
Abstract
Aims/hypothesis
Diabetic retinopathy is a serious complication of diabetes mellitus and can lead to blindness. A genetic component, in addition to traditional risk factors, has been well described although strong genetic factors have not yet been identified. Here, we aimed to identify novel genetic risk factors for sight-threatening diabetic retinopathy using a genome-wide association study.
Methods
Retinopathy was assessed in white Australians with type 2 diabetes mellitus. Genome-wide association analysis was conducted for comparison of cases of sight-threatening diabetic retinopathy (n = 336) with diabetic controls with no retinopathy (n = 508). Top ranking single nucleotide polymorphisms were typed in a type 2 diabetes replication cohort, a type 1 diabetes cohort and an Indian type 2 cohort. A mouse model of proliferative retinopathy was used to assess differential expression of the nearby candidate gene GRB2 by immunohistochemistry and quantitative western blot.
Results
The top ranked variant was rs3805931 with p = 2.66 × 10−7, but no association was found in the replication cohort. Only rs9896052 (p = 6.55 × 10−5) was associated with sight-threatening diabetic retinopathy in both the type 2 (p = 0.035) and the type 1 (p = 0.041) replication cohorts, as well as in the Indian cohort (p = 0.016). The study-wide meta-analysis reached genome-wide significance (p = 4.15 × 10−8). The GRB2 gene is located downstream of this variant and a mouse model of retinopathy showed increased GRB2 expression in the retina.
Conclusions/interpretation
Genetic variation near GRB2 on chromosome 17q25.1 is associated with sight-threatening diabetic retinopathy. Several genes in this region are promising candidates and in particular GRB2 is upregulated during retinal stress and neovascularisation.
Similar content being viewed by others

Introduction
Diabetic retinopathy is the most common cause of blindness in developed countries in patients under 75 years of age [1]. It is a common complication of both type 1 and type 2 diabetes mellitus. The overall prevalence of any retinopathy among patients with diabetes is 40.3%, with a prevalence of 8.2% for sight-threatening diabetic retinopathy [2]. The prevalence of diabetes itself is predicted to double between the years 2000 and 2030 [3], and thus the health burden of diabetic retinopathy is also expected to increase dramatically.
Diabetic retinopathy is defined by the presence of retinal microvascular lesions, including microaneurysms, retinal haemorrhages, lipid exudates, nerve fibre layer infarcts, intraretinal microvascular abnormalities and venous beading, collectively termed non-proliferative diabetic retinopathy [4]. The growth of abnormal new blood vessels is termed proliferative diabetic retinopathy [4]. Macular oedema is also a major cause of vision loss in diabetes and may occur in both non-proliferative and proliferative diabetic retinopathy [4].
It is well known that poor glycaemic control is a risk factor for diabetic retinopathy [5, 6]; however, tight control does not fully prevent the disease and intensive diabetic control can result in increased mortality [7]. Several studies have demonstrated familial clustering of diabetic retinopathy independent of glycaemic control [8–11]. The Family Investigation of Nephropathy and Diabetes (FIND) FIND-Eye study reported a significant heritability of 0.27 for diabetic retinopathy in families [12] and a study of a large cohort of American families with type 1 diabetes confirmed a strong familial risk for diabetic retinopathy, independent of duration of disease [13]. Numerous potentially associated genes have been reported in the literature [14] but few consistent findings have been reported.
Three recent genome-wide association studies (GWAS) for diabetic retinopathy have reported loci of interest that did not meet the statistical thresholds for genome-wide significance [15–17]. A study in a cohort of Taiwanese type 2 diabetes patients and a meta-analysis of Japanese patients identified several loci reaching significance, but these findings have not been validated in an independent cohort [18, 19]. None of the top loci in these studies appear to be replicated by other studies. Thus, no GWAS has yet identified robust genetic associations for diabetic retinopathy.
Here, we aimed to identify novel genetic risk loci for sight-threatening diabetic retinopathy using a GWAS in patients of European descent with type 2 diabetes and to demonstrate replication of the association in patients of the same ethnicity with type 1 and type 2 diabetes mellitus and in an Indian type 2 diabetes mellitus cohort.
