
Abstract
Viruses are considered to be potential key modulators of type 1 diabetes mellitus, with several possible mechanisms proposed for their modes of action. Here we discuss the evidence for virus involvement, including pancreatic infection and the induction of T cell-mediated molecular mimicry. A particular focus of this review is the further possibility that virus infection triggers bystander activation of pre-existing autoreactive lymphocytes. In this scenario, the virus triggers dendritic cell maturation and proinflammatory cytokine secretion by engaging pattern recognition receptors. These proinflammatory cytokines provoke bystander autoreactive lymphocyte activation in the presence of cognate autoantigen, which leads to enhanced beta cell destruction. Importantly, this mechanism does not necessarily involve pancreatic virus infection, and its virally non-specific nature suggests that it might represent a means commonly employed by multiple viruses. The ability of viruses specifically associated with type 1 diabetes, including group B coxsackievirus, rotavirus and influenza A virus, to induce these responses is also examined. The elucidation of a mechanism shared amongst several viruses for accelerating progression to type 1 diabetes would facilitate the identification of important targets for disease intervention.
Similar content being viewed by others

Introduction
Type 1 diabetes mellitus is a chronic autoimmune disease marked by the development of insulitis and the destruction of insulin-producing beta cells by autoreactive T cells [1]. Disease onset can be predicted by the inheritance of high-risk HLA genes in combination with the presence of circulating islet autoantibodies and islet-specific T cells [1]. Discordance between monozygotic twins suggests that high-risk genes alone cannot completely predict diabetes development [2]. Furthermore, high-risk HLA gene prevalence in patients is declining concurrently with increased diabetes incidence and a trend towards a younger age at onset, indicating a potentially important role for environmental factors [3].
Numerous environmental triggers have been linked to diabetes onset, including exposure to specific dietary antigens, intestinal microflora and infection [4]. Viruses are considered to be key potential diabetes modulators. For example, infection by members of the Enterovirus genus of the Picornaviridae family, or the Rotavirus genus in the Sedoreovirinae subfamily of the Reoviridae family, has been associated with altered diabetes development in humans and mice [5–8]. However, a direct causal relationship between a specific environmental stimulus and diabetes onset has proven difficult to establish. In part, this relates to the multiple potential environmental modulators experienced during the extended pre-clinical phase of diabetes. The effect of any putative causative agent may not be identifiable at diabetes diagnosis. Additionally, multiple mechanisms may trigger disease onset and environmental factors may modulate autoimmunity following an initial genetically determined trigger.
NOD mice are commonly used to investigate markers of diabetes development and virus roles. Like humans, diabetes onset in these mice is influenced by genetic and environmental factors and preceded by insulitis development [9]. Although aspects of disease development in NOD mice differ from that in humans, these mice are a valuable tool for understanding potential mechanisms of diabetes modulation that cannot be directly assessed in humans. Virus infections are hypothesised to contribute to diabetes development by three distinct but not mutually exclusive general mechanisms: pancreatic infection, T cell molecular mimicry and bystander activation. Increased intestinal inflammation and permeability have also been implicated in diabetes development [4], and certain bacterial infections and intestinal microbiota may modulate diabetes in these ways [10, 11]. Here we briefly discuss the potential roles of pancreatic infection and molecular mimicry, which have been extensively reviewed previously [12, 13]. The primary focus of this review is the potential role of bystander activation as a non-specific mechanism of autoimmune activation.
Detection of virus in the pancreas
Viral infection of pancreatic beta cells can result in cytolysis or cell damage [14, 15]. While cytolysis directly reduces beta cell mass, beta cell damage may contribute to diabetes progression through the release of sequestered autoantigens and induction of a local proinflammatory immune response [16]. A similar cascade of proinflammatory events indirectly leading to beta cell damage or death may also occur following infection of bystander pancreatic cells, such as alpha cells [17]. Current human studies are particularly directed towards the detection of virus-infected cells and viral nucleic acids in the pancreas during diabetes development or after diagnosis [18]. Pancreatic virus detection relies on the assumption that a particular virus infection occurs near the time of diabetes onset, occurs multiple times throughout pre-clinical diabetes development or has become chronic. However, if a virus infects only once, acutely, or is cleared by the time of diabetes diagnosis, then detection of pancreatic virus is unlikely. Furthermore, viruses might contribute to diabetes through extra-pancreatic infection at sites like the pancreatic lymph nodes (PLN) or the small intestine. Low-grade pancreatic enterovirus infection in new-onset diabetic patients was recently reported [18]. However, viral cytolytic activity was absent, precluding conclusions as to causality [18]. Although one study detected intestinal enterovirus infection more frequently in diabetic patients, associated with inflammation [19], a subsequent investigation did not support this observation [20]. Rather than a causative role, at least in some cases, higher enterovirus detection rates may indicate greater patient susceptibility to enterovirus infection. Establishing the nature of any association between pancreatic virus presence and type 1 diabetes is clearly a research priority. However, future studies should not discount the potential for viruses to alter diabetes development by exerting their effects in the PLN or intestine.
T cell molecular mimicry
In the context of viral infection, molecular mimicry occurs when viral peptide loaded onto MHC molecules at the antigen-presenting cell surface is recognised by autoreactive T cells, leading to their activation. This generally arises from sequence similarity between viral and self peptides. RIP-LCMV mice, which express particular lymphocytic choriomeningitis virus (LCMV) proteins under the control of the rat insulin promoter (RIP) in the pancreas [21], have been used extensively to understand the role of molecular mimicry in diabetes. In this model, diabetes occurs following infection with wild-type LCMV (which contains the exact self LCMV protein sequence), but not with LCMV variant strains or the cross-reactive Pichinde virus (which exhibits sequence similarity with the self LCMV protein). The ability of LCMV infection to induce diabetes in RIP-LCMV mice depends on virus replication in CD11c+ dendritic cells (DCs) that are refractory to type I IFN production due to Usp18 expression [22]. Type I IFNs are innate signalling molecules, and include IFNα and IFNβ. LCMV replication, LCMV-specific T cell expansion and diabetes incidence are reduced in the absence of CD11c+ DCs [22]. Interestingly, CD11c+ DC depletion or prevention of LCMV replication by ribavirin treatment in RIP-LCMV mice also diminishes serum IFNα production following LCMV infection [22]. Since Usp18-expressing CD11c+ DCs productively infected with LCMV would not produce IFNα, bystander CD11c+ DCs are the probable source of IFNα. Thus, both LCMV-specific T cell expansion and heightened type I IFN responses may contribute to diabetes progression. However, the inability of other viruses and non-replicating antigen to induce or accelerate diabetes in this model casts doubt on the ability of molecular mimicry alone to contribute to disease progression [22–24].
