
Abstract
Aims/hypothesis
While the underlying mechanisms in the development of insulin resistance remain inconclusive, metabolic dysfunction in both white adipose tissue (WAT) and skeletal muscle have been implicated in the process. Therefore, we investigated the independent and combined effects of α-linolenic acid (ALA) supplementation and exercise training on whole-body glucose homeostasis and mitochondrial bioenergetics within the WAT and skeletal muscle of obese Zucker rats.
Methods
We randomly assigned obese Zucker rats to receive a control diet alone or supplemented with ALA and to remain sedentary or undergo exercise training for 4 weeks (CON-Sed, ALA-Sed, CON-Ex and ALA-Ex groups). Whole-body glucose tolerance was determined in response to a glucose load. Mitochondrial content and bioenergetics were examined in skeletal muscle and epididymal WAT (eWAT). Insulin sensitivity and cellular stress were assessed by western blot.
Results
Exercise training independently improved whole-body glucose tolerance as well as insulin-induced signalling in muscle and WAT. However, the consumption of ALA during exercise training prevented exercise-mediated improvements in whole-body glucose tolerance. ALA consumption did not influence exercise-induced adaptations within skeletal muscle, insulin sensitivity and mitochondrial bioenergetics. In contrast, within eWAT, ALA supplementation attenuated insulin signalling, decreased mitochondrial respiration and increased the fraction of electron leak to reactive oxygen species (ROS).
Conclusions/interpretation
These findings indicate that, in an obese rodent model, consumption of ALA attenuates the favourable adaptive changes of exercise training within eWAT, which consequently impacts whole-body glucose homeostasis. The direct translation to humans, however, remains to be determined.
Similar content being viewed by others

Introduction
While the development of whole-body insulin resistance is not fully understood, the prominent working model suggests that high-fat diet-mediated reactive oxygen species (ROS) and inflammation rapidly attenuate insulin signalling within white adipose tissue (WAT), increasing lipolysis and NEFA release into the peripheral circulation [1, 2]. The increase in circulating NEFA results in excessive transport of lipids into muscle, and ultimately ectopic lipid accumulation and increased mitochondrial ROS emission, both of which contribute to skeletal muscle insulin resistance. Since muscle accounts for ~ 75% of insulin-stimulated glucose uptake [1], the functional effect of insulin on maintaining glucose homeostasis is indirectly influenced by WAT metabolism. Therefore, therapeutic modalities that improve insulin sensitivity in both WAT and skeletal muscle are probably required to preserve whole-body glucose tolerance.
It is well-established that exercise training improves whole-body glucose homeostasis and decreases oxidative stress, reactive lipid accumulation and markers of inflammation [3,4,5]. While the mechanisms responsible for these observations remain poorly defined, exercise training is known to increase mitochondrial content in both skeletal muscle [6] and WAT [7, 8], which probably contributes to the observed reduction in both reactive lipid accumulation and oxidative stress (ROS). In support of this, mitochondrial content (skeletal muscle and WAT) is reduced in several models of insulin resistance [9,10,11,12], while genetic and therapeutic approaches that increase mitochondrial density are associated with improvements in insulin sensitivity [11, 13,14,15,16,17]. Altogether, these reports suggest that intervention strategies that increase mitochondrial content may be particularly beneficial in managing and/or preventing the development of insulin resistance and type 2 diabetes.
In addition to exercise, the use of n-3 polyunsaturated fatty acids (PUFAs) as a treatment modality has gained considerable interest [18,19,20]. The n-6/n-3 ratio in a typical Western diet has increased ~ 15-fold in recent years [21] and is thought to contribute to the production of proinflammatory eicosanoids, primarily from arachidonic acid via the cyclooxygenase and lipoxygenase pathways. As a result, diets supplemented with n-3 PUFAs are thought to be beneficial with respect to several pathologies, including diet-induced insulin resistance/type 2 diabetes [18,19,20, 22]. While the mechanisms regulating the insulin-sensitising effects of n-3 PUFAs remain ambiguous, remodelling of mitochondrial membrane phospholipid composition [23, 24], reduction in intramuscular accumulation of reactive lipid intermediates [20] and increased transcription of gene targets involved in mitochondrial biogenesis and fatty acid oxidation [19, 20, 25] have all been proposed. In addition, n-3 PUFA supplementation in rodents under a high-fat challenge significantly reduces fat mass and adipocyte size [26], while in humans n-3 PUFAs increase adipocyte mitochondrial biogenesis and fatty acid oxidation [27] and limit fat cell proliferation [28]. Therefore, similar to exercise training, the prominent theory surrounding n-3 PUFA and insulin sensitivity suggests improvements in mitochondrial bioenergetics in both WAT and skeletal muscle.
Although evidence is mounting to suggest that n-3 PUFAs and exercise training independently improve whole-body glucose homeostasis, the additive effects remain unknown. Investigations into the combined effects appear warranted given that nutritional/exercise approaches have yielded disappointing results in the past. Specifically, antioxidant supplementations (vitamins C and E) have been shown to attenuate exercise-induced mitochondrial biogenesis [29,30,31], whereas resveratrol supplementation attenuates exercise-mediated improvement in cardiovascular health [32] and mitochondrial adaptations [33,34,35,36,37]. In addition, previous research has demonstrated that n-3 PUFA supplementation attenuates exercise-mediated splenic immune cell activation [38], raising the possibility that n-3 PUFA supplementation can attenuate exercise-mediated improvements in insulin sensitivity. This could be particularly relevant in WAT, a tissue that is not only highly regulated by immunity/stress processes, but that is also primarily influenced by n-3 PUFA supplementation [39]. In order to examine this possibility, we investigated the independent and combined effects of exercise training and dietary supplementation with the essential n-3 PUFA α-linolenic acid (ALA), within the WAT and skeletal muscle of obese Zucker rats, a widely used model of obesity and insulin resistance.