Methods
Participant recruitment
This study was approved by human research ethics committees of the relevant institutions, as listed in the electronic supplementary material (ESM). Written informed consent was obtained from each participant and the project conformed to the tenets of the Declaration of Helsinki.
Australian participants in the discovery cohort were recruited from the locations listed in the ESM between 2006 and 2011, as previously described [20]. Replication cohorts were recruited between 2011 and 2013 from these sites, as well as from additional sites listed in the ESM.
Discovery cohort
Patients of at least 18 years of age who were receiving medical treatment for type 2 diabetes mellitus for at least 5 years were invited to participate, as previously described [20]. Retinopathy status was determined from direct ophthalmic examination by the treating ophthalmologist and was graded according to modified Early Treatment in Diabetic Retinopathy Study (ETDRS) criteria [21]. Cases were defined as participants with sight-threatening diabetic retinopathy (severe non-proliferative diabetic retinopathy, proliferative diabetic retinopathy or clinically significant macular oedema) and were compared with controls with no diabetic retinopathy or minimal diabetic retinopathy. All participants meeting the inclusion criteria for case or control status with DNA available were included in the current analysis. Medical histories were obtained via a questionnaire. Biochemistry results were collected, including HbA1c, serum lipids and renal function tests. A detailed description of all clinical variables collected has been previously published [20]. DNA was extracted from whole blood using QIAamp Blood DNA Maxi Kits (Qiagen, Chadstone Centre, VIC, Australia).
Clinical traits were assessed for association with diabetic retinopathy in SPSS (v19.0, SPSS, Chicago, IL, USA) using a χ 2 test for dichotomous variables and binary logistic regression for quantitative variables.
Replication cohorts
The type 1 and type 2 diabetes mellitus white replication cohorts were recruited from Australian hospitals and Moorfields Eye Hospital London, UK, under an identical protocol to the discovery cohort. The white type 2 replication cohort consisted of 263 cases and 320 controls under the identical definition as the discovery cohort. In the type 1 cohort, cases were defined as participants with any severity of retinopathy in order to include as large a cohort as possible for this analysis. In total, 242 cases and 126 controls were included.
The Indian replication cohort was recruited from Aravind Eye Hospital in Tamil Nadu, India. Participants all had type 2 diabetes mellitus of at least 10 years duration. Clinical data were collected and diabetic retinopathy was classified according to ETDRS criteria on examination. Cases were included if they had sight-threatening diabetic retinopathy using the same definition as the white discovery cohort. In total, 334 cases and 365 controls were included in the Indian replication cohort. DNA was extracted from whole blood using the salt precipitation method [22].
SNP genotyping and association analysis
Genome-wide genotyping at single nucleotide polymorphisms (SNPs) of the discovery cohort was conducted using Human OmniExpress Beadchips (Illumina, San Diego, CA, USA) on a HiScan System (Illumina). Genotyping data were filtered to remove SNPs with genotyping rates <99%, minor allele frequencies <1% or Hardy–Weinberg equilibrium in controls p < 1 × 10−6. Relatedness between individuals was assessed using PLINK software (http://pngu.mgh.harvard.edu/~purcell/plink/) [23] and one of each pair of individuals with a PI-HAT >0.3 was excluded from further analysis (10 individuals). Principal components analysis was conducted using EIGENSOFT (https://data.broadinstitute.org/alkesgroup/EIGENSOFT/?) [24]. Ancestral outliers were identified by EIGENSOFT as any eigenvalue >6 SD from the mean and from visualising plots of principal components 1 and 2, and these were excluded from further analysis.
Association analyses were conducted using PLINK software [23]. Logistic regression under an additive model with adjustment for covariates (age, sex, duration of diabetes, HbA1c, hypertension, nephropathy and the first three principal components) was used to test association of the 602,755 SNPs. Individuals with missing clinical data were excluded from the fully adjusted logistic regression model. The Manhattan plot was generated using R (www.r-project.org) with code adapted from https://github.com/stephenturner/qqman. The detailed locus specific plot was generated using LocusZoom (http://locuszoom.sph.umich.edu/locuszoom/) [25] with recombination rates taken from the European component of the 1000 Genomes Project [26].