Potential mimics of islet autoantigens have been identified in several viruses, including the Enterovirus, group B coxsackievirus (CVB) and rotavirus [25, 26]. Peripheral blood mononuclear cells (PBMCs) from diabetic patients can concomitantly respond to an immunologically dominant peptide of glutamic acid decarboxylase 65 (GAD65), a known islet autoantigen, and a region of the CVB P2-C protein as a result of shared sequence similarity [25]. However, CVB4 does not induce diabetes in mice expressing the NOD-specific MHC II allele that allows GAD65 peptide presentation, but lacking all other NOD-specific diabetes susceptibility factors [16]. In addition, GAD65-specific T cell clones isolated from diabetic patients fail to respond to homologous CVB P2-C protein [27]. These data suggest that molecular mimicry alone is insufficient to induce diabetes onset. Similarly, human islet autoantigen-2 (IA-2)-specific CD4+ T cells previously stimulated with IA-2 peptide can be restimulated with a peptide mimic from the rotavirus protein VP7. The binding affinity of this VP7 peptide to high-risk HLA class II molecules is comparable with that of the related IA-2 peptide [28]. The authors postulate that this molecular mimicry may contribute to diabetes development synergistically with other mechanisms [28]. Compared with NOD mice, T cell receptor-transgenic 8.3-NOD mice develop diabetes more rapidly and have a larger reservoir of CD8+ T cells recognising the islet autoantigen islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP). Infection by Rhesus monkey rotavirus (RRV) in 8.3-NOD mice activates IGRP-specific CD8+ T cells independently of molecular mimicry [29]. Mimicry between IA-2 and RRV has not been investigated in mice. However, the potential RRV peptide mimic is not conserved in human rotaviruses, and MHC class II peptide presentation differs between mice and humans. We conclude that IA-2-specific molecular mimicry is unlikely to contribute to the accelerated diabetes onset observed following RRV infection in NOD mice. Overall, any accelerated diabetes onset by viruses through T cell molecular mimicry would seem subject to a strict set of important criteria that would be difficult to meet in non-transgenic animal models and humans.
Bystander activation
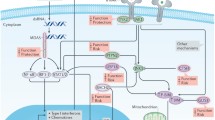
Bystander activation is characterised as T cell activation occurring independently of peptide presentation on MHCs to T cell receptors, or B cell activation without antigen recognition by the B cell receptor [30] (Fig. 1). The lack of antigen presentation on MHCs distinguishes bystander activation from pancreatic infection and molecular mimicry. It is distinct from bystander death, which requires direct pancreatic infection and the non-specific killing or damage of bystander beta cells by virus-specific T cells. Conversely, bystander activation involves DC activation by pattern recognition receptor (PRR) engagement and secretion of soluble cell-stimulating factors such as type I IFN (Fig. 1). These factors induce the activation of bystander lymphocytes, including autoreactive T cells, which contribute to beta cell death [31].

Bystander activation of lymphocytes by cytokine-secreting dendritic cells (DCs). (a) DCs (either conventional or plasmacytoid) encounter a stimulus, such as viral components, which interact with pattern recognition receptors (PRRs), such as Toll-like receptors, expressed either at the DC surface or intracellularly within endosomes. (b) This leads to DC maturation, shown by increased expression of MHC class I, MHC class II, CD80 and CD86. (c) Depending on the stimulus and PRRs, signalling pathways, including nuclear factor κB (NF-κB), are activated, ultimately resulting in the expression of proinflammatory cytokines. The cytokines produced depend on the DC subtype and signalling pathway activated, but may include type I IFNs and IL-12p70. (d) Heightened expression of MHC and co-factors also increases the ability of DCs to present antigen to CD4+ and CD8+ T cells. If this occurs in the PLN or islets where autoantigen accumulates, then autoantigen can be presented to and activate autoreactive T cells. (e) Secreted type I IFN and IL-12p70 directly induces non-specific bystander lymphocyte activation. This may include the activation and expansion of T cells, including autoreactive T cells, as determined by upregulation of activation markers, secretion of proinflammatory cytokines, such as TNF and IFNγ, or increases in cytolytic activity. Additionally, B cells may be activated, as shown by their elevated MHC class I, MHC class II, CD80 and CD86 expression, leading to increased autoantigen presentation to CD4+ and CD8+ T cells and further activation and expansion of autoreactive T cells
As bystander activation is expected to depend on a population of pre-existing autoreactive cells, it is likely to occur where autoreactive cells and cognate autoantigen accumulate, and depend critically on the extent of autoimmunity (i.e. age). This explains why bystander activation in type 1 diabetes is pancreas-specific rather than systemic. Autoreactive cells accumulate with age in the PLN and pancreas of NOD mice [32], the PLN being an important site for priming with autoantigen [33]. Autoreactive T cells are present in human pancreas [34] and can be expanded from the PLN of diabetic patients [35]. Whether autoreactive T cells accumulate at these sites during human diabetes development or are present prior to initial beta cell damage is unknown. Bystander activation might occur independently of pancreatic infection and instead contribute to T cell activation in the PLN [33]. These findings could help explain why virus modulation of diabetes is not always associated with virus presence in the pancreas, and can depend on mouse age at infection [7, 16, 36]. The requirement for pre-existing autoreactive cells also suggests that bystander activation accelerates disease progression rather than initiating beta cell damage. Pancreatic virus infection or other responses, such as beta cell stress or dysfunction, may produce such damage. Murine physiological beta cell death triggers autoantigen presentation by DCs and is required for subsequent innate immune cell activation and IFNα secretion in NOD mice [37, 38].
Studies increasingly address bystander activation as a possible mechanism triggered by virus infection to accelerate diabetes onset. Although most are animal studies, bystander activation in humans is feasible. Whilst some points may be unresolved, the data implicate cytokine-secreting DCs and PRRs. Although direct evidence is lacking to date, this area of research is worthy of further consideration.
Do cytokine-secreting DCs contribute to diabetes development?