Methods
Animals and diet
Six-week-old male obese Zucker rats (n = 32) were purchased from Charles River Laboratories (Wilmington, MA, USA). The animals were housed in wire cages in pairs in a temperature-regulated room on a 12:12 h light/dark cycle with water provided ad libitum. After 6 days of acclimatisation, the animals were randomly assigned by simple randomisation to receive a control (CON) diet (no. AIN-93G; 20% protein, 64% carbohydrate and 16% fat; Research Diets, New Brunswick, NJ, USA) or a diet supplemented with ALA (no. AIN-93G + 10% flaxseed oil; 20% protein, 54% carbohydrate and 26% fat), and then randomly divided to undergo exercise (Ex) training (CON-Ex and ALA-Ex) or remain sedentary (Sed; CON-Sed and ALA-Sed) for a total of 4 weeks (n = 8/group). Animals on the CON diet were given unrestricted access to food, while ALA-supplemented animals were pair-fed to maintain caloric intake. Body weight was measured every week, while glucose tolerance tests were performed 48 h before animals were anaesthetised with isoflurane (4% in O2) to conduct experiments described below. We have previously reported on the effects of these interventions with respect to cardiovascular function and hepatic lipid content [40, 41]. Experimenters were not blind to group assignment during the conduction of experiments and all experiments were conducted in accordance with protocols approved by the University of Guelph Animal Care Committee.
Exercise training
The Ex groups were first familiarised with treadmill running for 10 min sessions at 10 m/min on a 5% incline on three separate occasions. Thereafter, the animals were exercised five times a week for 45 min per session according to the following protocol: week 1, 15 m/min (5% incline); week 2, 15 m/min (10% incline); and week 3–4, 15 m/min (15% incline).
Glucose tolerance test
Tolerance to glucose was evaluated after 4 h of fasting as previously described [42]. Briefly, blood glucose was measured before and repeatedly after animals were i.p. injected with glucose (2 g/kg body weight). Thereafter, blood glucose vs time was plotted and the AUC calculated for each animal.
Muscle- and eWAT-specific insulin signalling
To determine the phosphorylation of proteins involved in insulin-mediated signalling by western blotting (described below), muscle and epididymal WAT (eWAT) was excised before (basal) and 15 min after an intravenous insulin injection (1.0 U/kg body weight) and rapidly frozen in liquid nitrogen.
Mitochondrial respiration in permeabilised muscle fibres and eWAT
Mitochondrial respiration was determined in permeabilised muscle fibres (PmFbs) using high-resolution respirometry at 37°C as previously described [43, 44]. Mitochondrial respiration was also determined in permeabilised eWAT at 37°C as previously described [45] except for the addition of 2,4-dinitrophenol (DNP; 100 μmol/l) at the end of state III respiration. Exogenous cytochrome c (10 μmol/l) was added at the end of all respiration experiments to ensure outer mitochondrial membrane integrity.
Mitochondrial H2O2 emission
Mitochondrial H2O2 emission was measured in PmFbs fluorometrically (Lumina; Thermo Scientific, Fisher, Waltham, MA, USA) at 37°C as previously described [44, 46]. In permeabilised eWAT, mitochondrial H2O2 emission was measured at 37°C similar to previously described methodology [45] except mitochondrial H2O2 emission rates were initiated with either pyruvate (10 mmol/l) + malate (5 mmol/l) or l-carnitine (2 mmol/l) + palmitoyl-CoA (P-CoA; 50 μmol/l).
mtDNA copy number in adipose tissue
Mitochondrial DNA (mtDNA) copy number was determined in eWAT using real-time PCR as previously described [15].
Western blot
Whole muscle and eWAT was homogenised as previously described [24]. Standard SDS-PAGE procedures were followed. All antibodies were commercially available and validated by manufacturers for western blotting. These included: total and phosphorylated (Thr308 and Ser473) Akt; total and phosphorylated c-Jun N-terminal kinase (JNK) 1 and 2; total and phosphorylated extracellular signal-regulated kinase (ERK) 1 and 2; total and phosphorylated p38 MAP kinase (p38); monocyte chemoattractant protein 1 (MCP1); MitoProfile oxidative phosphorylation (OXPHOS) antibody cocktail; cytochrome c oxidase IV (COXIV); IRS1; pyruvate dehydrogenase subunit E1-alpha (PDHE1α); adenine nucleotide translocase (ANT) 1 and 2; and trans-4-hydroxy-2-nonenal (4-HNE) with α-tubulin as the loading control. See electronic supplementary material (ESM) Table 1 for details of the antibodies.
Statistics
All data were analysed using a two-way ANOVA followed by Tukey’s honest significant difference (HSD) post hoc test where appropriate. For body weight and blood glucose measurements during the glucose challenge, a two-way ANOVA was used to analyse differences within each week. One control animal was humanely euthanised prior to study completion because of an infected skin lesion. A single animal was removed from the analysis of muscle and WAT insulin signalling, as the administration of insulin resulted in a 50% decline in Akt phosphorylation, which we deemed a failed experiment as the Akt phosphorylation was more then 2 SD from the mean. No other data was removed from the analysis. In some other experiments the sample size is smaller because of tissue limitations preventing a measurement from being performed, as opposed to the removal of data from the analysis. All values are expressed as mean ± SEM. Significance was determined at p < 0.05. Statistical tests were completed using Graph Pad version 6.2 (La Jolla, CA, USA).