The type 2 diabetes replication cohort was genotyped at SNPs of interest using iPlex Gold chemistry (Sequenom Inc, San Diego, CA, USA) visualised on a MassArray Analyzer (Sequenom Inc.). The type 1 replication cohort was typed on the Human OmniExpress Beadchips (Illumina Inc) in parallel with the discovery cohort. All SNPs with a p < 1 × 10−5 and a selection with p < 1 × 10−4 in the discovery cohort that were compatible with multiplexing on the MassArray platform were assessed in these two cohorts. Association analysis in the replication cohorts was conducted in PLINK under an allelic model as well as under logistic regression with adjustment for covariates. The type 1 replication cohort was subjected to the identical quality control procedures as the discovery cohort typed on the same SNP array. This cohort had an overall λ of 0.994, thus the principal components were not included as covariates in the analysis. Genotyping of the SNP of interest in the Indian replication cohort was conducted using a pre-designed TaqMan Assay (Life Technologies, Mulgrave, VIC, Australia) according to manufacturer’s protocols on an ABI 7900 HT Fast Real Time PCR System (Life Technologies) and analysis was conducted in PLINK under logistic regression with adjustment for all clinical covariates.
Meta-analyses of multiple cohorts were undertaken in METAL (http://csg.sph.umich.edu/abecasis/Metal/download/) with weighting for the number of people in each cohort [27] using the fully adjusted association statistics.
Sequencing of candidate genes
Seventy-two cases with the most severe retinopathy were selected from the discovery and type 2 replication cohorts for direct sequencing of the coding regions GRB2 (GenBank accession NM_002086.4) and MIR3678 (GenBank accession NR_037449.1) using standard Sanger sequencing as described in the ESM.
RNA extraction and RT-PCR for GRB2 and MIR3678
Cadaveric human retina was obtained from the Eye Bank of South Australia with approval from the Southern Adelaide Clinical Human Research Ethics Committee. RNA was extracted from homogenised tissue in TRIzol (Life Technologies) and DNA was removed using Turbo DNA-free kit (Life Technologies). cDNA was generated by reverse-transcription and transcripts amplified by PCR and visualised on 1% agarose as described in the ESM.
Immunohistochemical labelling in humans
Eye tissue was fixed in buffered formalin and embedded in paraffin. For immunolabelling, sections were blocked with 5% normal goat serum and incubated with the rabbit anti-GRB2 primary antibody (1:2,000, Abcam#32111; Abcam, Melbourne, VIC, Australia) at +4°C overnight. The bound GRB2 antibody was detected with the Novolink Polymer detection kit (Leica Microsystems, North Ryde, NSW, Australia) and Chromogen substrate colouration (Dako, North Sydney, NSW, Australia). Sections were counterstained with haematoxylin and mounted in dePeX (Merck, Frenchs Forest, NSW, Australia). Light microscopy was performed on Olympus BX50 brightfield upright microscope attached with a Q-Imaging colour CCD camera; images were taken using the Q-Capture software (Q-Imaging Corporate, Surrey, BC, Canada).
Transgenic mice with selective Müller cell ablation
This study was performed in accordance with the Association for Research in Vision and Ophthalmology statement and approved by The University of Sydney Animal Ethics Committee. The principles of laboratory animal care were followed. Male and female Rlbp1-CreER-DTA transgenic mice (Save Sight Institute, the University of Sydney, Sydney, NSW, Australia) were produced for selective and inducible Müller cell ablation as described previously [28]. Müller cell ablation was induced by daily intraperitoneal injection of tamoxifen (TMX, 3 mg/day, dissolved in sunflower oil) for 4 consecutive days as described previously [28]. Mice not carrying the Rlbp1 promoter but carrying the DTA176 gene and receiving TMX treatment in the same way were used as controls. Both Rlbp1-CreER-DTA and Rlbp1-CreER-LacZ mice carry mixed CBA/CaH and C57BL/6J genetic background, were kept in a 12 h dark and light cycle and fed with standard chow and water. Mice at 10–12 weeks of age were used in this study.
