh mRNA tended to increase (50%, p = 0.10) with Ad-ATGL (data not shown). Adenoviral overexpression of Atgl in the liver did not affect food intake (Table 1). Similarly, body mass and fat mass in Ad-ATGL (Ad-GFP, 33.7 ± 1.2% body mass; Ad-ATGL, 32.7 ± 1.8%, n = 4, p = 0.69) were not different from mice treated with Ad-GFP (Table 1). Serum ALT levels were not different between groups (Table 1).

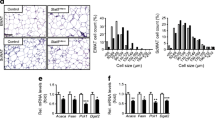
Hepatic Atgl expression reduces lipid deposition in livers of obese mice. Adenovirus containing GFP (white bars) or ATGL (black bars) was intravenously administered to mice and tissues were collected after 14 days. a Atgl mRNA was determined by qRT-PCR (n = 10–13). b Representative immunoblot showing ATGL (54 kDa) and ATGL-GFP (76 kDa). c Sectioned livers stained with Oil Red O demonstrate marked steatosis in Ad-GFP mice and a reduction in staining with Ad-ATGL. d Liver triacylglycerol was assessed biochemically (n = 8–9 per group). e Srebf1, Scd1 and Fas mRNA were assessed by qRT-PCR (n = 3–8 per group). † p = 0.06. Liver diacylglycerol (DAG) (f) and ceramide (g) were determined in Ad-GFP and Ad-ATGL mice (n = 10–13 per group). *Different from Ad-GFP, p < 0.05
Overproduction of ATGL reduced liver steatosis in obese mice as denoted by less Oil Red O staining of neutral lipids (Fig. 4c). The lipid droplets containing TAG appeared larger in Ad-GFP compared with Ad-ATGL mice and quantitative biochemical analysis of liver revealed a marked reduction in the intracellular TAG content of Ad-ATGL mice (Fig. 4d). Mediators of fatty acid storage, including sterol regulatory element-binding protein 1, stearoyl-coenzyme A desaturase 1 and fatty acid synthase were concomitantly decreased with Ad-ATGL (Fig. 4e). The lipids diacylglycerol (32%) and ceramide (34%) were reduced with Ad-ATGL (Fig. 4f, g). Interestingly, fasting plasma NEFA was increased in Ad-ATGL (Table 1) despite no change in basal and β-adrenergic lipolytic capacity as assessed in adipose tissue explants ex vivo (data not shown). While an increase in plasma NEFA positively correlates with TAG accumulation in ectopic tissues [29], we detected no increase in intracellular TAG deposition in skeletal muscle or heart with Ad-ATGL (Table 1). TAG secretion rate and plasma TAGs were not increased in Ad-ATGL compared with Ad-GFP (Table 1). Plasma β-hydroxybutyrate was elevated in Ad-ATGL mice (Table 1), suggesting enhanced β-oxidation in the liver.
Hepatic ATGL overproduction mildly increases hepatic insulin sensitivity
Because Ad-ATGL induced a marked reduction in hepatic steatosis, we postulated that insulin sensitivity may be improved in these mice. Fasting blood glucose tended to be lower in Ad-ATGL (12%, p = 0.08) and plasma insulin was not different between groups (decreased by 33% in Ad-ATGL, p = 0.17, Table 1) 2 weeks after adenovirus administration. Whole-body insulin sensitivity was mildly improved with Ad-ATGL (Fig. 5a), but these changes were not accompanied by enhanced skeletal muscle insulin responsiveness assessed ex vivo (Fig. 5b). The mild changes in whole-body glucose metabolism were accompanied by the suppression of hepatic Pcg-1α (also known as Ppargc1a; Fig. 5c), which stimulates the production of gluconeogenic enzymes [31]. Consistent with this effect, Pepck mRNA was suppressed in livers of Ad-ATGL mice (Fig. 5c). There was no difference in G6Pase mRNA. We tested the role of Ad-ATGL in modulating gluconeogenesis in vivo by examining glucose levels after the i.p. administration of pyruvate. The glucose area under the curve tended (p = 0.09) to be reduced in Ad-ATGL after a high pyruvate load (2 mg/kg), suggestive of reduced gluconeogenesis (Fig. 5d). We repeated this experiment with a lower pyruvate dose (0.35 mg/kg) to determine the sensitivity of the gluconeogenic response and detected a reduced glucose excursion with Ad-ATGL (Fig. 5e). Also, the phosphorylation of Akt and its downstream target forkhead box O (FOXO)1 (Fig. 5f–h) were increased in Ad-ATGL mice vs Ad-GFP. Collectively, these data demonstrate that enhanced hepatic insulin sensitivity in Ad-ATGL mice contributes to a subtle enhancement of whole-body insulin action.