Results
Whole-body glucose tolerance and body mass in response to ALA and exercise
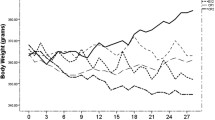
We have previously reported in these same animals that ALA supplementation appropriately increased the ALA, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) contents of lipids within the blood and the liver, verifying the dietary intervention [41]. In the present study, we report that the consumption of ALA independently did not alter body weight throughout the experimental protocol (Fig. 1a). In contrast, regardless of diet, exercise training significantly decreased body weight after 3 and 4 weeks of the intervention (Fig. 1a), probably because of the conserved caloric intake between groups despite the increased caloric expenditure. Next we examined whole-body glucose tolerance. While 4 weeks of ALA supplementation was not sufficient to improve glucose tolerance (Fig. 1b, c), exercise independently decreased the AUC by ~ 30% (Fig. 1b, c, p < 0.05), a response prevented by the simultaneous consumption of ALA (Fig. 1b, c).

Effect of 4 week ALA supplementation and exercise training on body weight and whole-body blood glucose homeostasis. (a) Body weight, (b) blood glucose and (c) calculated AUC in obese Zucker rats. Arrow, start of intervention; white circles/bars, CON; black circles/bars, ALA; solid line, Sed; dashed line, Ex. Mean ± SEM, n = 6–8. *p < 0.05 Ex vs Sed; † p < 0.05 vs all other groups
Skeletal muscle insulin signalling in response to ALA and exercise
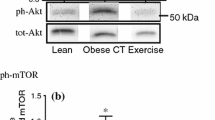
Given the observation that ALA consumption prevented the normal exercise training-mediated improvement in glucose tolerance, we examined skeletal muscle insulin sensitivity in an attempt to gain insight into the tissues affected by the combined intervention. Specifically, we determined the ability of insulin to phosphorylate proteins involved in the insulin signalling cascade. Regardless of diet, insulin failed to stimulate phosphorylation of Akt at Ser473 (Fig. 2a, b) and Thr308 (Fig. 2a, c) above basal levels in sedentary animals, demonstrating skeletal muscle insulin resistance in our model. In contrast, there was a main effect for exercise training to significantly increase Akt phosphorylation at both Ser473 and Thr308 (Fig. 2a–c). In further support of these findings, exercise training, irrespective of diet, significantly increased total IRS1 protein content (Fig. 2d). Taken together, these findings confirm that 4 weeks of exercise training improves insulin signalling in skeletal muscle and suggest that the impairments in glucose clearance in the ALA-Ex group cannot be explained by decreases in skeletal muscle insulin sensitivity.

Skeletal muscle insulin signalling is increased following 4 weeks of exercise training. (a) Representative blots of insulin signalling from muscle under basal and insulin-stimulated conditions. Muscle insulin-stimulated phosphorylated Ser473 (b) and Thr308 (c) Akt fold change over basal. (d) Total IRS1 content in muscle was averaged for basal and insulin-stimulated muscles, as these values represent a duplicate within an animal. Representative blot of IRS1 is also shown with α-tubulin as loading control. White bars, CON; black bars, ALA; dashed horizontal line, basal. Mean ± SEM, n = 6–8. **p < 0.01, ***p < 0.001 Ex vs Sed. Bsl, basal; Ins, insulin-stimulated
Skeletal muscle mitochondrial respiration and kinetics in response to ALA and exercise
To further establish that ALA supplementation did not augment training responses within skeletal muscle we determined various classical adaptations in mitochondrial bioenergetics. Specifically, we first analysed maximal complex I- and II-supported respiration. ALA supplementation did not alter respiration in any condition (Fig. 3a), while in contrast, regardless of diet, exercise training increased all aspects of mitochondrial respiration measured (Fig. 3a), further indicating that ALA does not affect exercise-induced adaptations in skeletal muscle. Surprisingly, whole-muscle OXPHOS and PDHE1α proteins were not changed (Fig. 3b, c), suggesting that exercise training was not sufficient to induce large changes in mitochondrial content.

Skeletal muscle mitochondrial respiration and content. (a) Basal and ADP-stimulated mitochondrial respiration in muscle. White bars, CON; black bars, ALA. (b) Mitochondrial content measured by an OXPHOS antibody in muscle. (c) Representative blots for mitochondrial OXPHOS content. Dashed horizontal line, CON-Sed; black bars, ALA-Sed; light-grey bars, CON-Ex; dark-grey bars, ALA-Ex. Mean ± SEM, n = 6–8. *p ≤ 0.05, ***p < 0.001 Ex vs Sed. CC, cytochrome c; CI, complex I; CII, complex II; CIII, complex III; CIV-1, complex IV subunit 1; CIV-4, complex IV subunit 4; G, glutamate; JO2, oxygen flux; PM, pyruvate + malate; RCR, respiratory control ratio; S, succinate
Next we determined if ALA supplementation and exercise training altered ADP respiratory kinetics by performing ADP titrations in the presence and absence of creatine. ALA did not affect the apparent ADP K m (Fig. 4b, d) in the presence or absence of creatine. In contrast, for exercise, there was a non-significant increase (p = 0.06) in the sensitivity of mitochondria to ADP in the presence of creatine (decreased apparent K m; Fig. 4a, b), whereas in the absence of creatine, exercise significantly decreased the sensitivity of mitochondria to ADP (increased apparent ADP K m; Fig. 4c, d). Furthermore, exercise training significantly increased ANT1 content by ~ 25% irrespective of diet (Fig. 4e, f), but did not alter the content of ANT2 (Fig. 4e, f). Altogether, exercise training, but not ALA supplementation, alters ADP respiratory kinetics and the expression of ANT1 but not ANT2 in skeletal muscle after 4 weeks.