Western blotting
Proteins were extracted from retinas and their concentrations were determined by detergent compatible protein assay. Equal amounts of protein were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis then transferred to a polyvinylidene difluoride membrane. Membranes were probed with the rabbit anti-GRB2 primary antibody (1:1,000, Abcam no. 32111) or to glial fibrillary acidic protein (GFAP, goat polyclonal, 1:500, Abcam no. ab53554) and then incubated with a secondary antibody conjugated with horseradish peroxidase. Protein bands were visualised using the G:Box BioImaging system (Syngene, Frederick, MD, USA). α/β-Tubulin (rabbit polyclonal, 1:2,000; Cell Signalling Technology no. 2148; Genesearch, Arundel, QLD, Australia), served as a loading control. Quantitative analysis was conducted after normalisation of the protein band densitometry to α/β-tubulin.
Fluorescence immunolabelling in mice
Mouse eyes were fixed in 4% paraformaldehyde and embedded in optimal cutting temperature compound as previously described [28]. For immunolabelling, frozen sections were blocked with 5% normal goat serum and incubated with the GRB2 primary antibody (1:100, Abcam no. 32111) and GFAP (rabbit polyclonal, 1:250, Dako no. Z0334) at +4°C overnight. After nuclear counterstaining with Hoechst, the bound GRB2 antibody was detected with an Alexa Fluor 488-conjugated secondary antibody diluted 1:1,000 (Invitrogen Life Technologies, Mulgrave, VIC, Australia).
Results
Genome-wide genotyping was completed for 442 cases and 582 controls. Principal components analysis identified 69 participants (46 cases and 23 controls) as ethnic outliers and these were excluded from further analysis. A further 111 individuals had missing clinical covariate measures and were also excluded, leaving 336 cases and 508 controls in the final analysis. Clinical and demographic details for the discovery cohort are given in ESM Table 1. As expected, duration of diabetes, HbA1c, nephropathy and hypertension were all significantly associated with the presence of sight-threatening diabetic retinopathy, as were age and sex. Genetic association analysis was conducted in PLINK using logistic regression adjusted for age, sex, duration of diabetes, hypertension, nephropathy HbA1c and three principal components. The genomic inflation factor (λ) was 1.002 and the Q–Q plot is shown in ESM Fig. 1.
We first assessed loci reported in previous GWAS of diabetic retinopathy [15–19] for association in the current study in an attempt to replicate previous findings. A p value of <0.002 was required to account for the 29 loci assessed. No previously reported SNPs showed replication at this level of significance, although several loci demonstrate nominal associations (p < 0.05) and may be worthy of further investigation (ESM Table 2).
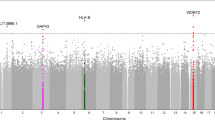
No SNP reached genome-wide significance in the GWAS analysis (Fig. 1a). The top ranked SNP was rs3805931 on chromosome 6 (p = 2.66 × 10−7, OR 0.50 [95% CI 0.39, 0.65]), with a second SNP at the same locus, rs1537638, showing similar significance (Table 1). Eight SNPs at five loci demonstrated p < 1 × 10−5 (Table 1).

Results of GWAS for sight-threatening diabetic retinopathy in type 2 diabetes mellitus. (a) Manhattan plot showing association results for sight-threatening diabetic retinopathy. The black dashed horizontal line indicates genome-wide significance level of p = 5 × 10−8 and the grey line represents suggestive association at p = 1 × 10−6. (b) The associated chromosome 17 locus at SNP rs9896052 showing -log10 (p values) in the discovery cohort (dots, left hand axis), average recombination rates in white patients (solid lines, right hand axis) and annotated protein coding transcripts in the region below
All SNPs with p < 1 × 10−5 and a selection with p < 1 × 10−4 were assessed in the white type 2 replication cohort comprising 263 cases and 320 controls (ESM Table 3). Neither of the top ranked chromosome 6 SNPs showed association in the type 2 cohort and thus replication of the primary study findings was not achieved (Table 2). Of the other SNPs, only rs9896052 (p in discovery = 6.55 × 10−5, OR 1.67 [95% CI 1.30,2.15]) was associated in the type 2 replication cohort (p = 0.035, OR 1.45 [95% CI 1.03, 2.18]; Table 2). This SNP was also assessed in a white type 1 cohort and an Indian type 2 cohort (ESM Table 3). An association with sight-threatening diabetic retinopathy was shown in the type 1 cohort (p = 0.041, OR 1.56 [95% CI 1.02, 2.38]) in the same direction as that of the discovery and type 2 replication cohorts (Table 2). In the Indian cohort, rs9896052 was also associated with the disease (p = 0.016, OR 1.47 [95% CI 1.07, 2.02]; Table 3). Meta-analysis of all four cohorts gave a p value of 4.15 × 10−8 (Table 3), which met the criteria for genome-wide significance (p < 5 × 10−8).