Ad-ATGL improves insulin sensitivity in mice. a Mice treated with Ad-GFP or Ad-ATGL were injected i.p. with 1 U/kg insulin and blood glucose was monitored for 120 min. White circles, Ad-GFP; black circles, Ad-ATGL (n = 11–13 per group).**p < 0.05 for main effect for treatment. b Glucose uptake into soleus muscle was determined ex vivo under basal or insulin stimulated (10 nmol/l) conditions (n = 6–8 per condition); *p < 0.05 vs basal within the same treatment. c Pepck, G6Pase and Pgc-1α (also known as Ppargc1a) mRNA in the liver of mice after a 4–6 h fast. (d, e) Mice were injected i.p. with a high (2 g/kg) (d) or low (0.35 g/kg) (e) pyruvate load, and blood glucose was assessed for 60–90 min. White circles, Ad-GFP; black circles, Ad-ATGL (n = 3–6 per group). f–h Mice were anaesthetised with isoflurane and injected i. v. with 5 U/kg dose insulin. Livers were freeze clamped after 10 min and insulin sensitivity was assessed by measuring (f) pAkt (S473) and (g) pFOXO1 (S256). *p < 0.05, † p = 0.058 vs Ad-GFP. Representative blots are shown in (h) (n = 7–8 per group)
Hepatic ATGL overproduction has no effect on inflammatory signalling
Hepatic steatosis can be accompanied by inflammation that is likely to contribute to insulin resistance [32]. We examined several markers of intracellular inflammation in the livers of mice. The mRNA expression of the pro-inflammatory cytokine TNF-α was reduced (p = 0.06) in Ad-ATGL vs Ad-GFP (ESM Fig. 1a), whereas Il6 and suppressor of cytokine signalling 3 expression were not different between groups (ESM Fig. 1a). There was no difference in CD68 protein content, suggesting no effect of Ad-ATGL on Kupffer cell numbers (ESM Fig. 1a).
Increased TNFα [33] and lipid accumulation leads to activation of serine/threonine kinases that can decrease insulin signalling [32]. Despite the reduced hepatic TNFα content and lipid accumulation in Ad-ATGL, the phosphorylation levels of c-Jun N-terminal kinase (JNK; ESM Fig. 1b) and activation of the nuclear factor-kappa B (NFκB) pathway, as indicated by nuclear factor of kappa B inhibitor alpha (IκBα) degradation (ESM Fig. 1c), were not different. Protein kinase C (PKC)ε is activated by diacylglycerol and has been implicated in the development of hepatic insulin resistance [34]. There was no difference in the membrane to cytosolic ratio (marker of PKC activity) of PKCε (ESM Fig. 1d) or PKCθ (data not shown). Thus, the improvement in hepatic insulin action with ATGL overproduction was not a consequence of diminished inflammatory pathway activation in the liver.
Discussion
ATGL is a critical regulator of triacylglycerol hydrolysis in most metabolic tissues [19] and truncated ATGL induces clinical symptoms including hepatic TAG accumulation and hepatomegaly [21]. The goal of this study was to directly assess the role of ATGL in hepatic lipid metabolism and insulin sensitivity. We show that ablation of Atgl in hepatocytes increased TAG storage by reducing the capacity to mobilise TAG-derived fatty acids for oxidation, without affecting TAG secretion. Surprisingly, insulin sensitivity was not impaired in Atgl-null hepatocytes despite the marked lipid accumulation. Hepatic overproduction of ATGL in obese mice reduced hepatic lipid content without influencing macrophage infiltration or local inflammation. ATGL overproduction led to subtle improvements in hepatic insulin responsiveness that did not translate into notable changes in whole-body insulin sensitivity, fasting blood glucose or insulin levels. These findings provide novel insights into the role of ATGL in hepatic TAG metabolism and provide specific evidence that ATGL is not a major mediator of hepatic insulin sensitivity.