DCs are a diverse population of innate immune cells with functionally distinct subsets. In the context of type 1 diabetes, their functionality varies with age and stage of autoimmune development [39]. This suggests that DCs might contribute to both delayed and accelerated diabetes onset. In broad terms there are two main DC types. Conventional DCs (cDCs) commonly present antigen on MHCs to T cells but can also express high levels of IL-12p70. This active form of IL-12 is a heterodimer of IL-12p35 and IL-12p40, encoded by IL-12A and IL-12B, respectively. IL-12p70 is a T cell-stimulating factor that enhances proinflammatory cytokine expression and favours cytotoxic T cell activity [40]. The expression of HLA-DR/MHC II and CD11c can be used to identify cDCs in humans and mice. Plasmacytoid DCs (pDCs) produce and secrete high levels of type I IFNs upon activation, which possess potent anti-viral activity and help shape innate and adaptive immune responses [41]. Murine pDCs can be classified by their low to intermediate levels of expression of MHC II and CD11c and co-expression of CD45R and plasmacytoid dendritic cell antigen-1 (PDCA-1). Human pDCs are characterised as HLA-DR+CD11c− with co-expression of blood dendritic cell antigen-2 (BDCA-2), blood dendritic cell antigen-4 (BDCA-4) and CD123.
Ablation of all cDC subsets in NOD mice reduces the insulitis and T cell activation induced following adoptive transfer of diabetogenic CD4+ T cells, suggesting that the cDC population plays a role in inducing disease [42]. However, expanding CD8+ cDCs alone protects mice from diabetes [43]. This implies the existence of functional diversity in the contributions of cDC subsets to diabetes development. NOD mouse cDCs are considered to contribute to diabetes primarily by capturing autoantigens in the PLN and presenting them to autoreactive T cells. However, these cDCs also secrete IL-12p70 to a greater extent than cDCs from non-diabetes-prone BALB/c mice [44]. In NOD mice, IL-12p70 is dispensable for diabetes development, whereas IL-12p70 treatment of mice with established insulitis accelerates diabetes onset [45, 46]. Thus, IL-12p70 probably influences the rate of progression to diabetes but is redundant for disease onset. No consistent correlation between cDC number or frequency and diabetes development has been observed in humans, although a polymorphism in IL-12B is associated with onset [47–49]. Therefore, cDCs may contribute to diabetes by presenting autoantigen and secreting IL-12p70.
Diabetes development in NOD mice is associated with increased numbers of IFNα-producing pDCs in the PLN at 3–4 weeks of age [50]. This temporally correlates with elevated levels of type I IFN-induced genes in islets [51]. Transient blockade of the type I IFN receptor, pDC depletion or blockade of pDC IFNα expression prior to insulitis development greatly reduces diabetes incidence [38, 50, 52]. IFNα production by pDCs in NOD mice requires B-1a cells and neutrophils and is associated with functionally impaired macrophages, accumulation of DNA complexes and activation of the PRR, Toll-like receptor (TLR) 9 [38]. When insulitis is established, type I IFN responses again peak in the pancreas but the pDCs appear to be tolerogenic [42, 51, 53]. Interestingly, NOD mice lacking a functional type I IFN receptor develop insulitis and diabetes similarly to wild-type NOD mice [51]. This suggests that type I IFN affects diabetes onset only if transiently induced at critical points during diabetes development. However, it remains possible that a transient blockade in type I IFN responses may trigger other unknown but beneficial modifications. Like cDCs, pDCs may play a dual role in diabetes development. For pDCs this duality may involve autoantigen presentation and CD4+ T cell activation [47]. Blood of at-risk and diabetic patients shows either increased or decreased numbers or frequencies of pDCs compared with controls. However, earlier reports of reduced pDC levels in diabetic patients may result from technical issues [47, 48, 54, 55]. Furthermore, diabetes development is preceded by the development of a transient type I IFN signature [56, 57].
Role of PRRs in diabetes development
DCs use PRRs such as TLRs to recognise pathogen-associated molecular patterns, including lipopolysaccharide and nucleic acids. Signalling through these receptors predominantly leads to activation of the nuclear factor κB (NF-κB) pathway and production of proinflammatory cytokines such as type I IFNs. These responses are critical for clearance of bacterial and virus infections [41], and multiple studies have suggested that these pathways contribute to type 1 diabetes development [58–60]. Therefore, infections that trigger specific PRRs might also inadvertently alter autoimmune responses.
Apart from TLR3, all TLRs commonly signal through the adaptor protein known as myeloid differentiation primary response protein 88 (MyD88) [61]. Some TLRs, including TLR2 and TLR4, are found on the cell surface, while others, like TLR3, TLR7 and TLR8, are located within intracellular endosomes. TLR2, TLR3 and TLR4, which recognise lipopeptides, double-stranded RNA and bacterial lipopolysaccharide, are mainly expressed in cDCs. In contrast, TLR7 and TLR9, which detect single-stranded RNA and DNA, are predominantly found in pDCs. This explains the biases of cDC and pDC responses towards bacterial and virus infection, respectively.
NOD mice lacking MyD88 are completely protected from diabetes [11]. This protection depends on the presence of commensal bacteria, as MyD88 knockout NOD mice bred under germ-free conditions still develop diabetes [11]. Importantly, as knockout of MyD88 in NOD mice under specific-pathogen-free conditions alters the intestinal microflora, altered abundance of a particular bacterial species may contribute to diabetes protection. Although an exact mechanism for this protection is not yet identified, this finding shows the importance of MyD88-dependent responses to intestinal microorganisms for autoimmune progression but not initiation in NOD mice. Interestingly, MyD88-dependent autoimmune activation is localised to the PLN in mice housed under specific-pathogen-free conditions [11].
Blockade of specific TLRs has variable affects on diabetes development. NOD mice lacking TLR3 or TLR4 remain susceptible to diabetes [11]. However, Tlr2 knockout can delay diabetes onset [62]. Tlr9 knockout NOD mice show reduced IFNα expression, pDC numbers and autoreactive CD8+ T cells, with delayed diabetes onset, compared with heterozygous littermates [62]. Although diabetes development has not been assessed in Tlr7 knockout NOD mice, treatment of 8.3-NOD mice with the TLR7 antagonist IRS661 inhibits diabetes onset [63]. Further supporting a role for endosomal TLRs, the prevention of endosomal acidification delays diabetes onset in NOD mice [62]. Human polymorphisms in genes encoding TLR2 and TLR3 are associated with an elevated risk of diabetes [64]. However, a role for TLR7 or TLR9 in human diabetes development has not been documented.