Although the top ranked locus on chromosome 6 did not show replication in the type 2 cohort, the type 1 cohort did show a trend towards association at this locus (rs1537638 p = 0.025, Table 2) and this region remains a candidate for further study in relation to diabetic retinopathy. Meta-analysis of the discovery cohort with the type 1 cohort gave a p value of 3.67 × 10−8 at this SNP and it was the top ranked SNP in the full meta-analysis of all SNPs typed on the array. However, as noted, this SNP did not show association in the type 2 replication cohort. No other SNPs reached genome-wide significance in the meta-analysis (ESM Table 4).
SNP rs9896052 on chromosome 17 is within a 150 kb block of linkage disequilibrium, which contains several genes: KIAA0195, CASKIN2, TSEN54 and LLGL2 (Fig. 1b). All of these transcripts are expressed in the retina (the Ocular Tissue Database: https://genome.uiowa.edu/otdb/, date last accessed: October 2013), although little is known about their function. According to ENCODE data (available at the University of California, Santa Cruz genome browser, http://genome.ucsc.edu/cgi-bin/hgGateway, date last accessed: October 2013), the SNP rs9896052 is located between two H3K27Ac marks at 73,401 Mbp and 73,425 Mbp. At 17 kb downstream of rs9896052, just outside this large linkage disequilibrium (LD) block are two additional genes GRB2 (Fig. 1b) and the microRNA gene MIR3678, located immediately 5′ to GRB2. Both these genes are excellent functional candidates for diabetic retinopathy and thus we evaluated them in more detail. RT-PCR of human retina showed expression of mature MIR3678 as well as both reported isoforms of GRB2, with the full length isoform 1 (NM_002086.4) more abundant than isoform 2 (NM_203506.2), which lacks exon 4 (Fig. 2). To further investigate the retinal expression pattern, we undertook immunohistochemistry for GRB2 in normal human retina. The protein is expressed throughout all layers of the retina, including in blood vessel wall (Fig. 2).

Expression and distribution of candidate genes in normal human retina. (a) RT-PCR for MIR3678. (b) RT-PCR for two transcripts of GRB2. (c) Immunohistochemistry with the anti-GRB2 antibody showing ubiquitous positive immuno-labelling through the retinal tissue and in retinal blood vessel endothelium (arrow). (d) Negative control. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; OS, outer segment. Scale bar, 20 μm
Direct sequencing of the coding regions of GRB2 and MIR3678 in 72 cases (ESM Table 5) identified known intronic SNPs (rs200248899 in one individual and rs12946365 with allele frequency of 0.257 as expected), but no coding variations, suggesting that coding mutations are unlikely to account for disease.
The response of GRB2 to retinal stress was explored in retina from a transgenic model of selective Müller cell ablation. In these mice, patchy loss of Müller cells leads to photoreceptor degeneration, blood–retinal barrier breakdown and the development of intraretinal neovascularisation, mimicking the features of diabetic retinopathy [28]. Retinal stress is demonstrated by a significant increase in GFAP labelling (Fig. 3). Increased GRB2 labelling was qualitatively observed in the retinal ganglion cell and both inner and outer nuclear layers of the Müller cell knockout mice, particularly at 3 months post Müller cell ablation, and this difference was significantly different as demonstrated by quantitative western blot (Fig. 3).

