
Abstract
Aims/hypothesis
Genetic predisposition to type 2 diabetes is well-established, and genetic risk scores (GRS) have been developed that capture heritable liabilities for type 2 diabetes phenotypes. However, the proteins through which these genetic variants influence risk have not been thoroughly investigated. This study aimed to identify proteins and pathways through which type 2 diabetes risk variants may influence pathophysiology.
Methods
Using a proteomics data-driven approach in a discovery sample of 7241 White participants in the Atherosclerosis Risk in Communities Study (ARIC) cohort and a replication sample of 1674 Black ARIC participants, we interrogated plasma levels of 4870 proteins and four GRS of specific type 2 diabetes phenotypes related to beta cell function, insulin resistance, lipodystrophy, BMI/blood lipid abnormalities and a composite score of all variants combined.
Results
Twenty-two plasma proteins were identified in White participants after Bonferroni correction. Of the 22 protein–GRS associations that were statistically significant, 10 were replicated in Black participants and all but one were directionally consistent. In a secondary analysis, 18 of the 22 proteins were found to be associated with prevalent type 2 diabetes and ten proteins were associated with incident type 2 diabetes. Two-sample Mendelian randomisation indicated that complement C2 may be causally related to greater type 2 diabetes risk (inverse variance weighted estimate: OR 1.65 per SD; p=7.0 × 10−3), while neuropilin-2 was inversely associated (OR 0.44 per SD; p=8.0 × 10−3).
Conclusions/interpretation
Identified proteins may represent viable intervention or pharmacological targets to prevent, reverse or slow type 2 diabetes progression, and further research is needed to pursue these targets.
Graphical abstract

Similar content being viewed by others


Introduction
Type 2 diabetes is characterised by complex disruptions in cell signalling and metabolic homeostasis across multiple organ systems. Among these, peripheral and hepatic insulin resistance, pancreatic beta cell dysfunction and adipose tissue accumulation are typical, and individuals with type 2 diabetes often present with various combinations of these phenotypes [1,2,3]. While lifestyle risk factors such as physical inactivity and hyper-caloric diets are well-established in disease development [4, 5], genetic liabilities also influence risk [6,7,8,9] and have been shown to contribute to the phenotypic heterogeneity of type 2 diabetes [9, 10].
Genome-wide association studies (GWAS) have identified over 450 gene variants related to type 2 diabetes [7,8,9,10,11,12]—most showing modest magnitudes of association with disease risk, typically <10% greater risk per allele [12,13,14]. Numerous additive genetic risk scores (GRS) for type 2 diabetes have been generated to integrate risk information across identified gene variants [6, 9]. Refining this approach to address the heterogeneity of type 2 diabetes, Goodarzi et al [10] developed four type 2 diabetes GRS for the distinct phenotypes of beta cell function, insulin resistance, lipodystrophy and BMI/aberrant blood lipids, which were comprised of independent clusters of gene variants. These type 2 diabetes risk variants may have cis or trans effects on gene expression, and the proteins and networks through which they affect metabolic dysfunction are unknown.
The present study aimed to identify proteins and pathways through which type 2 diabetes risk variants may collectively influence pathophysiology. To strengthen causal inference, a stepwise approach was applied in which: (1) plasma proteins associated with the type 2 diabetes phenotype GRSs were identified from a platform comprised of 4870 protein measures; (2) cross-sectional and prospective associations with type 2 diabetes were examined for identified proteins; and (3) a two-sample Mendelian randomisation (MR) analysis was conducted to investigate potential causal relationships between these proteins and type 2 diabetes. Collectively, these findings aim to provide greater understanding of genetic liability to type 2 diabetes.
Methods
Study population
The Atherosclerosis Risk in Communities Study (ARIC) of 15,792 individuals was originally designed to identify risk factors for cardiovascular disease and atherosclerosis and has been described in detail previously [15]. Male and female participants aged 45–64 years were recruited from four communities in the USA (Washington County, MD; the northwest suburbs of Minneapolis, MN; Jackson, MS; Forsyth County, NC). Information about risk factors was obtained at baseline (visit 1; 1987–1989) and in follow-up examinations: visit 2 (1990–1992), visit 3 (1993–1995), visit 4 (1996–1998), visit 5 (2011–2013), visit 6 (2016–2017) and visit 7 (2018–2019). Visit 3 served as the baseline for this analysis. Information on risk factors and use of medications was also obtained through follow-up phone calls to participants (annually before 2012; twice-yearly thereafter). Institutional review boards at each site approved the study protocol, and written informed consent was obtained from all participants.
Genetic risk scores for type 2 diabetes
GRS for type 2 diabetes were constructed from SNP dosages and clustered into four mutually exclusive phenotypes of beta cell function (50 SNPs), insulin resistance (29 SNPs), lipodystrophy (12 SNPs) and BMI/lipid abnormalities (12 SNPs), using SNPs and weights provided by Goodarzi et al [10]. Two additional GRS were generated from SNPs that remained uncategorised by Goodarzi et al in terms of diabetes phenotype (339 SNPs) and all SNPs combined (442 SNPs) [10]. For each participant, a weighted GRS was generated by multiplying the number of risk alleles at each variant by its corresponding diabetes-related risk value (logOR) and then summing the products. For variants unavailable in the ARIC GWAS data, a proxy variant in tight linkage disequilibrium (r2≥0.80) was substituted. In Black participants, no suitable proxy variants were available for rs145904381 (insulin resistance GRS) or rs1005752 (beta cell function GRS).