The converse of TLR blockade, TLR stimulation, can contribute to disease progression. For example, activation of TLR3 following administration of the double-stranded RNA viral mimic polyinosinic-polycytidylic acid can induce diabetes in BALB/c mice given insulin self-peptide [65]. Furthermore, TLR7 or TLR9 activation by agonist treatment accelerates diabetes onset in NOD mice [62, 63]. Neither TLR2 nor TLR9 appears to affect type I IFN-dependent signalling in the pancreas. However, CD8+ T cell activation and IFNα production in the PLN can be induced in NOD and 8.3-NOD mice following stimulation of TLR7 or TLR9 [51, 62, 63]. Diabetes still occurs, in some cases delayed, in single-TLR-knockout mice, suggesting a potential redundancy of these receptors for diabetes development. However, signalling through these pathways in the context of an infection might accelerate diabetes onset.
Importantly, heightened pDC responses to TLR stimulation are observed in NOD mice but not in diabetes-resistant C57BL/6 mice [66]. A similar increase in pDC-dependent IFNα secretion in response to TLR stimulation is found in PBMCs from diabetic patients compared with healthy controls [48]. Additionally, TLR-activated human pDCs from diabetic patients have a greater capacity than control pDCs to induce the differentiation of naive CD4+ T cells into IFNγ-secreting CD4+ T cells [48]. Together, these results suggest that humans and mice pre-disposed to diabetes may be more likely to show a heightened response to TLR stimulation, in particular, TLR7 or TLR9. Interestingly, most viruses linked to diabetes development contain RNA genomes (e.g. CVB and rotavirus). Moreover, several signal via TLR7, suggesting a potential link between viral induction of pDC-mediated IFNα expression and the ability of these viruses to accelerate diabetes onset.
The cytoplasmic receptors, melanoma differentiation-associated protein 5 (MDA-5) and retinoic acid-inducible gene 1 (RIG-I), also detect viral RNA. MDA-5 recognises long double-stranded RNA segments while RIG-I recognises short double-stranded RNA and single-stranded RNA molecules. Both receptors signal through the mitochondrial antiviral-signalling (MAVS) protein and are expressed by human islets [67]. Polymorphisms in the MDA-5 gene (also known as IFIH1) in humans are associated with type 1 diabetes [68, 69]. One study found higher levels of MDA-5 expression in individuals with diabetes-susceptibility alleles over those with diabetes-resistance alleles [70]. Therefore, innate signalling pathways apart from those triggered by TLRs may also contribute to diabetes development.
How might bystander lymphocyte activation contribute to diabetes onset?
B and T cells play important roles in diabetes development in NOD mice [31, 71]. T cells inducing proinflammatory responses are considered to be the main effector cells contributing to beta cell destruction [72]. In NOD mice, T cells are primed in the PLN and potentially the mesenteric lymph nodes (MLN) prior to trafficking to the pancreas [33, 73]. Furthermore, B cells accumulate in the PLN and show increased activation in an age-dependent manner [74]. Although islet autoantibody production is associated with diabetes progression, there is increasing evidence to suggest that antigen presentation by B cells on MHC class I and MHC class II molecules to autoreactive T cells is critical for autoreactive T cell expansion and diabetes development in mice [75, 76]. Autoreactive T cells are detected in the pancreas of some type 1 diabetic patients and show a proinflammatory phenotype [34, 77]. Additionally, B cell depletion in newly diagnosed patients partially preserves beta cell function [78].
IL-12p70 and type I IFNs can directly influence the activation and expansion of T cells. IL-12p70 secretion, together with high CD86 expression by TLR-activated DCs, is known to induce the development of IFNγ-secreting T helper-1 cells [79]. In concert with appropriate T cell receptor and co-stimulatory signals, type I IFN also contributes to the expansion and differentiation of CD8+ T cells [41]. This implies that the presence of autoantigen at the site of bystander activation would be critical for autoreactive CD8+ T cell activation. Virus-induced bystander activation in the PLN and pancreas in the presence of autoantigen would favour autoreactive CD8+ T cell activation. In the absence of autoantigen, type I IFN would promote an antiviral state rather than trigger cell expansion. This may also explain why virus-induced diabetes modulation seems highly dependent on the timing of infection, as it is considered that autoantigen and autoreactive T cells progressively accumulate in the PLN and pancreas as autoimmunity develops [33]. Type I IFNs can also directly increase the ability of B cells to present antigen to T cells, promote B cell survival and differentiation and increase expression of endogenous TLR7 [80]. Thus, pDC-dependent type I IFN expression, particularly within the PLN, may increase autoantigen presentation, responses of B cells to viruses and antibody production. Overall, bystander lymphocytes, activated by exposure to proinflammatory cytokines secreted by DC, are likely to contribute to accelerated diabetes onset.
Viruses that may trigger bystander activation to accelerate diabetes development
CVB
This single-stranded RNA virus spreads via the faecal–oral route to cause a diverse range of disease symptoms. CVB strains have a long-standing history of association with type 1 diabetes [5, 81]. In humans, infection is associated with either an increased risk (as with CVB1 infection) or a decreased risk (as with CVB3 and CVB6 infection) [82, 83]. CVB infects isolated human and murine beta cells [84–86]. In human islets, beta cell infection is associated with IFNα production [17]. However, in this context, type I IFN expression within islets prevents CVB replication and rapid beta cell death [17]. In the blood of diabetic children, the detection of IFNα and CVB mRNA is also linked [87]. Stimulation of PBMCs from non-diabetes-prone individuals with some but not all human CVB1 isolates increases IFNα expression, as does stimulation of PBMCs from diabetic patients with CVB4 [48, 88]. The latter depends on the presence of pDCs [48]. Although PRRs are not specifically implicated in this process by these studies, it is probable that the pDC-dependent expression of IFNα is mediated by TLR7. MDA-5 is also important for induction of type 1 IFN responses following murine CVB infection [89]. Any link between human diabetes susceptibility and enhanced MDA-5 expression might lead to enhanced type 1 IFN responses to CVB infection in genetically susceptible children. Further analysis of this possibility is required.