Skeletal muscle mitochondrial ADP kinetics and ADP transport proteins. (a) ADP titration and (b) mitochondrial ADP sensitivity (apparent K m) in the presence of 20 mmol/l creatine. (c) ADP titration and (d) mitochondrial ADP sensitivity (apparent K m) in the absence of creatine. (e) ANT1 and ANT2 content in muscle. (f) Representative blot of ANT1 and ANT2 with α-tubulin as loading control. White circles/bars, CON; black circles/bars, ALA; solid line, Sed; dashed line, Ex. Mean ± SEM, n = 6–8. *p < 0.05, **p < 0.01, † p = 0.06 Ex vs Sed. JO2, oxygen flux
Mitochondrial H2O2 emission and oxidative stress in skeletal muscle
ALA supplementation for 12 weeks has been previously shown to increase mitochondrial H2O2 emission in obese Zucker rats [40]. Given that mitochondrial-derived ROS have been linked to insulin resistance [46], we examined mitochondrial H2O2 emission rates in the presence and absence of a submaximal ADP concentration. The independent provision of ALA did not alter maximal H2O2 emission rates, the fraction of electron leak to H2O2 or H2O2 rates in the presence of ADP (Fig. 5a–d). Exercise training did not affect maximal H2O2 emission; however, there was a main effect for exercise training to significantly decrease H2O2 emission, as well as the fraction of electron leak to ROS, in the presence of ADP (Fig. 5c, d). Altogether, the consumption of ALA during exercise training did not alter any variable in skeletal muscle to account for the observed attenuation in glucose tolerance.

Skeletal muscle mitochondrial ROS emission rates. (a) Succinate-supported mitochondrial ROS emission rates. (b) Electron leak. (c) Succinate-supported mitochondrial ROS emission rates in the presence of 100 μmol/l ADP. (d) Electron leak in the presence of 100 μmol/l ADP. White bars, CON; black bars, ALA. Mean ± SEM, n = 5–8. **p < 0.01 Ex vs Sed. JO2, oxygen flux
eWAT insulin signalling in response to ALA and exercise
Given that whole-body glucose homeostasis is also influenced by WAT [2], we investigated the effect of ALA consumption within eWAT. Specifically, we examined the ability of insulin to phosphorylate proteins involved in the insulin signalling cascade. In contrast to skeletal muscle, in the sedentary animals, there was a main effect for ALA to significantly decrease insulin phosphorylation of Akt at Ser473 and Thr308 (Fig. 6a–c), indicating that ALA impaired eWAT insulin signalling. Importantly, in the exercise-trained animals, ALA prevented the ability of exercise to stimulate Akt phosphorylation at both Ser473 and Thr308 (Fig. 6a–c). These data suggest that ALA mitigated the favourable effects of exercise in eWAT in a similar fashion to the observed attenuation in whole-body glucose homeostasis observed in the ALA-Ex animals. In support of this, regardless of exercise training, there was a main effect for ALA increasing JNK1 and JNK2 phosphorylation (Fig. 7a) and a non-significant increase in ERK1 phosphorylation (Fig. 7b, p = 0.08). In contrast, no differences in the phosphorylation of p38 or MCP1 content were observed irrespective of diet (Fig. 7c, d). Therefore, the impairments in insulin signalling within eWAT of ALA-supplemented animals coincided with the activation of stress signalling pathways but not overt inflammation.

ALA attenuates insulin signalling in eWAT after 4 weeks of supplementation. (a) Representative blots of insulin signalling from eWAT under basal (Bsl) and insulin-stimulated (Ins) conditions. eWAT insulin-stimulated phosphorylated (b) Ser473 and (c) Thr308 Akt fold change over basal, where all groups of animals were quantified from the same immunoblot. White bars, CON; black bars, ALA; dashed horizontal line, basal. Mean ± SEM, n = 6–8. * p < 0.05 ALA vs CON. Bsl, basal; Ins, insulin

ALA activates stress signalling pathways in eWAT after 4 weeks of supplementation but does not lead to overt inflammation. (a) Phosphorylated JNK1/2 content. (b) Phosphorylated ERK1/2 content. (c) Phosphorylated p38 content. (d) MCP1 content. Representative blots area also shown. White bars, CON; black bars, ALA. Mean ± SEM, n = 6–8. *p < 0.05, **p < 0.01, † p = 0.08 ALA vs CON
eWAT mitochondrial respiration in response to ALA and exercise
Next we investigated mitochondrial respiration in permeabilised eWAT. Similar to the approach used in skeletal muscle, we first analysed maximal complex I and II carbohydrate (pyruvate)-supported respiration. ALA supplementation significantly decreased all aspects of mitochondrial respiration irrespective of exercise training (Fig. 8a). We also analysed mitochondrial respiration in the presence of maximal P-CoA, a naturally occurring lipid that represents the majority of lipid available for oxidation in vivo, but found no differences between groups (Fig. 8b).