Genotyping
Genotyping, imputation and quality control procedures have been described previously [16]. Genome-Wide Human SNP array 6.0 (Affymetrix, USA) was used to genotype genomic DNA extracted from whole blood, and race-specific imputation of variant dosages to the TOPMed reference panel (freeze 5b) was performed to increase the number of genetic markers beyond this array [17]. Individuals who were first-degree relatives of each other, genetic outliers and those whose genotypes did not match genotype data from other platforms were removed prior to imputation. Based on the GWAS data, principal components reflecting population substructure or genetic ancestry were generated using EIGENSTRAT [18].
Proteomics measurement
Proteins were measured in plasma collected at visit 3 (1993–1995) using a modified DNA aptamer-based array that measures 4870 proteins (SomaScan version 4.0, SomaLogic, USA), described previously [19, 20]. Samples that had been stored at −70°C and not previously thawed were transferred to the SomaLogic laboratory and incubated with proprietary reagents. Protein levels were quantified using single-stranded DNA-based modified aptamers that bind to specific protein epitopes. Protein concentrations are reported as relative fluorescent units.
Proteomics quality control
Protein measurements by SomaScan were standardised and normalised as previously described [21, 22]. Briefly, hybridisation control normalisation was applied to each sample based on a set of hybridisation control sequences to correct for systematic biases during hybridisation. Median signal normalisation was applied to measures within plates to remove sample or assay biases due to variations in pipetting, reagent concentrations, assay timing and other sources of systematic variability within single plate runs. Each plate contained calibrators for each aptamer reagent to correct for plate-to-plate variation based on global reference materials. ARIC investigators previously conducted a pilot study of SomaScan version 3 in 42 ARIC participants and reported excellent metrics of assay reproducibility: median coefficient of variance (Q1–Q3) of 5.0 (4.1–6.9) and median intraclass correlation (Q1–Q3) of 0.96 (0.92–0.98) [22].
Logarithmic transformations (base 2) were applied to all proteins prior to analysis. A total of 422 blind duplicate plasma aliquots were included, and the median inter-assay Bland–Altman coefficient of variation was 6.3%. The median split sample reliability coefficient was 0.85 after applying the following quality control filters on 5284 available aptamer measurements: Bland–Altman coefficient of variation >50% or a variance of <0.01 on the log scale (n=94 excluded) and non-specific binding to non-proteins (n=313 excluded). After all quality control measures were completed, 4870 aptamer measurements remained, corresponding to 4697 unique human proteins or protein complexes.
Assessment of kidney function
The eGFR at visit 3 was included as a covariate to account for the influences of kidney function on plasma proteins. The Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation was used to estimate GFR (ml/min per 1.73 m2) [23]. A creatinase enzymatic method using a Roche Modular P Chemistry Analyzer measured serum creatinine (Roche Diagnostics, USA) and was standardised to isotope-dilution mass spectrometry as described previously [24]. A turbidimetric immunoassay (Gentian, Norway) calibrated and standardised to the International Federation of Clinical Chemistry and Laboratory Medicine reference was used to measure serum cystatin C level [25].
Diabetes ascertainment
Prevalent diabetes at visit 3 was defined by a fasting blood glucose level ≥7.0 mmol/l, non-fasting blood glucose ≥11.1 mmol/l, self-reported physician diagnosis of diabetes, or self-reported use of diabetes medications. Among those without prevalent diabetes at visit 3, incident diabetes was obtained by self-reported physician diagnosis of diabetes or self-reported use of diabetes medications at visit 4 (between 1996 and 1998) and in annual or semi-annual follow-up telephone interviews between 1999 and 2019.
Statistical analyses
A total of 12,887 participants attended visit 3. After excluding those with missing genetic data (n=2737), proteomic data (n=1041) or covariate data (n=194), 1674 Black and 7241 White participants were available for the main analysis of GRS–protein associations. Race-specific linear regression models were used to estimate associations between each of the six GRSs and proteins. Covariate adjustments were made for age, sex, field centre, eGFR at visit 3, and ten principal components of ancestry (unadjusted analyses shown in electronic supplementary material [ESM] Table 1). For each protein, participants with values that were more than 6 SD from the population mean were excluded. Bonferroni correction was applied to account for multiple testing: the 4870 protein outcomes and six GRSs stipulated a significance threshold of p<1.71 × 10−6 in White participants. Analyses in Black participants were treated as a replication. Significant replication was defined as having the same direction of association observed in White participants and meeting a Bonferroni-corrected significance threshold that accounts for the number of proteins tested for each GRS in Black participants.