The role of type I IFN in the acceleration of murine diabetes by CVB is less well understood. Strains of CVB1, CVB3 and CVB4 that productively infect islets in NOD mice with established insulitis are capable of diabetes acceleration [36, 90, 91]. For CVB4, this acceleration requires a threshold number of autoreactive T cells and therefore the presence of pre-existing autoimmunity [92]. Based on mouse studies, CVB4 is thought to induce diabetes not by inducing beta cell death or molecular mimicry, but instead by evoking beta cell damage, release of sequestered antigens and presentation of these antigens by resident macrophages to autoreactive T cells [14, 16, 93]. Type I IFN expression within islets prevents CVB replication and CVB-induced diabetes acceleration [94]. Little is known regarding CVB presence within PLN and the type I IFN response at this site following CVB infection. Thus, it is unknown whether type I IFN-mediated bystander activation following CVB infection in diabetes-prone hosts plays a role in diabetes acceleration by CVB.
Rotavirus
These viruses are a leading cause of gastroenteritis in young children and animals. The triple-layered, infectious rotavirus particle contains a genome comprising 11 double-stranded RNA segments. Rotavirus shows a natural tropism for the differentiated enterocytes of the small intestine, but also spreads extraintestinally and commonly causes viraemia in children and mouse models [95, 96]. In an Australian study of children with a high genetic risk of developing type 1 diabetes, a temporal correlation was observed between serum anti-rotavirus antibodies and the presence of antibodies against the islet autoantigens insulin, GAD65 and IA-2 [6]. More recently, rotavirus infection before 6 months of age in combination with exposure to cow’s milk was also associated with an increase in autoantibodies to GAD65 [97].
Infection of older NOD mice with established insulitis with the rotavirus strain RRV accelerates diabetes onset [7]. This acceleration is dependent on the presence of insulitis, as diabetes development is delayed or unaffected following RRV infection of infant and young adult NOD mice [98]. Diabetes acceleration by RRV is associated with a minimal degree of intestinal inflammation and is independent of pancreatic infection [7, 29]. Instead, it involves virus spread to the MLN and PLN, where virus associates with antigen presenting cells, including DCs, inducing cellular maturation [99]. Rotavirus has been detected within the MLN during human infection [100]. However, rotavirus presence in human PLN has not been reported to date. While RRV does not directly associate with B or T cells in NOD mice, these cells in the lymph nodes, islets and spleen show markers of increased activation [29]. Using an ex vivo model, we have demonstrated that rotavirus-exposed pDCs contribute to the activation of B and T cells, including autoreactive T cells, through TLR7 and type I IFN signalling [30]. These data indicate that rotavirus can induce bystander lymphocyte activation of NOD mouse cells. Whereas CVB induction of type I IFN is strain-specific in human PBMCs [48, 88], stimulation of murine lymphocytes by rotavirus seems independent of virus strain or replicative ability. Instead, RRV-mediated diabetes acceleration by bystander activation appears to depend on the ability of the virus to first spread to particular lymph nodes where autoreactive lymphocytes accumulate. Indeed, although it replicates in the intestine, the porcine rotavirus CRW-8 neither efficiently spreads to the MLN or PLN nor modulates type 1 diabetes onset in NOD mice [99]. Although these NOD mouse studies suggest that TLR7 is critical for bystander activation, signalling through MDA-5 plays a role in rotavirus-induced type I IFN expression [101]. Therefore, a role for enhanced signalling through MDA-5 in rotavirus acceleration of diabetes, like that proposed for CVB, cannot be ruled out.
Infection of non-diabetes-prone mice with murine rotavirus induces type I IFN-dependent B cell activation [102]. This suggests that type I IFN-dependent activation of autoreactive lymphocytes in NOD mice may occur and contribute to diabetes acceleration. Furthermore, rotavirus induces pDC-dependent activation of human B cells, suggesting that bystander activation by rotavirus might occur in humans [102]. Whether type I IFN responses are heightened in PBMCs of diabetic patients in response to rotavirus, as observed for CVB, remains to be determined. However, this hypothesis is supported by the observation that exposure to RRV ex vivo induces significantly greater activation of B cells from NOD mice than C57BL/6 mice [30].
Other viruses
Influenza A virus is the type species of the Influenzavirus A genus, family Orthomyxoviridae. These single-stranded RNA viruses primarily replicate in the respiratory tract. Influenza A viruses also replicate in isolated primary human pancreatic islets, and spread to the pancreas in turkeys, causing exocrine and endocrine tissue damage [103]. Recent studies identified a possible link between pandemic H1N1 influenza A virus infection and diabetes development in humans [104, 105]. However, their involvement in human diabetes remains speculative. A recent history of upper respiratory tract infections has been associated with a transient type I IFN response prior to diabetes development in children [56]. Additionally, influenza A virus induced significantly more pDC-dependent IFNα secretion by PBMCs from diabetic patients compared with PBMCs from healthy controls [48]. Higher IFNα secretion correlated with increased numbers of pDCs vs cDCs [48]. Other single-stranded RNA viruses, such as those causing rubella and mumps from the Paramyxoviridae family, have also been linked to childhood diabetes development [106]. Rubella and mumps viruses can infect and replicate in beta cells [107, 108]. Some evidence suggests that rubella virus may elicit antibody- or T cell-mediated molecular mimicry [109, 110]. It would be interesting to determine whether these viruses also induce heightened IFNα responses in diabetic patients, as this might represent a common mechanistic pathway for accelerated diabetes onset by such viruses.