ALA decreases mitochondrial respiration in eWAT after 4 weeks of supplementation. (a) Pyruvate- and (b) lipid-supported mitochondrial respiration. (c) mtDNA content and mitochondrial OXPHOS content summed. (d) Pyruvate- and (e) lipid-supported mitochondrial respiration normalised to mtDNA content. White bars, CON; black bars, ALA. Mean ± SEM, n = 7–8 respiration; n = 6–7 content. *p < 0.05, **p < 0.01, ***p < 0.001 ALA vs CON or Ex vs Sed; † p = 0.09 Ex vs Sed. AU, arbitrary units; CC, cytochrome c; G, glutamate; JO2, oxygen flux; PM, pyruvate + malate; RCR, respiratory control ratio; S, succinate
Given the dynamic nature of WAT (e.g. the ability of adipocytes to undergo both hypertrophy and hyperplasia), the number of adipocytes, and consequently mitochondria, will inevitably vary per milligram wet weight [47]. Therefore, we measured the amount of mitochondria in eWAT. ALA supplementation did not alter mtDNA in any condition (Fig. 8c), while in contrast, to our surprise, exercise training was associated with significantly lower mtDNA regardless of diet (Fig. 8c). Similarly, exercise training coincided with reduced OXPHOS protein content within eWAT (Fig. 8c and ESM Fig. 1), further suggesting that mitochondrial content is lower within adipocytes at this moment in time. We therefore expressed mitochondrial respiration relative to mtDNA, which revealed a significant main effect of exercise irrespective of diet, whereby maximal pyruvate- and P-CoA-supported respiration were increased (Fig. 8d, e), suggesting improvements in mitochondrial respiratory function with exercise training in eWAT of obese Zucker rats. However, in contrast to lipid-supported respiration, there was a significant main effect for carbohydrate-supported mitochondrial respiration to be lower in the presence of ALA (Fig. 8d). These findings provide evidence that mitochondrial respiratory function, regardless of how the data is normalised, is compromised in eWAT of obese Zucker rats following the consumption of ALA, and may also contribute to the observed attenuation in insulin signalling after the combined intervention.
Mitochondrial H2O2 emission and oxidative stress in eWAT
Given the observation that ALA consumption decreased mitochondrial respiration in eWAT, we determined rates of mitochondrial H2O2 emission. Across all groups, ALA supplementation irrespective of exercise had no effect on pyruvate-supported mitochondrial H2O2 emission in eWAT (Fig. 9a). In contrast, ALA significantly decreased P-CoA-supported mitochondrial H2O2 emission in eWAT of both groups (Fig. 9a). However, when rates of mitochondrial H2O2 emission were expressed relative to mtDNA, there was a main effect of exercise training to increase pyruvate- and P-CoA-supported mitochondrial H2O2 emission irrespective of diet (Fig. 9b). In contrast, ALA significantly increased the fraction of electron leak to H2O2 with pyruvate, while with P-CoA, there was no difference in any of the groups regardless of diet or exercise (Fig. 9c). Furthermore, despite an increase in rates of mitochondrial H2O2 emission with exercise training when expressed relative to mtDNA, 4-HNE (a marker of lipid peroxidation) was decreased with ALA and unaltered by exercise (Fig. 9d), suggesting improvements in cellular redox state of eWAT with 4 weeks of ALA supplementation.

Mitochondrial ROS emission rates and oxidative stress in eWAT. (a) Pyruvate- and lipid-supported mitochondrial ROS emission rates. (b) Mitochondrial ROS emission rates normalised to mitochondrial DNA content. (c) Electron leak. (d) 4-HNE content with representative blot. White bars, CON; black bars, ALA. Mean ± SEM, n = 6–8 ROS emission; n = 7–8 blot. *p < 0.05, **p < 0.01 ALA vs CON or Ex vs Sed; † p = 0.05 ALA vs CON. PM, pyruvate + malate
Discussion
In the present study we provide evidence that ALA supplementation prevents exercise-induced improvements in glucose homeostasis in a rodent model of obesity. Moreover, the mechanism of action appears independent of changes in skeletal muscle insulin sensitivity and mitochondrial bioenergetics. In contrast, within eWAT, ALA supplementation (1) increased certain markers of cellular stress (JNK, ERK); (2) attenuated insulin-induced Akt phosphorylation; (3) decreased mitochondrial respiration; and (4) increased the fraction of electron leak to ROS. Altogether, while the present data highlights the beneficial responses to exercise training in diverse tissues, it has also revealed that the simultaneous consumption of ALA with training mitigates some of these favourable adaptive changes.
The effects of ALA and exercise on glucose homeostasis and insulin sensitivity
In the present study, whole-body glucose tolerance was improved following exercise training, and, unexpectedly, ALA supplementation independently or in combination with exercise training did not improve glucose tolerance. These findings are in stark contrast to our previous reports, which have shown that whole-body glucose tolerance is improved in obese Zucker rats following a diet supplemented with ALA [39, 40]. The main difference between the present study and our previous reports showing a beneficial response in glucose homeostasis following consumption of n-3 PUFAs is the length of time consuming the PUFA fortified diets. Specifically, in the present study animals consumed a diet high in ALA for 4 weeks, while in previous studies animals were supplemented for 12 weeks [39, 40]. In humans, a previous report also showed that diets enriched in n-3 PUFAs take ~ 4 weeks to alter the lipid composition of muscle [48] and therefore it is likely that a slightly longer duration of dietary consumption would have resulted in a beneficial response to the independent intake of ALA. Moreover, to our surprise, the consumption of ALA coincided with an attenuated insulin-induced Akt phosphorylation within eWAT and not skeletal muscle. While the importance of adipose tissue for glucose homeostasis is well understood [2], it is unlikely that eWAT alone accounted for these findings in the ALA-Ex group as glucose tolerance was not different between ALA-Sed and CON-Sed animals despite a decrease in insulin signalling in the ALA-Sed animals. Indeed, recent work published from our lab found that insulin sensitivity in the liver was lower in the ALA-Ex compared with the CON-Ex group [41], and thus it is possible that the ability of insulin to suppress hepatic glucose output was also attenuated in the ALA-Ex group, which would explain the profound impact of the combined intervention on glucose tolerance in spite of increased skeletal muscle insulin sensitivity. Therefore, taken together, we provide clear evidence that ALA prevents exercised-induced improvements in whole-body glucose homeostasis in an obese rodent model, an effect that appears to involve, at least in part, alterations within eWAT.