Steatosis is characterised by TAG deposition and a major contributor is enhanced fatty acid availability from dysregulated adipose tissue lipolysis [35]. The susceptibility of the liver to fatty acid fluxes is hardly surprising, as visceral fat directly drains fatty acids into the liver via the portal vein. While this is less studied, a growing body of evidence indicates that the modification of TAG synthesis or degradation processes contributes to steatosis development [11, 22, 36–38].
Unlike a previous study [22], we closely examined ATGL loss of function in isolated hepatocytes to avoid the potential for confounding regulation introduced by neural, hormonal and inflammatory factors. Atgl deletion induced several alterations in fatty acid handling that resulted in TAG accumulation. An important implication of our studies is that decreased fatty acid oxidation contributes to steatosis in Atgl −/− hepatocytes. Gene expression of proteins involved in mitochondrial fatty acid oxidation was actually increased in Atgl −/− hepatocytes, suggesting that the defect in hepatic fatty acid oxidation was independent of defective protein production. Instead, the oxidation of fatty acids derived from intra- and extracellular sources was reduced by 58% and 85%, respectively, supporting the premise that most of the fatty acids entering the liver are first incorporated into TAG prior to oxidation. Thus, the critical and novel observation is that an inability to mobilise TAG-derived fatty acids contributes to the reduction in oxidation and steatosis. In support of this idea, ATGL overproduction in rat hepatoma cells decreased cellular TAG and increased fatty acid oxidation [22]. Also, downregulation of the ATGL co-activator CGI-58 reduced fatty acid oxidation whereas CGI-58-mediated TAG hydrolysis was coupled to enhanced fatty acid oxidation in HepG2 cells [23]. The concept that plasma-derived fatty acids first enter the TAG pool within intracellular lipid droplets before their eventual oxidation was suggested by Gibbons et al. [14] and experimentally shown in skeletal muscle [39]. A second implication from these studies is that hepatocytes continue to take up fatty acids despite marked steatosis. The absence of a powerful negative feedback loop may reflect the requirement of the liver to utilise fatty acid substrate for gluconeogenesis, which occurs during starvation with concurrent hepatic lipid accumulation [40], or may reflect the requirement of the liver to clear and package excess plasma NEFA that are invariably toxic to most tissues [2].
A previous study reported reduced TAG content in obese mice after massive (200-fold) ATGL and HSL adenovirus administration [22]. We also found that modest ATGL overproduction reduced TAG content in the livers of obese mice. The decrease in TAG content was most likely due to increased TAG lipolysis and oxidation of the liberated fatty acids. Consistent with increased rates of fatty acid oxidation in livers of Ad-ATGL mice, we detected elevated fasting plasma β-hydroxybutyrate. Our results showing no increase in circulating TAG or lipid deposition in other ectopic tissues (skeletal muscle, heart, adipose tissue) with Ad-ATGL are consistent with the notion that TAG export is unaffected by hepatic ATGL overproduction in vivo (Table 1) [22] or hepatic ATGL ablation in vitro (Fig. 2i). We were unable to complete these measures in Atgl −/− mice because of unacceptable mortality. Collectively, these data indicate that ATGL directs TAG-derived fatty acids towards oxidation. In contrast, the other major TAG lipase, TGH, preferentially directs fatty acids from TAG towards VLDL production [41]. Thus, while ATGL and TGH both possess TAG lipase activity, they appear to channel the liberated fatty acids towards oxidation and VLDL packaging, respectively.