Conclusions
Much current research is focused on virus detection within the pancreas of patients with type 1 diabetes. Detection at diabetes onset is likely to be a rare occurrence for many viruses. Furthermore, pancreatic virus may not necessarily indicate a role in diabetes, as this may result from increased susceptibility to infection owing to pancreatic injury. Despite shortcomings, pancreatic virus detection is important for understanding virus-induced pancreatic cell death and inflammation. Bystander activation as a mechanism for diabetes acceleration by viruses is influenced by the timing and location of virus infection and may operate remotely from the pancreas. This would help explain why viruses are not always detectable in the pancreas and potentially allow identification of additional diabetogenic viruses and their infections. Future human studies should determine whether autoreactive T cells accumulate and become activated in the PLN and other remote sites, as well as the pancreas, and attempt to identify the role of bystander activation at these sites in diabetes development. More efforts should be directed towards virus detection and inflammation analysis in the PLN and other remote sites in humans progressing towards diabetes. As bystander activation is non-specific and does not require presentation of peptide on MHCs, it may represent a common mechanism employed by multiple viruses. Therefore, unbiased virus detection studies should be conducted in patients and potential links between the immune responses induced by these viruses should be identified. Evidence indicates that diabetes-prone mice and humans may be more susceptible to these bystander responses to viruses, supporting a role for the interaction between virus infection and genetic susceptibility. Therefore, understanding the phenotype of specific genetic variants, such as the MDA-5 polymorphisms associated with diabetes, may allow identification of important virus-specific immune responses that are heightened in at-risk patients. Overall, future studies should investigate the possibility that infections inducing bystander lymphocyte activation may contribute to diabetes progression in at-risk children.
Abbreviations
- CVB:
-
Group B coxsackievirus
- cDC:
-
Conventional dendritic cell
- DC:
-
Dendritic cell
- IA-2:
-
Islet autoantigen-2
- LCMV:
-
Lymphocytic choriomeningitis virus
- MDA-5:
-
Melanoma differentiation-associated protein 5
- MLN:
-
Mesenteric lymph nodes
- MyD88:
-
Myeloid differentiation primary response protein 88
- PBMC:
-
Peripheral blood mononuclear cell
- pDC:
-
Plasmacytoid dendritic cell
- PLN:
-
Pancreatic lymph nodes
- PRR:
-
Pattern recognition receptor
- RIG-I:
-
Retinoic acid-inducible gene 1
- RRV:
-
Rhesus monkey rotavirus
- TLR:
-
Toll-like receptor
References
van Belle TL, Coppieters KT, von Herrath MG (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91:79–118
Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J (2003) Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes 52:1052–1055
Fourlanos S, Varney MD, Tait BD et al (2008) The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care 31:1546–1549
Knip M, Simell O (2012) Environmental triggers of type 1 diabetes. Cold Spring Harb Perspect Med 2:a007690
Yeung WC, Rawlinson WD, Craig ME (2011) Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ 342:d35
Honeyman MC, Coulson BS, Stone NL et al (2000) Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 49:1319–1324
Graham KL, Sanders N, Tan Y, Allison J, Kay TW, Coulson BS (2008) Rotavirus infection accelerates type 1 diabetes in mice with established insulitis. J Virol 82:6139–6149
Tracy S, Drescher KM (2007) Coxsackievirus infections and NOD mice: relevant models of protection from, and induction of, type 1 diabetes. Ann N Y Acad Sci 1103:143–151
Anderson MS, Bluestone JA (2005) The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 23:447–485
Lee AS, Gibson DL, Zhang Y, Sham HP, Vallance BA, Dutz JP (2010) Gut barrier disruption by an enteric bacterial pathogen accelerates insulitis in NOD mice. Diabetologia 53:741–748
Wen L, Ley RE, Volchkov PY et al (2008) Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455:1109–1113
Schneider DA, von Herrath MG (2014) Potential viral pathogenic mechanism in human type 1 diabetes. Diabetologia 57:2009–2018
Coppieters KT, Wiberg A, von Herrath MG (2012) Viral infections and molecular mimicry in type 1 diabetes. APMIS 120:941–949
Horwitz MS, Ilic A, Fine C, Balasa B, Sarvetnick N (2004) Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin Immunol 110:134–144
Yoon JW, McClintock PR, Onodera T, Notkins AL (1980) Virus-induced diabetes mellitus. XVIII. Inhibition by a nondiabetogenic variant of encephalomyocarditis virus. J Exp Med 152:878–892
Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N (1998) Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med 4:781–785
Chehadeh W, Kerr-Conte J, Pattou F et al (2000) Persistent infection of human pancreatic islets by coxsackievirus B is associated with α interferon synthesis in beta cells. J Virol 74:10153–10164
Krogvold L, Edwin B, Buanes T et al (2014) Detection of a low-grade enteroviral infection in the islets of Langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes. doi:10.2337/db14-1370
Oikarinen M, Tauriainen S, Oikarinen S et al (2012) Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes 61:687–691
Mercalli A, Lampasona V, Klingel K et al (2012) No evidence of enteroviruses in the intestine of patients with type 1 diabetes. Diabetologia 55:2479–2488
Ohashi PS, Oehen S, Buerki K et al (1991) Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 65:305–317
Honke N, Shaabani N, Zhang DE et al (2013) Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PLoS Pathog 9:e1003650
Christen U, Edelmann KH, McGavern DB et al (2004) A viral epitope that mimics a self antigen can accelerate but not initiate autoimmune diabetes. J Clin Invest 114:1290–1298
Sevilla N, Homann D, von Herrath M et al (2000) Virus-induced diabetes in a transgenic model: role of cross-reacting viruses and quantitation of effector T cells needed to cause disease. J Virol 74:3284–3292
Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK (1994) Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest 94:2125–2129
Honeyman MC, Stone NL, Harrison LC (1998) T cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA-2: potential for mimicry with rotavirus and other environmental agents. Mol Med 4:231–239
Schloot NC, Willemen SJ, Duinkerken G, Drijfhout JW, de Vries RR, Roep BO (2001) Molecular mimicry in type 1 diabetes mellitus revisited: T cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Hum Immunol 62:299–309
Honeyman MC, Stone NL, Falk BA, Nepom G, Harrison LC (2010) Evidence for molecular mimicry between human T cell epitopes in rotavirus and pancreatic islet autoantigens. J Immunol 184:2204–2210