The effects of ALA and exercise on cellular homeostasis within the eWAT
The induction of insulin resistance within WAT has been proposed to involve a number of mechanisms including, but not limited to, reductions in mitochondrial content [9, 10], increased mitochondrial ROS [45], oxidative stress [2] and inflammation [2]. In the present study, while ALA did not alter mtDNA or OXPHOS content within eWAT, ALA supplementation decreased maximal respiration and increased the phosphorylation of JNK and ERK stress kinases, responses that may contribute to the apparent maladaptive response to exercise training while consuming ALA. The apparent induction of cellular stress in the combined intervention group is supported by the previous finding that n-3 PUFA supplementation attenuates exercise-mediated splenic immune cell activation [38]. Despite the apparent negative effects of consuming ALA while training, ALA decreased P-CoA induced H2O2 emission and 4-HNE content within eWAT, a situation normally associated with an improvement in insulin sensitivity in diverse tissues [46, 49]. However, considering the observation that ALA prevented exercise-induced improvements in glucose homeostasis and insulin-mediated phosphorylation of Akt in eWAT; alternatively, ROS may be required for cellular adaptations to exercise and thus the observed reduction in P-CoA-mediated ROS and 4-HNE may not actually represent an optimal cellular response. This possibility is supported by previous research, which has shown that consumption of antioxidants attenuates exercise-induced gene transcription [29,30,31]. Altogether, the present data highlights that ALA consumption attenuates exercise-induced improvements in eWAT insulin sensitivity in association with markers of cellular stress and decreased mitochondrial respiration and ROS emission. However, the consumption of ALA during training did not alter markers of mitochondrial content, suggesting ALA did not mitigate all responses to training. Indeed, the reduction in mitochondrial content with training was surprising considering numerous reports showing the contrary [7, 8]. While it is tempting to speculate that exercise decreased mitochondrial content because of the induction of adipogenesis (i.e. an increase in nuclear DNA relative to mtDNA), an alternative explanation could be that exercise improved the intrinsic function of mitochondria (demonstrated by an increase in respiration/mtDNA), negating the requirement for sustained mitochondrial content. Clearly, detailed temporal responses in concert with histology are required to delineate if adipogenesis contributes to the unique responses observed. Regardless, the present data highlights that mitochondrial respiratory function is compromised with combined ALA consumption and exercise training in obese Zucker rats.
Translation to humans
While the present data provides evidence that ALA supplementation is detrimental with respect to exercise-mediated improvements in glucose homeostasis, caution is warranted before extrapolating these findings to humans. The present study supplemented a standard diet with 10% flaxseed oil, which is physiologically impossible to achieve in humans. For example, a supplementation of 3 g/day of n-3 PUFAs in humans represents ~ 0.02% of daily lipids, assuming a diet consisting of 40% fat. In addition, in the present study animals did not train overly aggressively, as obese Zucker rats run at a low intensity. This probably explains the lack of robust changes in mitochondrial biogenesis in the skeletal muscle of these animals. Since humans display greater adaptations to exercise training and consume a smaller amount of n-3 PUFAs in a dietary supplement, it is possible that a detrimental effect would not be observed in a combined intervention. Moreover, in the present study ALA was utilised as a fatty acid but most n-3 PUFA supplements are fortified with EPA and DHA. While ALA is converted to these lipids, the desaturation process is relatively minor [50], and therefore EPA and DHA might exert a different biological effect. Nevertheless, previous research using a diet low in ALA and fortified in EPA and DHA has shown that the beneficial responses of n-3 PUFAs with respect to immune cell activation is prevented with exercise [38], suggesting the responses observed in the present study will not be isolated to ALA. Clearly, future work in humans is required to fully delineate the possible interaction between n-3 PUFAs and exercise training.
In conclusion, we assessed the combined effect of 4 week ALA supplementation and exercise training on whole-body glucose homeostasis in obese Zucker rats. We show that ALA attenuated exercise-mediated improvements in whole-body glucose tolerance in concert with select markers of cellular stress, impaired mitochondrial bioenergetics and insulin resistance within eWAT. In contrast, exercise displayed beneficial responses within muscle regardless of ALA consumption. Taken together, in a model of obesity, ALA supplementation prevents exercise-mediated improvements in glucose tolerance. However, since eWAT contributes minimally to direct glucose tolerance, the definitive mechanism of action for the impairment remains to be elucidated.