Proteins identified in the main GRS–protein analysis were tested for associations with prevalent and incident diabetes in a secondary analysis, and a schematic of analytical sample sizes is shown in ESM Fig. 1. Logistic regression was used to estimate ORs for each plasma protein with prevalent diabetes at visit 3 among those with all covariate data (n=11,063 participants and 1733 cases), with adjustment for age, sex, field centre/race, physical activity based on the ARIC physical activity sport and exercise index [26], BMI, cigarette smoking status and eGFR (all assessed at the visit 3 time point). Field centre and race variables were combined as only Black participants were recruited at the field centre in Jackson, MS. A sensitivity analysis was conducted in which the definition of prevalent type 2 diabetes was limited to self-reported physician diagnosis or use of diabetes medications (n=10,995 participants and 1067 cases), since including laboratory values of fasting glucose may inflate the type I error rate. For prospective analysis, Cox regression was used to estimate HRs for incident diabetes over a median follow-up of 23.9 years (n=6224 participants, 2354 incident cases) with the same adjustments described above. Date of diagnosis was defined as the date of the interview at which diabetes was first reported; individuals who remained diabetes-free were censored at the time of their last available interview. For both cross-sectional and prospective analyses, Bonferroni correction was applied to correct for multiple testing: 22 proteins were tested, stipulating a significance threshold of p≤2.3 × 10−3. Unadjusted protein–type 2 diabetes analyses shown in ESM Table 2.
Two-sample MR was performed using the MR-Base web application [27] and was restricted to proteins found to be related to both prevalent and incident type 2 diabetes in the secondary analysis. Instrumental variables consisted of protein quantitative trait loci (pQTLs) identified in White ARIC participants by GWAS analyses between the proteins and genetic dosages imputed based on the TOPMed (freeze 5b) reference, with adjustment for age, sex, field centre and ten principal components of ancestry. Clumping was used to prune pQTLs that did not reach the significance threshold (p≤5 × 10−8), were in linkage disequilibrium (r2 for linkage disequilibrium <0.2) or were within a clumping distance threshold of 10,000 kb. Identified pQTLs in the ARIC study were confirmed using published data from the INTERVAL [28] and AGES-Reykjavik [29] studies. For identified pQTLs, type 2 diabetes summary statistics were obtained from a large consortium study available through the MR-Base platform [12]; this study was selected based on the numbers of type 2 diabetes cases (n for events=62,892) and overlap with the TOPMed imputation panel used in the ARIC study. For pQTLs that were unavailable in the outcome summary data, proxy SNPs in tight linkage disequilibrium (r2≥0.8) were substituted. For proteins with only one pQTL, Wald ratios were calculated. Two and three pQTLs were identified for neurofascin and complement C2, respectively; inverse variance weighted (IVW) meta-analyses of Wald ratios were performed, and an MR-Egger sensitivity analysis [30] was conducted for C2. For significant two-sample MR results, reverse causality was evaluated. Type 2 diabetes-related SNPs were derived from the above consortium study [12]. Type 2 diabetes-related SNPs from this study were tested for their associations with neuropilin-2 and complement C2, and their associations with these proteins were obtained from GWAS by Sun et al [28] and Suhre et al [31], respectively. MR-Egger and IVW estimates were generated.
Canonical pathway enrichment was examined using the Ingenuity Pathway Analysis platform (Qiagen, USA) to determine potential mechanisms and signalling cascades through which GRSs may influence respective pathogenic phenotypes [32]. Entrez Gene IDs, log expression ratios (i.e. variant–protein association estimates) and false discovery rate q values from primary analyses were uploaded. To control for multiple comparisons, a Benjamini–Hochberg false discovery rate was applied and proteins with q values ≤0.05 were deemed significant. Core analysis was performed, and direct and indirect experimentally confirmed relationships across species were evaluated.
Results
The analytical sample comprised 8915 participants with a mean age of 60.1 years, of whom 54.1% were female. To assess the potential for selection bias, all 12,887 participants who attended visit 3 were compared to the analytical sample with complete genotype, proteomic and covariate data (Table 1). The analytical sample had a smaller proportion of Black participants than the visit 3 cohort; however, demographic, clinical and lifestyle characteristics were otherwise similar. Upon stratification into Black (n=1674) and White (n=7241) participant groups, it was found that, compared with White participants, Black participants showed higher proportions of current smokers (17.2% vs 21.7%) and never smokers (38.5% vs 45.4%), and a greater prevalence of type 2 diabetes (12.7% vs 27.7%).
Identification of GRS-related proteins
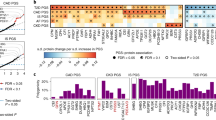
As shown in Fig. 1, the 12-variant type 2 diabetes GRS defining the BMI/lipid phenotype was associated with 14 plasma proteins in White participants. Of these, eight were replicated in Black participants: leucine-rich repeat neuronal protein 1 (p=2.1 × 10−14), MOB-like protein phocein (p=9.1 × 10−6), dehydrogenase/reductase SDR family member 9 (p=9.0 × 10−7), ADP-ribosylation factor-like protein 2 (p=1.2 × 10−4), d-3-phosphoglycerate dehydrogenase (p=1.5 × 10−13), neurofilament light polypeptide (p=4.3 × 10−14), tubulin-specific chaperone A (p=9.2 × 10−15) and protein S100-A13 (p=1.0 × 10−13).