Pane JA, Webster NL, Zufferey C, Coulson BS (2014) Rotavirus acceleration of murine type 1 diabetes is associated with increased MHC class I-restricted antigen presentation by B cells and elevated proinflammatory cytokine expression by T cells. Virus Res 179:73–84
Pane JA, Webster NL, Coulson BS (2014) Rotavirus activates lymphocytes from non-obese diabetic mice by triggering toll-like receptor 7 signaling and interferon production in plasmacytoid dendritic cells. PLoS Pathog 10:e1003998
Phillips JM, Parish NM, Bland C, Sawyer Y, de la Pena H, Cooke A (2009) Type 1 diabetes development requires both CD4+ and CD8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev Diabet Stud 6:97–103
Trudeau JD, Kelly-Smith C, Verchere CB et al (2003) Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 111:217–223
Gagnerault MC, Luan JJ, Lotton C, Lepault F (2002) Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med 196:369–377
Coppieters KT, Dotta F, Amirian N et al (2012) Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 209:51–60
Kent SC, Chen Y, Bregoli L et al (2005) Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 435:224–228
Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S (2004) Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology 329:381–394
Turley S, Poirot L, Hattori M, Benoist C, Mathis D (2003) Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med 198:1527–1537
Diana J, Simoni Y, Furio L et al (2013) Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med 19:65–73
Van Belle TL, Juntti T, Liao J, von Herrath MG (2010) Pre-existing autoimmunity determines type 1 diabetes outcome after Flt3-ligand treatment. J Autoimmun 34:445–452
Hamza T, Barnett JB, Li B (2010) Interleukin 12 a key immunoregulatory cytokine in infection applications. Int J Mol Sci 11:789–806
Stetson DB, Medzhitov R (2006) Type I interferons in host defense. Immunity 25:373–381
Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD (2007) The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol 179:5041–5053
O’Keeffe M, Brodnicki TC, Fancke B et al (2005) Fms-like tyrosine kinase 3 ligand administration overcomes a genetically determined dendritic cell deficiency in NOD mice and protects against diabetes development. Int Immunol 17:307–314
Poligone B, Weaver DJ Jr, Sen P, Baldwin AS Jr, Tisch R (2002) Elevated NF-κB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J Immunol 168:188–196
Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L (1995) Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med 181:817–821
Trembleau S, Penna G, Gregori S et al (1999) Pancreas-infiltrating Th1 cells and diabetes develop in IL-12-deficient nonobese diabetic mice. J Immunol 163:2960–2968
Allen JS, Pang K, Skowera A et al (2009) Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T cells through immune complex capture. Diabetes 58:138–145
Xia CQ, Peng R, Chernatynskaya AV et al (2014) Increased IFN-α-producing plasmacytoid dendritic cells (pDCs) in human Th1-mediated type 1 diabetes: pDCs augment Th1 responses through IFN-α production. J Immunol 193:1024–1034
Morahan G, Huang D, Ymer SI et al (2001) Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet 27:218–221
Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO (2008) Interferon-α initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A 105:12439–12444
Quah HS, Miranda-Hernandez S, Khoo A et al (2014) Deficiency in type I interferon signaling prevents the early interferon-induced gene signature in pancreatic islets but not type 1 diabetes in NOD mice. Diabetes 63:1032–1040
Li Q, McDevitt HO (2011) The role of interferon α in initiation of type I diabetes in the NOD mouse. Clin Immunol 140:3–7
Welzen-Coppens JM, van Helden-Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA (2013) The kinetics of plasmacytoid dendritic cell accumulation in the pancreas of the NOD mouse during the early phases of insulitis. PLoS One 8:e55071
Chen X, Makala LH, Jin Y et al (2008) Type 1 diabetes patients have significantly lower frequency of plasmacytoid dendritic cells in the peripheral blood. Clin Immunol 129:413–418
Vuckovic S, Withers G, Harris M et al (2007) Decreased blood dendritic cell counts in type 1 diabetic children. Clin Immunol 123:281–288
Ferreira RC, Guo H, Coulson RM et al (2014) A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 63:2538–2550
Kallionpaa H, Elo LL, Laajala E et al (2014) Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes 63:2402–2414
Zhao Y, Krishnamurthy B, Mollah ZU, Kay TW, Thomas HE (2011) NF-κB in type 1 diabetes. Inflamm Allergy Drug Targets 10:208–217
Zipris D (2010) Toll-like receptors and type 1 diabetes. Adv Exp Med Biol 654:585–610
Devendra D, Eisenbarth GS (2004) Interferon α—a potential link in the pathogenesis of viral-induced type 1 diabetes and autoimmunity. Clin Immunol 111:225–233
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Zhang Y, Lee AS, Shameli A et al (2010) TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes. J Immunol 184:5645–5653
Lee AS, Ghoreishi M, Cheng WK, Chang TY, Zhang YQ, Dutz JP (2011) Toll-like receptor 7 stimulation promotes autoimmune diabetes in the NOD mouse. Diabetologia 54:1407–1416
Trejo-de la OA, Hernandez-Sancen P, Maldonado-Bernal C (2014) Relevance of single-nucleotide polymorphisms in human TLR genes to infectious and inflammatory diseases and cancer. Genes Immun 15:199–209
Moriyama H, Wen L, Abiru N et al (2002) Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci U S A 99:5539–5544
Peng RH, Paek E, Xia CQ, Tennyson N, Clare-Salzler MJ (2006) Heightened interferon-α/β response causes myeloid cell dysfunction and promotes T1D pathogenesis in NOD mice. Ann N Y Acad Sci 1079:99–102
Hultcrantz M, Huhn MH, Wolf M et al (2007) Interferons induce an antiviral state in human pancreatic islet cells. Virology 367:92–101
Nejentsev S, Walker N, Riches D, Egholm M, Todd JA (2009) Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324:387–389
Smyth DJ, Cooper JD, Bailey R et al (2006) A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 38:617–619
Liu S, Wang H, Jin Y et al (2009) IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet 18:358–365
Hu CY, Rodriguez-Pinto D, Du W et al (2007) Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest 117:3857–3867
Katz JD, Benoist C, Mathis D (1995) T helper cell subsets in insulin-dependent diabetes. Science 268:1185–1188
Jaakkola I, Jalkanen S, Hanninen A (2003) Diabetogenic T cells are primed both in pancreatic and gut-associated lymph nodes in NOD mice. Eur J Immunol 33:3255–3264
Marino E, Batten M, Groom J et al (2008) Marginal-zone B cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T cells. Diabetes 57:395–404