The fatty liver of insulin-resistant mice is attributed in part to increased production and activity of sterol regulatory element binding protein (SREBP)-1c and subsequent upregulation of lipogenic gene expression [42]. Our findings of reduced SREBP-1c, and its downstream targets FAS and SCD1, and a tendency for reduced fasting plasma insulin support the possibility that TAG synthesis rates were reduced with Ad-ATGL. A major issue of clinical relevance is whether providing metabolic relief by directing lipids away from the liver is a sound approach for the treatment of steatosis/fatty liver and insulin resistance, or whether such an approach would give rise to compensatory lipid storage in other tissues and the induction of secondary complications [43]. A critical finding from this study is that hepatic ATGL overproduction does not result in the redistribution/partitioning of lipids into other tissues, supporting the practicality of enhancing ATGL production by pharmacological manipulation for treating fatty liver.
The role of ATGL in hepatic insulin action is unresolved, with reports of increased [19] and decreased [30] insulin sensitivity in whole-body Atgl −/− mice. Here, we directly examined the effect of ATGL on hepatic insulin sensitivity. Studies in isolated hepatocytes demonstrated normal insulin sensitivity in Atgl −/− hepatocytes despite marked TAG accumulation. This is consistent with recent studies demonstrating a mismatch between steatosis and insulin sensitivity [10–12, 22] and suggests that lipid accumulation per se is not sufficient to induce insulin resistance. In contrast, increasing liver ATGL mildly improved hepatic insulin action in obese insulin-resistant mice. These functional outcomes were mechanistically underpinned by suppression of PGC-1α and phosphoenolpyruvate kinase (PEPCK) in fasting mice, improved insulin signal transduction and suppression of gluconeogenesis following pyruvate exposure. These changes were not sufficient to reduce fasting glycaemia and insulinaemia.
Hepatic steatosis is closely linked to insulin resistance; however, TAGs are unlikely to directly cause insulin resistance because they are stored within lipid droplets. Also, TAG accumulates during fasting when insulin sensitivity is not impaired, refuting the likelihood of a causal negative relationship. Other metabolically active lipid intermediates, such as diacylglycerol and ceramide, are mechanistically linked to defective insulin signal transduction. There is considerable associative evidence to support a role for diacylglycerol in mediating hepatic insulin resistance, with the demonstration of concurrent reductions in diacylglycerol, PKCε activity and improved insulin receptor substrate signalling following various experimental manipulations [34, 36, 37]. While we showed that ATGL overproduction reduced liver diacylglycerol by ~30%, PKCε membrane translocation was unaffected. Other serine/threonine kinases such as JNK and inhibitor of kappa kinase are implicated in the development of insulin resistance [44, 45], yet neither kinase was attenuated with ATGL overproduction. Ceramide inhibits insulin signalling at Akt by dephosphorylation mediated by protein phosphatase 2A [46] and by preventing the translocation of Akt [47]. Pharmacological and genetic interference of ceramide biosynthesis is associated with enhanced insulin sensitivity [48]. Our studies show that ATGL overproduction reduced ceramide content and increased Akt phosphorylation in the liver of obese mice, supporting the concept that ATGL influences distal hepatic insulin signalling [30]. Interestingly, ceramide was not increased in Atgl −/− hepatocytes, which may explain the maintenance of insulin sensitivity in these cells.
In summary, this study demonstrates that hepatic ATGL production is reduced in several models of rodent obesity and that ATGL ablation leads to pronounced steatosis that is characterised by an impairment in fatty acid oxidation. Increasing ATGL production in the liver reduces hepatic steatosis and mildly enhances liver insulin sensitivity. These effects are not sufficient to improve fasting glycaemia or insulinaemia in rodent obesity, suggesting that ATGL represents a pharmacological therapeutic target for fatty liver disease but not type 2 diabetes.