Abbreviations
- ALA:
-
α-linolenic acid
- ANT:
-
Adenosine nucleotide translocase
- CON:
-
Control (diet)
- COXIV:
-
Cytochrome c oxidase IV
- DHA:
-
Docosahexaenoic acid
- DNP:
-
2,4-Dinitrophenol
- EPA:
-
Eicosapentaenoic acid
- ERK:
-
Extracellular signalling-related kinase
- eWAT:
-
Epididymal white adipose tissue
- Ex:
-
Exercise (group)
- 4-HNE:
-
trans-4-Hydroxy-2-nonenal
- JNK:
-
c-Jun N-terminal kinase
- MCP1:
-
Monocyte chemotactic protein 1
- mtDNA:
-
Mitochondrial DNA
- OXPHOS:
-
Mitochondrial oxidative phosphorylation
- P-CoA:
-
Palmitoyl-CoA
- PDHE1α:
-
Pyruvate dehydrogenase subunit E1-alpha
- PmFbs:
-
Permeabilised muscle fibres
- PUFA:
-
Polyunsaturated fatty acid
- ROS:
-
Reactive oxygen species
- Sed:
-
Sedentary (group)
- WAT:
-
White adipose tissue
References
Meo SD, Iossa S, Venditti P (2017) Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol 233:R15–R42. https://doi.org/10.1530/JOE-16-0598
Cusi K (2010) The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr Diab Rep 10:306–315. https://doi.org/10.1007/s11892-010-0122-6
Ploug T, Stallknecht BM, Pedersen O et al (1990) Effect of endurance training on glucose transport capacity and glucose transporter expression in rat skeletal muscle. Am J Phys 259:E778–E786
Caponi PW, Lehnen AM, Pinto GH et al (2013) Aerobic exercise training induces metabolic benefits in rats with metabolic syndrome independent of dietary changes. Clinics 68:1010–1017. https://doi.org/10.6061/clinics/2013(07)20
Flynn MG, McFarlin BK, Markofski MM (2007) The anti-inflammatory actions of exercise training. Am J Lifestyle Med 1:220–235. https://doi.org/10.1177/1559827607300283
Holloszy JO, Booth FW (1976) Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol 38:273–291. https://doi.org/10.1146/annurev.ph.38.030176.001421
Sutherland LN, Bomhof MR, Capozzi LC et al (2009) Exercise and adrenaline increase PGC-1α mRNA expression in rat adipose tissue. J Physiol 587:1607–1617. https://doi.org/10.1113/jphysiol.2008.165464
Stallknecht B, Vinten J, Ploug T, Galbo H (1991) Increased activities of mitochondrial enzymes in white adipose tissue in trained rats. Am J Phys 261:E410–E414
Choo H-J, Kim J-H, Kwon O-B et al (2006) Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 49:784–791. https://doi.org/10.1007/s00125-006-0170-2
Wilson-Fritch L, Nicoloro S, Chouinard M et al (2004) Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114:1281–1289. https://doi.org/10.1172/JCI21752
Chomentowski P, Coen PM, Radiková Z et al (2010) Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab 96:494–503. https://doi.org/10.1210/jc.2010-0822
Asterholm IW, Mundy DI, Weng J et al (2012) Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab 15:171–185. https://doi.org/10.1016/j.cmet.2012.01.004
Beaudoin M-S, Snook LA, Arkell AM et al (2013) Resveratrol supplementation improves white adipose tissue function in a depot-specific manner in Zucker diabetic fatty rats. Am J Phys Regul Integr Comp Phys 305:R542–R551. https://doi.org/10.1152/ajpregu.00200.2013
Benton CR, Holloway GP, Han X-X et al (2010) Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia 53:2008–2019. https://doi.org/10.1007/s00125-010-1773-1
Benton CR, Nickerson JG, Lally J et al (2008) Modest PGC-1α overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. J Biol Chem 283:4228–4240. https://doi.org/10.1074/jbc.M704332200
Boden G, Homko C, Mozzoli M et al (2005) Thiazolidinediones upregulate fatty acid uptake and oxidation in adipose tissue of diabetic patients. Diabetes 54:880–885
Rong JX, Qiu Y, Hansen MK et al (2007) Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 56:1751–1760. https://doi.org/10.2337/db06-1135
Storlien LH, Kraegen EW, Chisholm DJ et al (1987) Fish oil prevents insulin resistance induced by high-fat feeding in rats. Science 237:885–888
Neschen S, Morino K, Dong J et al (2007) n-3 Fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-alpha-dependent manner. Diabetes 56:1034–1041. https://doi.org/10.2337/db06-1206
Lanza IR, Blachnio-Zabielska A, Johnson ML et al (2013) Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet. Am J Physiol Endocrinol Metab 304:E1391–E1403. https://doi.org/10.1152/ajpendo.00584.2012
Simopoulos AP (2002) The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed Pharmacother Biomed Pharmacother 56:365–379
Popp-Snijders C, Schouten JA, Heine RJ et al (1987) Dietary supplementation of omega-3 polyunsaturated fatty acids improves insulin sensitivity in non-insulin-dependent diabetes. Diabetes Res Edinb Scotl 4:141–147
Khairallah RJ, Kim J, O’Shea KM et al (2012) Improved mitochondrial function with diet-induced increase in either docosahexaenoic acid or arachidonic acid in membrane phospholipids. PLoS One 7:e34402. https://doi.org/10.1371/journal.pone.0034402
Herbst EAF, Paglialunga S, Gerling C et al (2014) Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J Physiol 592:1341–1352. https://doi.org/10.1113/jphysiol.2013.267336
Jelenik T, Rossmeisl M, Kuda O et al (2010) AMP-activated protein kinase α2 subunit is required for the preservation of hepatic insulin sensitivity by n-3 polyunsaturated fatty acids. Diabetes 59:2737–2746. https://doi.org/10.2337/db09-1716
Ruzickova J, Rossmeisl M, Prazak T et al (2004) Omega-3 PUFA of marine origin limit diet-induced obesity in mice by reducing cellularity of adipose tissue. Lipids 39:1177–1185
Flachs P, Horakova O, Brauner P et al (2005) Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce beta-oxidation in white fat. Diabetologia 48:2365–2375. https://doi.org/10.1007/s00125-005-1944-7
Hensler M, Bardova K, Jilkova ZM et al (2011) The inhibition of fat cell proliferation by n-3 fatty acids in dietary obese mice. Lipids Health Dis 10:128. https://doi.org/10.1186/1476-511X-10-128
Strobel NA, Peake JM, Matsumoto A et al (2011) Antioxidant supplementation reduces skeletal muscle mitochondrial biogenesis. Med Sci Sports Exerc 43:1017–1024. https://doi.org/10.1249/MSS.0b013e318203afa3