Plasma proteins that were significantly associated with the type 2 diabetes GRS defining the BMI/lipid phenotype in 7241 White participants, with replication in 1674 Black participants. Results for a regression model examining the BMI GRS and protein outcomes (effect size expressed as difference in log2 protein level per SD in the GRS), adjusted for age, sex, field centre, ten principal components of ancestry and eGFR at ARIC study visit 3. The Bonferroni correction for multiple testing was applied with a significance threshold of p<1.7 × 10−6 in White participants; Black individuals were treated as a replication cohort with a significance threshold of p<4.2 × 10−3 for correction of 12 tests. Asterisks indicate replication in Black participants
As shown in ESM Table 3, the 29-variant type 2 diabetes GRS defining the insulin resistance phenotype was related to plasma chymotrypsinogen B2 (p=1.1 × 10−12) and SLIT and NTRK-like protein 3 (p=1.2 × 10−6) in White participants; the latter result was replicated in Black participants (p=0.02). The 12-variant type 2 diabetes GRS defining the lipodystrophy phenotype was related to plasma neuromedin-B (p=7.7 × 10−7) in White participants but this result was not replicated in Black participants (p>0.05). The 50-variant type 2 diabetes GRS defining the beta cell function phenotype was related to cartilage intermediate layer protein 2 in White participants (p=2.2 × 10−10), but the association was not replicated in Black participants.
Over 300 GWAS-identified gene variants were previously associated with type 2 diabetes but remain uncategorised with respect to a distinct type 2 diabetes phenotype. When we combined these 339 variants into an ‘uncategorised’ phenotype GRS, one significant plasma protein association was observed in White participants: lactase-phlorizin hydrolase (p=8.5 × 10−27); this result was replicated in Black participants (p=0.02). Finally, combining all GWAS-derived variants generated a 442-variant GRS that was related to seven proteins in White participants, as shown in Fig. 2. Of these, no associations were replicated in Black participants following Bonferroni correction (p>7.0 × 10−3), but all were directionally consistent. Point estimates, 95% CIs and p values for these associations are provided in ESM Table 3. Associations between GRS exposure variables and metabolic characteristics are shown in ESM Table 4.

Plasma proteins that were significantly associated with the overall type 2 diabetes genetic risk score in 7241 White participants, with replication in 1674 Black participants. Results for a regression model examining the GRS of all diabetes-related variants and protein outcomes (effect size expressed as difference in log2 protein level per SD in the GRS), adjusted for age, sex, field centre, ten principal components of ancestry, and eGFR at ARIC study visit 3. The Bonferroni correction for multiple testing was applied with a significance threshold of p<1.7 × 10−6 in White participants; Black individuals were treated as a replication cohort with a significance threshold of p<7.1 × 10−3 for correction of seven tests
A sensitivity analysis was performed in which 555 participants with physician-diagnosed diabetes or those taking diabetes medications were excluded from the GRS–protein analysis; the results are shown in ESM Table 5. Most associations in the subset without diagnosed diabetes were similar in magnitude to those observed in the full analytic sample.
Associations of identified proteins and type 2 diabetes
In the secondary analysis, the 22 proteins identified in the main analysis were tested for associations with prevalent and incident diabetes. Among 11,063 Black and White participants at visit 3, there were 1733 individuals with type 2 diabetes. As shown in Fig. 3a, cross-sectional analysis showed that 18 of the 22 proteins were significantly related to prevalent diabetes (p≤2.2 × 10−3). Four of these 22 proteins were moderately to strongly correlated with one another. Specifically, neurofilament light polypeptide was correlated with tubulin-specific chaperone protein A (r=0.79; p<0.001), d-3-phosphoglycerate-dehydrogenase (r=0.69; p<0.001) and protein-S100-A13 (r=0.65; p<0.001). Despite these correlations, all proteins were tested for their associations with type 2 diabetes outcomes.

Associations between identified proteins and (a) prevalent type 2 diabetes, among 11,063 participants at visit 3 (OR and 95% CI are shown; n for events=1733), or (b) incident type 2 diabetes, among 6224 ARIC participants over a median of 23.9 years of follow-up (HR ratios and 95% CI are shown; n=2354 events). Models were adjusted for age, sex, field centre/race, physical activity, BMI, cigarette smoking status and eGFR at visit 3
A sensitivity analysis was performed in which prevalent type 2 diabetes was defined as self-reported physician diagnosis or use of type 2 diabetes medications (n for cases=1069). The results are shown in ESM Table 6 and are consistent with the previous model that used a more expansive definition of type 2 diabetes that included laboratory values of fasting glucose obtained at visit 3, indicating robust associations.
As shown in Fig. 3b, 10 of the 22 plasma proteins were significantly related to incident diabetes over a median follow-up of 23.9 years among 6224 Black and White participants without diabetes at visit 3 (n=2354 incident cases). ESM Table 7 provides point estimates, 95% CIs and p values for cross-sectional and prospective analyses.
ESM Table 8 provides a summary of our main findings: protein–GRS associations (0 or 1), replication in Black participants, and protein associations with prevalent and incident type 2 diabetes.
Two-sample MR analysis
For the ten proteins associated with both prevalent and incident type 2 diabetes, we performed a two-sample MR analysis to examine relationships between genetically determined protein levels and type 2 diabetes outcomes. Five of the ten proteins were found to have available genetic instruments. The results of this analysis are presented in Table 2, showing the target protein, corresponding pQTL(s) and r2 value specifying the percentage variance in protein level explained by the pQTL(s) in the ARIC study. A significant and inverse association was observed for neuropilin-2, suggesting that low neuropilin-2 levels promote type 2 diabetes development (OR per SD 0.44; p=0.008). Complement C2 was found to be positively related to type 2 diabetes (OR per SD 1.65; p=0.007); however, the MR-Egger results for complement C2 were not significant (OR per SD 1.07; p=0.96) and the test for directional horizontal pleiotropy was null (p=0.72). Further details of pQTLs are shown in ESM Table 9.