Abbreviations
- ATGL:
-
Adipose triacylglycerol lipase
- ALT:
-
Alanine aminotransferase
- CGI:
-
Comparative gene identification
- JNK:
-
c-Jun N-terminal kinase
- FOXO:
-
Forkhead box O
- G6Pase:
-
Glucose-6-phosphatase
- HGP:
-
Hepatic glucose production
- HSL:
-
Hormone sensitive lipase
- NAFLD:
-
Non-alcoholic fatty liver disease
- PEPCK:
-
Phosphoenolpyruvate kinase
- PKC:
-
Protein kinase C
- SREBP:
-
Sterol regulatory element binding protein
- TAG:
-
Triacylglycerol
- TGH:
-
Triacylglycerol hydrolase
- Wt:
-
Wild type
References
Friedman J (2002) Fat in all the wrong places. Nature 415:268–269
Unger RH (2003) Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 144:5159–5165
Almeda-Valdes P, Cuevas-Ramos D, Aguilar-Salinas CA (2009) Metabolic syndrome and non-alcoholic fatty liver disease. Ann Hepatol 8(Suppl 1):S18–S24
Biddinger SB, Kahn CR (2006) From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol 68:123–158
Bock G, Chittilapilly E, Basu R et al (2007) Contribution of hepatic and extrahepatic insulin resistance to the pathogenesis of impaired fasting glucose: role of increased rates of gluconeogenesis. Diabetes 56:1703–1711
Gastaldelli A, Baldi S, Pettiti M et al (2000) Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes 49:1367–1373
Halaas JL, Gajiwala KS, Maffei M et al (1995) Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269:543–546
Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL (2000) Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 6:77–86
Oakes ND, Cooney GJ, Camilleri S, Chisholm DJ, Kraegen EW (1997) Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes 46:1768–1774
Kantartzis K, Machicao F, Machann J et al (2009) The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin Sci (Lond) 116:531–537
Monetti M, Levin MC, Watt MJ et al (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6:69–78
Solinas G, Vilcu C, Neels JG et al (2007) JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab 6:386–397
Nagle CA, Klett EL, Coleman RA (2009) Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res 50(Suppl):S74–S79
Gibbons GF, Wiggins D, Brown AM, Hebbachi AM (2004) Synthesis and function of hepatic very-low-density lipoprotein. Biochem Soc Trans 32:59–64
Francone OL, Kalopissis AD, Griffaton G (1989) Contribution of cytoplasmic storage triacylglycerol to VLDL-triacylglycerol in isolated rat hepatocytes. Biochim Biophys Acta 1002:28–36
Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A et al (2008) Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab 7:496–507
Koonen DP, Jacobs RL, Febbraio M et al (2007) Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 56:2863–2871
Zimmermann R, Strauss JG, Haemmerle G et al (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306:1383–1386
Haemmerle G, Lass A, Zimmermann R et al (2006) Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312:734–737
Lass A, Zimmermann R, Haemmerle G et al (2006) Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin–Dorfman Syndrome. Cell Metab 3:309–319
Kobayashi K, Inoguchi T, Maeda Y et al (2008) The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J Clin Endocrinol Metab 93:2877–2884
Reid BN, Ables GP, Otlivanchik OA et al (2008) Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem 283:13087–13099
Brown JM, Chung S, Das A, Shelness GS, Rudel LL, Yu L (2007) CGI-58 facilitates the mobilization of cytoplasmic triglyceride for lipoprotein secretion in hepatoma cells. J Lipid Res 48:2295–2305
Caviglia JM, Sparks JD, Toraskar N et al (2009) ABHD5/CGI-58 facilitates the assembly and secretion of apolipoprotein B lipoproteins by McA RH7777 rat hepatoma cells. Biochim Biophys Acta 1791:198–205
Kato M, Higuchi N, Enjoji M (2008) Reduced hepatic expression of adipose tissue triglyceride lipase and CGI-58 may contribute to the development of non-alcoholic fatty liver disease in patients with insulin resistance. Scand J Gastroenterol 43:1018–1019
Schoenborn V, Heid IM, Vollmert C et al (2006) The ATGL gene is associated with free fatty acids, triglycerides, and type 2 diabetes. Diabetes 55:1270–1275
Scott JW, van Denderen BJ, Jorgensen SB et al (2008) Thienopyridone drugs are selective activators of AMP-activated protein kinase β1-containing complexes. Chem Biol 15:1220–1230