Differential expression of GRB2 in retinae of transgenic mice after selective Müller cell ablation. (a, c, e) GRB2 immunolabelling (green) with Hoechst nuclear counterstaining (red). (b, d, f) GFAP immunolabelling (green) with Hoechst nuclear counterstaining (red). (a, b) Control mice. Transgenic mice (c, d) 2 weeks and (e, f) 3 months after Müller cell ablation. INL, inner nuclear layer; ONL, outer nuclear layer. Arrows in (c) and (d) point to areas of protrusion of degenerated photoreceptors after patchy Müller cell ablation. Profound activation of surviving Müller cells after patchy loss of Müller cells in (d) and (f). (a–f) Scale bar, 50 μm. (g) Western blots for GRB2 and GFAP at 3 months after Müller cell ablation; α/β tubulin is included as a loading control. (h) Significant upregulation of GRB2 in transgenic mice compared with controls. (i) Confirms upregulation of GFAP as expected. Tg, transgenic, Ctl, control. **p < 0.01
Discussion
This study is the first to report an association of the 17q25.1 locus with diabetic retinopathy. We show an association between SNP rs9896052 and sight-threatening diabetic retinopathy in white and Indian patients with type 2 diabetes, as well as in patients with type 1 diabetes. A genome-wide significant p value of 4.15 × 10−8 was achieved in a meta-analysis of all cohorts. This suggests that the association with retinopathy is independent of the type of diabetes and is a risk factor for retinopathy itself. The minor allele frequencies for rs9896052 for controls in both the white and Indian cohorts are similar to those in relevant HapMap populations (HapMap Centre d'Etude du Polymorphisme [CEU] white: 0.321, HapMap Gujarati Indians in Houston: 0.431) and the OR is similar between all three replication cohorts (ranging from 1.45 to 1.56) demonstrating the robustness of this finding. Several additional SNPs at this locus were associated with diabetic retinopathy in the discovery cohort; however, no association was seen in the replication cohorts. Evaluation of the LD patterns in each data set revealed that the discovery cohort demonstrates reasonably strong LD between rs9896052 and the two adjacent SNPs rs6501801 and rs9892171 (r 2 = 0.46 and 0.33, respectively). However, the type 2 replication cohort has less LD (r 2 = 0.29 and 0.20, respectively), thus explaining why the association is not seen at these nearby SNPs in this cohort (ESM Fig. 2). It is not clear why the LD patterns differ; however, the association statistics observed are consistent with the LD patterns.
Several other GWAS for diabetic retinopathy have been published [15–19], although none report a genome-wide significant signal at 17q25. Of note, four of the previously published studies were conducted in non-European populations (Chinese [17], Taiwanese [18], Japanese [19] and Mexican American [15]), while one was predominantly in patients with type 1 diabetes mellitus [16]. These fundamental differences may explain the lack of replication observed not only with the current study, but also with each other. As our study demonstrates consistent association at 17q25 across multiple types of diabetes and in different ethnic groups, there may be some genetic factors that are independent of diabetes type and ethnicity and others that are specific. A limitation of this and previously published work that may affect the reproducibility of the result is the size of the cohort, and larger studies and meta-analyses are required to provide a clearer picture of the genetic architecture of diabetic retinopathy.
We restricted the current study to severe forms of diabetic retinopathy and compared them with contemporaneously recruited diabetic patients with no or minimal diabetic retinopathy. The inclusion of minimal diabetic retinopathy in the control group recognises that, at this end of the spectrum, differences in grading have limited meaning for clinical decision-making as even a single microaneurysm could constitute minimal diabetic retinopathy. The use of clinical grading rather than retinal photography could be considered a weakness of this study; however, the use of the extreme phenotype negates the need for detailed grading as there is strong agreement between clinical assessments and photographic grading for categorising diabetic retinopathy [29].
It is yet to be determined which of the genes in this region are involved in the pathogenesis of diabetic retinopathy; however, several annotated transcripts, notably MIR3678 and GRB2, make compelling candidates. Although our data indicate that coding mutations in these genes are unlikely to account for disease, it is possible that SNP rs9896052 or a SNP in LD with it, is in a regulatory region that alters the expression of these genes.
Little is known specifically about MIR3678, although among many predicted targets (miRbase.org) is EGLN3, which is a hypoxia inducible factor prolyl hydroxylase important in oxygen sensing and the response to hypoxia, a pathway highly relevant to diabetic retinopathy. MIR3678 is not annotated in rodent genomes adding complexity to the assessment of the role of this transcript in animal models of diabetic retinopathy. Further work in human tissues will be required to assess its role in this disease.