The potential for reverse causality was evaluated for neuropilin-2 and complement C2 (ESM Table 10). For neuropilin-2, 112 type 2 diabetes-related SNPs were available for analysis; estimates were null for IVW analyses (OR per SD 0.87; p=0.13) and MR-Egger sensitivity analyses (OR per SD 1.02; p=0.6) (ESM Fig. 2). For complement C2, 57 type 2 diabetes-related SNPs were available for analysis; estimates were null for IVW analyses (OR per SD 1.06; p=0.76) and MR-Egger sensitivity analyses (OR per SD 1.00; p=0.96) (ESM Fig. 3). Tests for horizontal pleiotropy were null for both proteins but approached significance for neuropilin-2 (p=0.054).
Pathway enrichment analysis
Pathway analysis was performed for proteins associated with each GRS. One canonical pathway was identified in which protein associations with the GRS defining the BMI/lipid phenotype and plasma proteins indicated retinoate biosynthesis I activation (z score 2.24; p=2.3 × 10−7). Pathway components included alcohol dehydrogenase 4 (q value=0.03), aldo-keto reductase family 1 member C4 (q0.02), dehydrogenase/reductase SDR family member 9 (q0.0002) and retinol binding protein 5 (q0.03).
Discussion
Proteomics research has examined associations with prevalent and incident T2D, its complications and responses to pharmacotherapies, but no proteomics study has examined the genetic liabilities captured by previously generated GRS specific for T2D phenotypes. Using a large-scale agnostic proteomics approach in White participants, we identified 22 unique plasma proteins associated with GRSs for BMI, insulin resistance, beta cell dysfunction and lipodystrophy, or a composite of all type 2 diabetes-related variants. Of these, ten proteins were replicated in 1674 Black participants, and 21 of the 22 associations were directionally consistent between the discovery and replication samples. Of these 22 proteins, 18 were statistically significantly related to prevalent type 2 diabetes, and ten of these were related to incident diabetes over a median of 24 years of follow-up. MR analysis showed that higher neuropilin-2 levels may be protective against type 2 diabetes, while higher complement C2 levels may be related to disease development based on the IVW estimate, although this was tempered by the MR-Egger result. Collectively, these findings suggest potential mechanisms through which the phenotype-specific GRSs confer greater risks of type 2 diabetes.
Of the identified proteins, plexin-B2, S100-A13 arfaptin-2, neurofascin and complement C2 have previously been shown to be associated with prevalent and/or incident type 2 diabetes [33]. The plexin-B2 receptor and its ligand, semaphorin 6A (with a significance level of p=1.1 × 10−5 in the fully adjusted model, above the Bonferroni-corrected threshold), have been shown to be associated with HbA1c levels in individuals with type 2 diabetes [34], and circulating levels of plexin-B2 have been shown to be positively related to greater risk of prevalent type 2 diabetes, but not incident type 2 diabetes [33]. In contrast, protein S100-A13 has been suggested to have a protective effect against adverse metabolic outcomes [33, 35]. As shown here and by Gudmundsdottir et al in the AGES-Reykjavik cohort [33], lower levels of protein S100-A13 were related to both prevalent and incident type 2 diabetes. In agreement with these findings, hypomethylation of the S100A13 gene has been shown to be associated with lower protein levels and prevalent diabetic retinopathy, potentially through NF-κB activation and greater hyperglycaemia-induced vascular damage [35]. We were unable to identify a suitable genetic instrument for these proteins for evaluation in MR analysis.
Apart from the above five proteins, the remaining identified proteins are novel observations, and few have been characterised in type 2 diabetes development—indeed, most have functions that are seemingly unrelated to metabolic health or disease. Leucine-rich repeat neuronal protein 1, neuromedin-B, neurofilament light polypeptide and neuropilin-2 have been characterised in gastric cancer [36], smooth muscle contraction [37], head and neck cancer [38] and angiogenesis [39], respectively. Despite this, we report here that the semaphorin receptor, neuropilin-2, may have a protective role against disease development based on MR results.
Arfaptin-2 and neurofascin do not have any known physiological roles in metabolic function or adipogenesis despite their associations with type 2 diabetes shown here. However, consistent with a potential protective effect, the ubiquitously expressed arfaptin-2 protein has been shown to inhibit NF-κB activity [40]. Whether arfaptin-2 suppression of NF-κB signalling influences adiposity or predisposition to adipose tissue accumulation has not been shown, but a central role for NF-κB has been reported in models of obesity [41] and insulin resistance [42].
Finally, we confirm that plasma levels of complement C2 are positively associated with risk of type 2 diabetes as shown previously [33], and the results of the MR analysis suggest a causal relationship. These findings are consistent with the established role of the complement system in inflammation and its deleterious effects on metabolic function. Recent studies have shown that activation of complement components likely promote insulin resistance [43, 44] and microvascular complications including nephropathy [45,46,47] and retinopathy [48, 49]. However, the IVW result must be considered in combination with the non-significant result of the MR-Egger sensitivity analysis. Based on the latter finding and the null directional horizontal pleiotropy finding, it appears that the instrumental variable for complement C2 likely violated the ‘instrument strength independent of direct effect’ assumption [50]. Critically, of the three pQTL components of the C2 instrumental variable, the pQTL proximal to the C2 gene (rs558702) is an intron variant, showed the largest magnitude of association with type 2 diabetes (OR per SD 2.46; 95% CI 1.45–4.18) and is more likely to be independent of horizontal pleiotropy. It may therefore provide a more reliable estimate of the effect of complement C2 on type 2 diabetes. Taken together, the potential causal effect of complement C2 on type 2 diabetes risk remains equivocal. Additional research is warranted to further characterise the role of C2 and other complement system components in type 2 diabetes.