Watt MJ, Steinberg GR, Chen ZP, Kemp BE, Febbraio MA (2006) Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J Physiol 574:139–147
Watt MJ, Hevener A, Lancaster GI, Febbraio MA (2006) Ciliary neurotrophic factor prevents acute lipid-induced insulin resistance by attenuating ceramide accumulation and phosphorylation of c-Jun N-terminal kinase in peripheral tissues. Endocrinology 147:2077–2085
Kienesberger PC, Lee D, Pulinilkunnil T et al (2009) Adipose triglyceride lipase (ATGL) deficiency causes tissue-specific changes in insulin signaling. J Biol Chem 284:30218–30229
Yoon JC, Puigserver P, Chen G et al (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413:131–138
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Fujishiro M, Gotoh Y, Katagiri H et al (2003) Three mitogen-activated protein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes. Mol Endocrinol 17:487–497
Samuel VT, Liu ZX, Wang A et al (2007) Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117:739–745
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115:1343–1351
Choi CS, Savage DB, Kulkarni A et al (2007) Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem 282:22678–22688
Neschen S, Morino K, Hammond LE et al (2005) Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab 2:55–65
Yu XX, Murray SF, Pandey SK et al (2005) Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 42:362–371
Kanaley JA, Shadid S, Sheehan MT, Guo Z, Jensen MD (2009) Relationship between plasma FFA, intramyocellular triglycerides and long-chain acylcarnitines in resting humans. J Physiol 587:5939–5950
Moller L, Stodkilde-Jorgensen H, Jensen FT, Jorgensen JO (2008) Fasting in healthy subjects is associated with intrahepatic accumulation of lipids as assessed by 1H-magnetic resonance spectroscopy. Clin Sci (Lond) 114:547–552
Lehner R, Vance DE (1999) Cloning and expression of a cDNA encoding a hepatic microsomal lipase that mobilizes stored triacylglycerol. Biochem J 343(Pt 1):1–10
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131
Hoy AJ, Watt MJ (2009) Lipid partitioning and insulin sensitivity: lessons learnt from genetic models. APJOE 1:35–47
Hirosumi J, Tuncman G, Chang L et al (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Yuan M, Konstantopoulos N, Lee J et al (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science 293:1673–1677
Schmitz-Peiffer C, Craig DL, Biden TJ (1999) Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated pkb pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 274:24202–24210
Cazzolli R, Carpenter L, Biden TJ, Schmitz-Peiffer C (2001) A role for protein phosphatase 2a-like activity, but not atypical protein kinase Cζ, in the inhibition of protein kinase b/akt and glycogen synthesis by palmitate. Diabetes 50:2210–2218
Holland WL, Brozinick JT, Wang LP et al (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5:167–179
Acknowledgements
We thank G. Haemmerle and R. Zechner (University of Graz) for the Atgl −/− mice and C. Schmitz-Peiffer for the PKCε antibody. These studies were supported by research grants from the National Health and Medical Research Council (NHMRC) of Australia and a Monash Fellowship. S. M. Turpin was supported by a NHMRC Dora Lush Biomedical Postgraduate Scholarship. A. J. Hoy is supported by a Biomedical Australian training fellowship and M. J. Watt a Senior Research Fellowship from the NHMRC.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 144 kb)
ESM Fig. 1
Inflammatory signalling is not affected by ATGL overproduction. a Tnfα, Il-6 and Socs3 mRNA was determined by qRT-PCR (n = 6–7). The representative immunoblot shows no changes in CD68 production between groups (n = 5 per group). (b) Phospho-JNK (Thr183/Tyr185) (p46 = JNK1 and p54 = JNK2) and (c) IκBα were assessed in liver lysates. d PKCε was assessed in membrane (denoted by M) and cytosolic (denoted by C) fractions. Representative immunoblots are shown above graphs. n = 7–8 for all figure parts (PDF 333 kb)
ESM Table 1
TaqMan gene expression assays (PDF 132 kb)
ESM Table 2
Insulin-stimulated Akt signalling in Wt and Atgl −/− mice in vivo (PDF 142 kb)
Rights and permissions
About this article
Cite this article
Turpin, S.M., Hoy, A.J., Brown, R.D. et al. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia 54, 146–156 (2011). https://doi.org/10.1007/s00125-010-1895-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1895-5