Noorchashm H, Lieu YK, Noorchashm N et al (1999) I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol 163:743–750
Marino E, Tan B, Binge L, Mackay CR, Grey ST (2012) B cell cross-presentation of autologous antigen precipitates diabetes. Diabetes 61:2893–2905
Arif S, Tree TI, Astill TP et al (2004) Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest 113:451–463
Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H et al (2009) Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 361:2143–2152
Lichtenegger FS, Mueller K, Otte B et al (2012) CD86 and IL-12p70 are key players for T helper 1 polarization and natural killer cell activation by Toll-like receptor-induced dendritic cells. PLoS One 7:e44266
Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A (2012) Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol 90:498–504
Gamble DR, Kinsley ML, FitzGerald MG, Bolton R, Taylor KW (1969) Viral antibodies in diabetes mellitus. BMJ 3:627–630
Oikarinen S, Tauriainen S, Hober D et al (2014) Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes 63:655–662
Laitinen OH, Honkanen H, Pakkanen O et al (2014) Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes 63:446–455
Yoon JW, Onodera T, Jenson AB, Notkins AL (1978) Virus-induced diabetes mellitus. XI. Replication of coxsackie B3 virus in human pancreatic beta cell cultures. Diabetes 27:778–781
Nair S, Leung KC, Rawlinson WD, Naing Z, Craig ME (2010) Enterovirus infection induces cytokine and chemokine expression in insulin-producing cells. J Med Virol 82:1950–1957
Yoon JW, Onodera T, Notkins AL (1978) Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with coxsackie virus B4. J Exp Med 148:1068–1080
Chehadeh W, Weill J, Vantyghem MC et al (2000) Increased level of interferon-α in blood of patients with insulin-dependent diabetes mellitus: relationship with coxsackievirus B infection. J Infect Dis 181:1929–1939
Hamalainen S, Nurminen N, Ahlfors H et al (2014) Coxsackievirus B1 reveals strain specific differences in plasmacytoid dendritic cell mediated immunogenicity. J Med Virol 86:1412–1420
Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW (2010) MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol 84:254–260
Larsson PG, Lakshmikanth T, Laitinen OH et al (2015) A preclinical study on the efficacy and safety of a new vaccine against Coxsackievirus B1 reveals no risk for accelerated diabetes development in mouse models. Diabetologia 58:346–354
Serreze DV, Wasserfall C, Ottendorfer EW et al (2005) Diabetes acceleration or prevention by a coxsackievirus B4 infection: critical requirements for both interleukin-4 and gamma interferon. J Virol 79:1045–1052
Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA (2000) Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T cells in pancreatic islets. Diabetes 49:708–711
Horwitz MS, Ilic A, Fine C, Rodriguez E, Sarvetnick N (2002) Presented antigen from damaged pancreatic beta cells activates autoreactive T cells in virus-mediated autoimmune diabetes. J Clin Invest 109:79–87
Flodstrom M, Maday A, Balakrishna D, Cleary MM, Yoshimura A, Sarvetnick N (2002) Target cell defense prevents the development of diabetes after viral infection. Nat Immunol 3:373–382
Chiappini E, Azzari C, Moriondo M, Galli L, de Martino M (2005) Viraemia is a common finding in immunocompetent children with rotavirus infection. J Med Virol 76:265–267
Blutt SE, Kirkwood CD, Parreno V et al (2003) Rotavirus antigenaemia and viraemia: a common event? Lancet 362:1445–1449
Lempainen J, Tauriainen S, Vaarala O et al (2012) Interaction of enterovirus infection and cow’s milk-based formula nutrition in type 1 diabetes-associated autoimmunity. Diabetes Metab Res Rev 28:177–185
Graham KL, O’Donnell JA, Tan Y et al (2007) Rotavirus infection of infant and young adult nonobese diabetic mice involves extraintestinal spread and delays diabetes onset. J Virol 81:6446–6458
Pane JA, Webster NL, Graham KL, Holloway G, Zufferey C, Coulson BS (2013) Rotavirus acceleration of murine type 1 diabetes is associated with a T helper 1-dependent specific serum antibody response and virus effects in regional lymph nodes. Diabetologia 56:573–582
Lappalainen S, Ylitalo S, Arola A, Halkosalo A, Rasanen S, Vesikari T (2012) Simultaneous presence of human herpesvirus 6 and adenovirus infections in intestinal intussusception of young children. Acta Paediatr 101:663–670
Holloway G, Coulson BS (2013) Innate cellular responses to rotavirus infection. J Gen Virol 94:1151–1160
Deal EM, Lahl K, Narvaez CF, Butcher EC, Greenberg HB (2013) Plasmacytoid dendritic cells promote rotavirus-induced human and murine B cell responses. J Clin Invest 123:2464–2474
Capua I, Mercalli A, Pizzuto MS et al (2013) Influenza A viruses grow in human pancreatic cells and cause pancreatitis and diabetes in an animal model. J Virol 87:597–610
Watanabe N (2011) Conversion to type 1 diabetes after H1N1 influenza infection: a case report. J Diabetes 3:103
Nenna R, Papoff P, Moretti C et al (2011) Detection of respiratory viruses in the 2009 winter season in Rome: 2009 influenza A (H1N1) complications in children and concomitant type 1 diabetes onset. Int J Immunopathol Pharmacol 24:651–659
Ramondetti F, Sacco S, Comelli M et al (2012) Type 1 diabetes and measles, mumps and rubella childhood infections within the Italian Insulin-dependent Diabetes Registry. Diabet Med 29:761–766
Prince GA, Jenson AB, Billups LC, Notkins AL (1978) Infection of human pancreatic beta cell cultures with mumps virus. Nature 271:158–161
Numazaki K, Goldman H, Wong I, Wainberg MA (1989) Infection of cultured human fetal pancreatic islet cells by rubella virus. Am J Clin Pathol 91:446–451
Karounos DG, Wolinsky JS, Thomas JW (1993) Monoclonal antibody to rubella virus capsid protein recognizes a beta-cell antigen. J Immunol 150:3080–3085
Ou D, Mitchell LA, Metzger DL, Gillam S, Tingle AJ (2000) Cross-reactive rubella virus and glutamic acid decarboxylase (65 and 67) protein determinants recognised by T cells of patients with type I diabetes mellitus. Diabetologia 43:750–762
Funding
Work in BSC’s laboratory is supported by Project Grants (APP1044868 and APP1085596) and Senior Research Fellowship ID628319 to BSC from the National Health and Medical Research Council of Australia.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
Both authors were responsible for the conception of the manuscript, its revision and its evaluation for important intellectual content. JP drafted and wrote the manuscript, and both authors approved the final version.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pane, J.A., Coulson, B.S. Lessons from the mouse: potential contribution of bystander lymphocyte activation by viruses to human type 1 diabetes. Diabetologia 58, 1149–1159 (2015). https://doi.org/10.1007/s00125-015-3562-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3562-3