An important consideration in evaluating the present findings is the degree of pathway convergence, which depends on the variant composition of the GRS. It has been suggested that a GRS may capture convergence in downstream disease pathways that originate with hundreds to thousands of genetic risk variants [33]. In that case, a GRS comprised of variants that operate through a shared pathway(s) would be expected to show moderate to strong associations with protein components of that pathway. Conversely, a GRS comprised of variants that operate through different pathways would be expected to show more modest associations with proteins specific to one pathway. The relatively small magnitudes of association observed here suggest that the latter is occurring, and a few gene variants within each GRS may be driving the significant findings. In addition, these GRSs are likely incomplete as not all variants contributing to a specific type 2 diabetes phenotype are currently known. Taken together, these observations may have implications for future studies to: (1) sub-classify known variants by shared pathways; and (2) identify new variants and epigenetic modifications to obtain a more complete representation of genetic liability, which may then inform the degree of pathway convergence for each diabetes phenotype.
Limitations
There are limitations in the present analysis that must be acknowledged. First, GRS construction assumed an additive genetic architecture of independent risk variants and did not account for the possibility of gene–gene or gene–environment interactions. Additionally, gold standard assessments of beta cell function, insulin resistance and lipodystrophy were not available in ARIC participants, and we were unable to assess relationships between identified proteins and these phenotypic manifestations of type 2 diabetes. Our two-sample MR analysis was limited by two factors: (1) no genetic instruments were found for cartilage intermediate layer protein-2 in the ARIC study; and (2) outcome summary statistics for the s100-A13, arfaptin-2, plexin-B2 and platelet activating factor-acetyl hydrolase pQTLs or identified proxy SNPs were unavailable in the study selected from MR-Base, which prevented MR analysis of these proteins. Limiting the generalisability of our findings, GRSs were comprised of variants identified from GWA studies conducted in individuals of European ancestry, and this may have contributed to the relatively moderate replication of GRS–protein associations among Black participants (ten of 22 proteins); however, the directional consistency in associations between groups was substantial: 21 of the 22 proteins identified in White participants were directionally consistent in Black participants. Finally, we had limited statistical power to identify pQTLs in Black participants and were therefore unable to perform MR analysis in that group; additional studies with larger samples of Black individuals are needed.
Conclusions
Using a large-scale agnostic proteomic approach, we identified 22 novel proteins associated with GRS for type 2 diabetes in the ARIC study. Most of these were found to be associated with prevalent or incident type 2 diabetes after adjustment for potential confounders. Two-sample MR analysis provided evidence that complement C2 and neuropilin-2 levels may be causally related to type 2 diabetes risk, although the former finding should be interpreted with caution. Additional research is warranted to further interrogate these proteins and characterise their possible roles in the pathophysiology of type 2 diabetes and as potential targets for pharmacological intervention.
Data availability
The datasets analysed for this study are available from the ARIC Coordinating Center (arichelp@unc.edu) on reasonable request, which may include completion of a data and material distribution agreement.
Abbreviations
- ARIC:
-
Atherosclerosis Risk in Communities
- GRS:
-
Genetic risk score
- GWAS:
-
Genome-wide association studies
- IVW:
-
Inverse variance weighted
- MR:
-
Mendelian randomisation
- pQTL:
-
Protein quantitative trait locus
References
Faerch K, Hulmán A, Solomon TP (2016) Heterogeneity of pre-diabetes and type 2 diabetes: implications for prediction, prevention and treatment responsiveness. Curr Diabetes Rev 12(1):30–41. https://doi.org/10.2174/1573399811666150416122903
Ahlqvist E, Storm P, Käräjämäki A et al (2018) Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 6(5):361–369. https://doi.org/10.1016/s2213-8587(18)30051-2
Ali S, Nafis S, Kalaiarasan P et al (2016) Understanding genetic heterogeneity in type 2 diabetes by delineating physiological phenotypes: SIRT1 and its gene network in impaired insulin secretion. Rev Diabet Stud 13(1):17–34. https://doi.org/10.1900/rds.2016.13.17
Jannasch F, Kröger J, Schulze MB (2017) Dietary patterns and type 2 diabetes: a systematic literature review and meta-analysis of prospective studies. J Nutr 147(6):1174–1182. https://doi.org/10.3945/jn.116.242552
Bennett DA, Du H, Bragg F et al (2019) Physical activity, sedentary leisure-time and risk of incident type 2 diabetes: a prospective study of 512 000 Chinese adults. BMJ Open Diabetes Res Care 7(1):e000835. https://doi.org/10.1136/bmjdrc-2019-000835