Gomez-Cabrera M-C, Domenech E, Romagnoli M et al (2008) Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr 87:142–149
Ristow M, Zarse K, Oberbach A et al (2009) Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A 106:8665–8670. https://doi.org/10.1073/pnas.0903485106
Gliemann L, Schmidt JF, Olesen J et al (2013) Resveratrol blunts the positive effects of exercise training on cardiovascular health in aged men. J Physiol 591:5047–5059. https://doi.org/10.1113/jphysiol.2013.258061
Scribbans TD, Ma JK, Edgett BA et al (2014) Resveratrol supplementation does not augment performance adaptations or fibre-type–specific responses to high-intensity interval training in humans. Appl Physiol Nutr Metab 39:1305–1313. https://doi.org/10.1139/apnm-2014-0070
Yoshino J, Conte C, Fontana L et al (2012) Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab 16:658–664. https://doi.org/10.1016/j.cmet.2012.09.015
Olesen J, Gliemann L, Biensø R et al (2014) Exercise training, but not resveratrol, improves metabolic and inflammatory status in skeletal muscle of aged men. J Physiol 592:1873–1886. https://doi.org/10.1113/jphysiol.2013.270256
Ringholm S, Olesen J, Pedersen JT et al (2013) Effect of lifelong resveratrol supplementation and exercise training on skeletal muscle oxidative capacity in aging mice; impact of PGC-1α. Exp Gerontol 48:1311–1318. https://doi.org/10.1016/j.exger.2013.08.012
Polley KR, Jenkins N, O’Connor P, McCully K (2016) Influence of exercise training with resveratrol supplementation on skeletal muscle mitochondrial capacity. Appl Physiol Nutr Metab Physiol Appl Nutr Metab 41:26–32. https://doi.org/10.1139/apnm-2015-0370
Robinson LE, Field CJ (1998) Dietary long-chain (n-3) fatty acids facilitate immune cell activation in sedentary, but not exercise-trained rats. J Nutr 128:498–504
Matravadia S, Zabielski P, Chabowski A et al (2016) LA and ALA prevent glucose intolerance in obese male rats without reducing reactive lipid content, but cause tissue-specific changes in fatty acid composition. Am J Phys Regul Integr Comp Phys 310:R619–R630. https://doi.org/10.1152/ajpregu.00297.2015
Miotto PM, Horbatuk M, Proudfoot R et al (2017) Alpha-linolenic acid supplementation and exercise training reveal independent and additive responses on hepatic lipid accumulation in obese rats. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.00438.2016
Barbeau P-A, Holloway TM, Whitfield J et al (2017) α-linolenic acid and exercise training independently, and additively, decreases blood pressure and prevents diastolic dysfunction in obese Zucker rats. J Physiol. https://doi.org/10.1113/JP274036
Matravadia S, Herbst EAF, Jain SS et al (2014) Both linoleic and α-linolenic acid prevent insulin resistance but have divergent impacts on skeletal muscle mitochondrial bioenergetics in obese Zucker rats. Am J Physiol Endocrinol Metab 307:E102–E114. https://doi.org/10.1152/ajpendo.00032.2014
Perry CGR, Kane DA, Lin C-T et al (2011) Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J 437:215–222. https://doi.org/10.1042/BJ20110366
Smith BK, Perry CGR, Herbst EAF et al (2013) Submaximal ADP-stimulated respiration is impaired in ZDF rats and recovered by resveratrol. J Physiol 591:6089–6101. https://doi.org/10.1113/jphysiol.2013.259226
Paglialunga S, Ludzki A, Root-McCaig J, Holloway GP (2015) In adipose tissue, increased mitochondrial emission of reactive oxygen species is important for short-term high-fat diet-induced insulin resistance in mice. Diabetologia 58:1071–1080. https://doi.org/10.1007/s00125-015-3531-x
Anderson EJ, Lustig ME, Boyle KE et al (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119:573–581. https://doi.org/10.1172/JCI37048
Kraunsøe R, Boushel R, Hansen CN et al (2010) Mitochondrial respiration in subcutaneous and visceral adipose tissue from patients with morbid obesity. J Physiol 588:2023–2032. https://doi.org/10.1113/jphysiol.2009.184754
McGlory C, Galloway SDR, Hamilton DL et al (2014) Temporal changes in human skeletal muscle and blood lipid composition with fish oil supplementation. Prostaglandins Leukot Essent Fat Acids 90:199–206. https://doi.org/10.1016/j.plefa.2014.03.001
Houstis N, Rosen ED, Lander ES (2006) Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440:944–948. https://doi.org/10.1038/nature04634
Brenna JT (2002) Efficiency of conversion of alpha-linolenic acid to long chain n-3 fatty acids in man. Curr Opin Clin Nutr Metab Care 5:127–132
Acknowledgements
We would like to thank D.C. Wright (Human Health & Nutritional Sciences, University of Guelph, Canada) for generously providing some of the antibodies used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
All relevant data are included in the article and/or the ESM files.
Funding
This work was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC, GPH, 03656). Infrastructure was purchased with assistance from the Canadian Foundation for Innovation/Ontario Research Fund (GPH, 25136). CMFM, PMM and EAFH were recipients of NSERC graduate scholarships.
Duality of interest
The authors declare that there is no duality of interest associated with this article.
Contribution statement
GPH designed the experiments. CMFM and GPH wrote the manuscript. CMFM, RP, PMM, EAFH and REKM performed experiments, and analysed and interpreted data. All authors edited the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All people designated as authors qualify for authorship, and all those who qualify for authorship are listed. CMFM is the guarantor of this work.
Electronic supplementary material
ESM
(PDF 1326 kb)
Rights and permissions
About this article

Cite this article
Monaco, C.M., Proudfoot, R., Miotto, P.M. et al. α-linolenic acid supplementation prevents exercise-induced improvements in white adipose tissue mitochondrial bioenergetics and whole-body glucose homeostasis in obese Zucker rats. Diabetologia 61, 433–444 (2018). https://doi.org/10.1007/s00125-017-4456-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4456-3