Functionally, GRB2 is a promising candidate for diabetic retinopathy susceptibility. It binds phosphorylated insulin receptor substrate 1 and subsequently activates the MAPK pathway via Ras in response to insulin [30]. This gene is also involved in vascular endothelial growth factor signalling leading to angiogenesis, which is characteristic of proliferative diabetic retinopathy [31]. GRB2 encodes two transcripts in humans [32]. Isoform 1 is expressed in most human tissues and isoform 2 is either absent or expressed at considerably lower levels relative to isoform 1 [32]. Isoform 2, known as GRB3-3, lacks a portion of the SH2 domain and thus does not bind to the phosphorylated tyrosine kinase receptor [32]. It appears to act as a negative regulator of isoform 1 and may be involved in inducing apoptosis [32]. The present study is the first to show expression of both isoforms in the human retina. The GRB2 protein was observed in all layers of the human and mouse retina and we demonstrated that retinal stress leading to neovascular retinopathy in a rodent model results in increased expression of the protein consistent with a role in diabetic retinopathy.
Further work assessing all the genes in this region is required to determine whether or not GRB2, MIR3678 or one of the other less well characterised genes at this locus is the primary contributor to diabetic retinopathy, and to determine the mechanism of action of the genetic predisposition identified in this study.
Abbreviations
- ETDRS:
-
Early Treatment in Diabetic Retinopathy Study
- GFAP:
-
Glial fibrillary acidic protein
- GWAS:
-
Genome-wide association study
- LD:
-
Linkage disequilibrium
- SNPs:
-
Single nucleotide polymorphisms
References
National Diabetes Data Group (1995) Diabetes in America, vol 2. National Institutes of Health; National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda
Kempen JH, O'Colmain BJ, Leske MC et al (2004) The prevalence of diabetic retinopathy among adults in the United States. Arch Ophthalmol 122:552–563
Wild S, Roglic G, Green A, Sicree R, King H (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053
Australian Diabetes Society (2008) Guidelines for the management of diabetic retinopathy. National Health and Medical Research Council, Canberra
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
UK Prospective Diabetes Study (UKPDS) Group (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Gerstein HC, Miller ME, Byington RP et al (2008) Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 358:2545–2559
The Diabetes Control and Complications Trial Research Group (1997) Clustering of long-term complications in families with diabetes in the diabetes control and complications trial. Diabetes 46:1829–1839
Rema M, Saravanan G, Deepa R, Mohan V (2002) Familial clustering of diabetic retinopathy in South Indian type 2 diabetic patients. Diabet Med 19:910–916
Hallman DM, Huber JC Jr, Gonzalez VH, Klein BE, Klein R, Hanis CL (2005) Familial aggregation of severity of diabetic retinopathy in Mexican Americans from Starr County, Texas. Diabetes Care 28:1163–1168
Hietala K, Forsblom C, Summanen P, Groop PH (2008) Heritability of proliferative diabetic retinopathy. Diabetes 57:2176–2180
Arar NH, Freedman BI, Adler SG et al (2008) Heritability of the severity of diabetic retinopathy: the FIND-Eye study. Invest Ophthalmol Vis Sci 49:3839–3845
Monti MC, Lonsdale JT, Montomoli C, Montross R, Schlag E, Greenberg DA (2007) Familial risk factors for microvascular complications and differential male-female risk in a large cohort of American families with type 1 diabetes. J Clin Endocrinol Metab 92:4650–4655
Abhary S, Hewitt AW, Burdon KP, Craig JE (2009) A systematic meta-analysis of genetic association studies for diabetic retinopathy. Diabetes 58:2137–2147
Fu YP, Hallman DM, Gonzalez VH et al (2010) Identification of diabetic retinopathy genes through a genome-wide association study among Mexican-Americans from Starr County, Texas. J Ophthalmol. doi:10.1155/2010/861291
Grassi MA, Tikhomirov A, Ramalingam S, Below JE, Cox NJ, Nicolae DL (2011) Genome-wide meta-analysis for severe diabetic retinopathy. Hum Mol Genet 20:2472–2481
Sheu WH, Kuo JZ, Lee IT et al (2013) Genome-wide association study in a Chinese population with diabetic retinopathy. Hum Mol Genet 22:3165–3173