Läll K, Mägi R, Morris A et al (2017) Personalized risk prediction for type 2 diabetes: the potential of genetic risk scores. Genet Med 19(3):322–329. https://doi.org/10.1038/gim.2016.103
Mahajan A, Wessel J, Willems SM et al (2018) Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet 50(4):559–571. https://doi.org/10.1038/s41588-018-0084-1
Hale PJ, López-Yunez AM, Chen JY (2012) Genome-wide meta-analysis of genetic susceptible genes for Type 2 Diabetes. BMC Syst Biol 6(Suppl 3):S16. https://doi.org/10.1186/1752-0509-6-s3-s16
Udler MS, McCarthy MI, Florez JC et al (2019) Genetic risk scores for diabetes diagnosis and precision medicine. Endocr Rev 40(6):1500–1520. https://doi.org/10.1210/er.2019-00088
Goodarzi MO, Palmer ND, Cui J et al (2020) Classification of type 2 diabetes genetic variants and a novel genetic risk score association with insulin clearance. J Clin Endocrinol Metab 105(4):1251–1260. https://doi.org/10.1210/clinem/dgz198
Scott RA, Scott LJ, Mägi R et al (2017) An expanded genome-wide association study of type 2 diabetes in europeans. Diabetes 66(11):2888–2902. https://doi.org/10.2337/db16-1253
Xue A, Wu Y, Zhu Z et al (2018) Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun 9(1):2941. https://doi.org/10.1038/s41467-018-04951-w
Morris AP (2018) Progress in defining the genetic contribution to type 2 diabetes susceptibility. Curr Opin Genet Dev 50:41–51. https://doi.org/10.1016/j.gde.2018.02.003
Prasad RB, Groop L (2015) Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel) 6(1):87–123. https://doi.org/10.3390/genes6010087
Wright JD, Folsom AR, Coresh J et al (2021) The ARIC (Atherosclerosis Risk In Communities) study: JACC focus seminar 3/8. J Am Coll Cardiol 77(23):2939–2959. https://doi.org/10.1016/j.jacc.2021.04.035
Pankow JS, Tang W, Pankratz N et al (2017) Identification of genetic variants linking protein C and lipoprotein metabolism: the ARIC study (Atherosclerosis Risk in Communities). Arterioscler Thromb Vasc Biol 37(3):589–597. https://doi.org/10.1161/atvbaha.116.308109
Taliun D, Harris DN, Kessler MD et al (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590(7845):290–299. https://doi.org/10.1038/s41586-021-03205-y
Byun J, Han Y, Gorlov IP et al (2017) Ancestry inference using principal component analysis and spatial analysis: a distance-based analysis to account for population substructure. BMC Genomics 18(1):789. https://doi.org/10.1186/s12864-017-4166-8
Gold L, Walker JJ, Wilcox SK et al (2012) Advances in human proteomics at high scale with the SOMAscan proteomics platform. New Biotechnol 29(5):543–549. https://doi.org/10.1016/j.nbt.2011.11.016
Kim CH, Tworoger SS, Stampfer MJ et al (2018) Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci Rep 8(1):8382. https://doi.org/10.1038/s41598-018-26640-w
Williams SA, Kivimaki M, Langenberg C et al (2019) Plasma protein patterns as comprehensive indicators of health. Nat Med 25(12):1851–1857. https://doi.org/10.1038/s41591-019-0665-2
Tin A, Yu B, Ma J et al (2019) Reproducibility and variability of protein analytes measured using a multiplexed modified aptamer assay. J Appl Lab Med 4(1):30–39. https://doi.org/10.1373/jalm.2018.027086
Inker LA, Schmid CH, Tighiouart H et al (2012) Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med 367(1):20–29. https://doi.org/10.1056/NEJMoa1114248
Ballew SH, Chen Y, Daya NR et al (2017) Frailty, kidney function, and polypharmacy: the atherosclerosis risk in communities (ARIC) study. Am J Kidney Dis 69(2):228–236. https://doi.org/10.1053/j.ajkd.2016.08.034
Grubb A, Blirup-Jensen S, Lindström V et al (2010) First certified reference material for cystatin C in human serum ERM-DA471/IFCC. Clin Chem Lab Med 48(11):1619–1621. https://doi.org/10.1515/cclm.2010.318
Porter AK, Schilsky S, Evenson KR et al (2019) The association of sport and exercise activities with cardiovascular disease risk: the atherosclerosis risk in communities (ARIC) study. J Phys Act Health 16(9):698–705. https://doi.org/10.1123/jpah.2018-0671
Hemani G, Zheng J, Elsworth B et al (2018) The MR-Base platform supports systematic causal inference across the human phenome. Elife 7:e34408. https://doi.org/10.7554/eLife.34408
Sun BB, Maranville JC, Peters JE et al (2018) Genomic atlas of the human plasma proteome. Nature 558(7708):73–79. https://doi.org/10.1038/s41586-018-0175-2
Emilsson V, Gudmundsdottir V, Gudjonsson A et al (2022) Coding and regulatory variants are associated with serum protein levels and disease. Nat Commun 13(1):481. https://doi.org/10.1038/s41467-022-28081-6
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44(2):512–525. https://doi.org/10.1093/ije/dyv080
Suhre K, Arnold M, Bhagwat AM et al (2017) Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun 8:14357. https://doi.org/10.1038/ncomms14357
Krämer A, Green J, Pollard J Jr et al (2014) Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 30(4):523–530. https://doi.org/10.1093/bioinformatics/btt703
Gudmundsdottir V, Zaghlool SB, Emilsson V et al (2020) Circulating protein signatures and causal candidates for type 2 diabetes. Diabetes 69(8):1843–1853. https://doi.org/10.2337/db19-1070