Huang YC, Lin JM, Lin HJ et al (2011) Genome-wide association study of diabetic retinopathy in a Taiwanese population. Ophthalmology 118:642–648
Awata T, Yamashita H, Kurihara S et al (2014) A genome-wide association study for diabetic retinopathy in a Japanese population: potential association with a long intergenic non-coding RNA. PLoS One 9:e111715
Kaidonis G, Abhary S, Daniell M et al (2014) Genetic study of diabetic retinopathy: recruitment methodology and analysis of baseline characteristics. Clin Experiment Ophthalmol 42:486–493
Early Treatment Diabetic Retinopathy Study Research Group (1991) Grading diabetic retinopathy from stereoscopic color fundus photographs—an extension of the modified Airlie House classification. ETDRS report number 10. Ophthalmology 98:786–806
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Pruim RJ, Welch RP, Sanna S et al (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26:2336–2337
Genomes Project Consortium, Abecasis GR, Auton A et al (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191
Shen W, Fruttiger M, Zhu L et al (2012) Conditional Mullercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J Neurosci 32:15715–15727
Gangaputra S, Lovato JF, Hubbard L et al (2013) Comparison of standardized clinical classification with fundus photograph grading for the assessment of diabetic retinopathy and diabetic macular edema severity. Retina 33:1393–1399
Skolnik EY, Lee CH, Batzer A et al (1993) The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: implications for insulin control of ras signalling. EMBO J 12:1929–1936
Anselmi F, Orlandini M, Rocchigiani M et al (2012) c-ABL modulates MAP kinases activation downstream of VEGFR-2 signaling by direct phosphorylation of the adaptor proteins GRB2 and NCK1. Angiogenesis 15:187–197
Fath I, Schweighoffer F, Rey I et al (1994) Cloning of a Grb2 isoform with apoptotic properties. Science 264:971–974
Acknowledgements
We thank the following people for technical assistance with patient recruitment, DNA extraction and genotyping: E. Souzeau, K. Dowell, A. Kuot and S. Thorpe (Department of Ophthalmology, Flinders University, Adelaide, SA, Australia), L. Kolmogorova (Save Sight Institute, University of Sydney, Sydney, NSW, Australia), J. Haariko (Ophthalmology, Australian National University, Canberra, ACT, Australia), F. Richter (Department of Ophthalmology, Royal Melbourne Hospital, Melbourne, VIC, Australia) and J. Hadler (Diamantina Institute, The University of Queensland, Brisbane, QLD, Australia).
Funding
This work was supported by the National Health and Medical Research Council (NHMRC) of Australia [no. 595918] and the Ophthalmic Research Institute of Australia. KPB is supported by a Senior Research Fellowship from the NHMRC and JEC by a Practitioner Fellowship from the NHMRC. Research conducted at Moorfields Eye Hospital was funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust (www.nihr.ac.uk) and the University College, London Institute of Ophthalmology. Research at the Centre for Eye Research Australia was funded by the Australian Research Council (www.arc.gov.au) grant LP0884108.
Duality of interest statement
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
KPB, SA, NP, MAB, JEC designed the study; SA, GK, AWH, MD, RWE, JHC, SRL, BP, AJ, PS, ELL, KR, MP, PGH, NP, MCG collected data; RDF, WS, GK, BA, SS, SK, GG conducted laboratory experiments; KPB, RDF, GK, MAB conducted statistical analysis; KPB, RDF, WS, SS wrote the paper; all authors reviewed and critiqued the paper, contributed to data interpretation, and reviewed the final version of the paper. KPB is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 346 kb)
ESM Fig. 1
(PDF 181 kb)
ESM Fig. 2
(PDF 178 kb)
ESM Table 1
(PDF 330 kb)
ESM Table 2
(PDF 390 kb)
ESM Table 3
(PDF 181 kb)
ESM Table 4
(PDF 190 kb)
ESM Table 5
(PDF 249 kb)
Rights and permissions
About this article

Cite this article
Burdon, K.P., Fogarty, R.D., Shen, W. et al. Genome-wide association study for sight-threatening diabetic retinopathy reveals association with genetic variation near the GRB2 gene. Diabetologia 58, 2288–2297 (2015). https://doi.org/10.1007/s00125-015-3697-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3697-2