Knight S, May H, Anderson J et al (2018) Semaphorin and plexin pathways associated with increases in hemoglobin A1c in individuals with diabetes mellitus. J Am Coll Cardiol 71(11, Supl):A1822. https://doi.org/10.1016/S0735-1097(18)32363-5
Li T, Xu Y, Shi Y et al (2020) Genome-wide analysis of DNA methylation identifies S100A13 as an epigenetic biomarker in individuals with chronic (≥ 30 years) type 2 diabetes without diabetic retinopathy. Clin Epigenetics 12(1):77. https://doi.org/10.1186/s13148-020-00871-z
Hou JY, Wang YG, Ma SJ et al (2017) Identification of a prognostic 5-Gene expression signature for gastric cancer. J Cancer Res Clin Oncol 143(4):619–629. https://doi.org/10.1007/s00432-016-2324-z
Ohki-Hamazaki H, Neuromedin B (2000) Prog Neurobiol 62(3):297–312. https://doi.org/10.1016/s0301-0082(00)00004-6
Chen B, Chen J, House MG et al (2012) Role of neurofilament light polypeptide in head and neck cancer chemoresistance. Mol Cancer Res 10(3):305–315. https://doi.org/10.1158/1541-7786.Mcr-11-0300
Alghamdi AAA, Benwell CJ, Atkinson SJ et al (2020) NRP2 as an emerging angiogenic player; promoting endothelial cell adhesion and migration by regulating recycling of α5 integrin. Front Cell Dev Biol 8:395. https://doi.org/10.3389/fcell.2020.00395
You DJ, Park CR, Furlong M et al (2015) Dimer of arfaptin 2 regulates NF-κB signaling by interacting with IKKβ/NEMO and inhibiting IKKβ kinase activity. Cell Signal 27(11):2173–2181. https://doi.org/10.1016/j.cellsig.2015.08.012
Zhang X, Zhang G, Zhang H et al (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 135(1):61–73. https://doi.org/10.1016/j.cell.2008.07.043
Cai D, Yuan M, Frantz DF et al (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11(2):183–190. https://doi.org/10.1038/nm1166
Moreno-Navarrete JM, Fernández-Real JM (2019) The complement system is dysfunctional in metabolic disease: Evidences in plasma and adipose tissue from obese and insulin resistant subjects. Semin Cell Dev Biol 85:164–172. https://doi.org/10.1016/j.semcdb.2017.10.025
Lim J, Iyer A, Suen JY et al (2013) C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J 27(2):822–831. https://doi.org/10.1096/fj.12-220582
Zheng JM, Ren XG, Jiang ZH et al (2018) Lectin-induced renal local complement activation is involved in tubular interstitial injury in diabetic nephropathy. Clin Chim Acta 482:65–73. https://doi.org/10.1016/j.cca.2018.03.033
Huang Y, Xu J, Wu X et al (2019) High expression of complement components in the kidneys of type 2 diabetic rats with diabetic nephropathy. Front Endocrinol (Lausanne) 10:459. https://doi.org/10.3389/fendo.2019.00459
Li XQ, Chang DY, Chen M et al (2019) Complement activation in patients with diabetic nephropathy. Diabetes Metab 45(3):248–253. https://doi.org/10.1016/j.diabet.2018.04.001
Mandava N, Tirado-Gonzalez V, Geiger MD et al (2020) Complement activation in the vitreous of patients with proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 61(11):39. https://doi.org/10.1167/iovs.61.11.39
Shahulhameed S, Vishwakarma S, Chhablani J et al (2020) A systematic investigation on complement pathway activation in diabetic retinopathy. Front Immunol 11:154. https://doi.org/10.3389/fimmu.2020.00154
Burgess S, Thompson SG (2017) Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 32(5):377–389. https://doi.org/10.1007/s10654-017-0255-x
Acknowledgements
The authors thank the staff and participants of the ARIC study for their important contributions.
Authors’ relationships and activities
ES is a member of the Editorial Board of Diabetologia. The authors declare that there are no other relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the final version for publication. JSP is responsible for the integrity of the work as a whole.
Funding
This work was supported by grant number 2T32HL007779-26 from the US National Heart, Lung, and Blood Institute, and by an award from the Hawley Foundation to BTS. The Atherosclerosis Risk in Communities study has been funded in whole or in part by federal funds from the US National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under contract numbers 75N92022D00001, 75N92022D00002, 75N92022D00003, 75N92022D00004, 75N92022D00005, R01HL087641, R01HL059367 and R01HL086694. Funding was also supported by US National Human Genome Research Institute contract U01HG004402, and US National Institutes of Health contract HHSN268200625226C. Infrastructure was partly supported by grant number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research. PLL was supported by K24 HL159246. SomaLogic Inc. performed the SomaScan assays in exchange for use of ARIC study data. This work was supported in part by US National Heart, Lung, and Blood Institute/National Institutes of Health grant R01HL134320. The measurement of serum creatinine and cystatin C was performed as part of an ancillary study supported by research funding from Kyowa Kirin (principal investigator: Kunihiro Matsushita).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Steffen, B.T., Tang, W., Lutsey, P.L. et al. Proteomic analysis of diabetes genetic risk scores identifies complement C2 and neuropilin-2 as predictors of type 2 diabetes: the Atherosclerosis Risk in Communities (ARIC) Study. Diabetologia 66, 105–115 (2023). https://doi.org/10.1007/s00125-022-05801-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05801-7