
Abstract
Rationale
Though interoceptive properties of antidepressants have been described, discriminative stimulus (DS) properties of mirtazapine, which does not affect monoamine reuptake, remain uncharacterized.
Objectives
The objectives of the study are to train rats to recognize a mirtazapine DS, then perform substitution studies with other antidepressants and drugs acting at sites occupied by mirtazapine.
Materials and methods
Using a two-lever, fixed-ratio 10 schedule, rats were trained to discriminate mirtazapine (2.5 mg/kg, i.p.) from saline.
Results
Sessions, 63 ± 8, were necessary to reach the criterion for 14 rats that all subsequently recognized (100%) mirtazapine at the training dose. Mirtazapine blocks serotonin (5-HT)2C receptors, and the 5-HT2C antagonists, SB242,084, SB243,213 and S32006, revealed dose-dependent and full (≥80%) substitution at doses of 2.5, 2.5, and 0.63 mg/kg, respectively. By contrast, the 5-HT2A antagonists, MDL100,907 and SR46349-B, the 5-HT2B antagonist, SB204,741, and the 5-HT3 antagonist, ondansetron, showed no significant substitution. Though mirtazapine indirectly recruits 5-HT1A receptors, the 5-HT1A agonists, buspirone and 8-OH-DPAT, did not substitute. Mirtazapine blocks α2-adrenoceptors, but several α2-adrenoceptor antagonists (yohimbine, RX821,002 and atipamezole) failed to substitute. Despite blockade by mirtazapine of histamine H1 receptors, no substitution was seen with the selective H1 antagonist, pyrilamine. Finally, the selective noradrenaline reuptake inhibitor, reboxetine (0.16), fully substituted for mirtazapine, whereas the 5-HT/noradrenaline reuptake inhibitors, duloxetine and S33005, several 5-HT reuptake inhibitors (citalopram, fluvoxamine, and paroxetine) and the dopamine reuptake inhibitors, bupropion and GBR12,935, did not substitute.
Conclusion
Mirtazapine elicits a DS in rats for which selective antagonists at 5-HT2C receptors display dose-dependent substitution, whereas drugs acting at other sites recognized by mirtazapine are ineffective.
Similar content being viewed by others


Introduction
Compared to drug discrimination assays with psychoactive agents like antipsychotics (see this Special Issue; Dekeyne and Millan 2003; Goudie et al. 2004; Alici et al. 2006), antidepressant discrimination studies remain comparatively uncommon. This is unfortunate since discriminative stimulus (DS) studies are potentially instructive in evaluating several interesting issues: first, in determining whether a training drug shares interoceptive effects with other classes of clinically active antidepressant; second, in characterizing mechanisms of action involved in the interoceptive effects of antidepressants using substitution tests with ligands selective for specific classes of receptor and transporter; third, in relating DS properties of antidepressants to their functional actions, including their influence upon mood. In fact, despite difficulties faced in training, these questions have been addressed in drug discrimination studies of the selective serotonin (5-HT) reuptake inhibitor (SSRI), citalopram, the selective noradrenaline (NA) reuptake inhibitor (NARI), reboxetine, and dopamine (DA) reuptake inhibitor, bupropion (Olivier et al. 1993; Terry and Katz 1997; Millan et al. 1999a, 1999b; Dekeyne et al. 2001a, 2001b; Young and Glennon 2002; Millan and Dekeyne 2007; see Dekeyne and Millan 2003 for review). By contrast, the poor tolerance of tricyclic agents upon long-term administration, has complicated their use as training drugs (Zhang and Barrett 1991; Dekeyne and Millan 2003). Furthermore, stimulus properties of clinically employed drugs like nefazodone and trazodone have only been indirectly characterized: that is, used in substitution studies but not employed as training drugs (Eckler et al. 2003). Furthermore, drug discrimination studies with the “atypical” antidepressant, mirtazapine, which differs from all other clinically active agents in not modifying monaomine reuptake (Davis and Wilde 1996; Tatsumi et al. 1997; Millan et al. 2000a; Anttila and Leinonen 2001), have never been documented.
Accordingly, the purpose of the present report was to evaluate the DS properties of mirtazapine. The facilitatory influence of mirtazapine upon ascending dopaminergic and adrenergic pathways provides a functional substrate for its beneficial influence in depressed states and can be attributed to antagonism of 5-HT2C receptors and α2(A)-adrenoceptors (ARs) which exert a tonic, inhibitory influence upon dopaminergic and adrenergic projections (Millan et al. 2000a; Di Matteo et al. 2002; Devoto et al. 2004; Invernizzi and Garattini 2004; Dekeyne et al. 2008). Blockade of 5-HT2C receptors improves anxious states and exerts a favorable influence on sexual function and sleep architecture which are perturbed in depressive states (Davis and Wilde 1996; Kent 2000; Anttila and Leinonen 2001; Millan 2006; Dekeyne et al. 2008). Mirtazapine also behaves as an antagonist at 5-HT2A receptors, blockade of which may also exert a beneficial influence upon mood, anxious symptoms, and sleep (Davis and Wilde 1996; Kent 2000; Anttila and Leinonen 2001; Millan 2006; Dekeyne et al. 2008). Furthermore, antagonism of 5-HT2A receptors by mirtazapine reduces the stress-induced activation of the hypothalamus–pituitary–adrenal axis by braking hypothalamic liberation of corticotropin-releasing factor (Schule et al. 2002; Fabricio et al. 2005; Millan 2006). While the antagonist actions of mirtazapine at 5-HT2B receptors are of unclear significance (Millan et al. 2000a), an interesting difference between mirtazapine and monoamine reuptake inhibitors consists in its pronounced antagonist properties at 5-HT3 receptors (Anttila and Leinonen 2001) which counter nausea and gastrointestinal side effects (Pedersen and Klysner 1997; Olivier et al. 2000; Costall and Naylor 2004; Ramamoorthy et al. 2008). Furthermore, while mirtazapine has no affinity for 5-HT1A receptors, it may indirectly activate postsynaptic 5-HT1A receptors via an increase in 5-HT release (De Boer 1996; Berendsen and Broekkamp 1997; Haddjeri et al. 1998; Rauggi et al. 2005). Indeed, in a cross-familiarization procedure in rats, pre-exposure to the prototypical 5-HT1A agonist, 8-OH-DPAT, interfered with a conditioned taste aversion (CTA) elicited by mirtazapine (Berendsen and Broekkamp 1997). Nonetheless, a role of enhanced 5-HT release and 5-HT1A receptors in the actions of mirtazapine has been questioned (Bengtsson et al. 2000; Millan et al. 2000a; Whale et al. 2000; Nakayama et al. 2004). Finally, in addition to modest antagonist properties at α1-adrenoceptors (ARs), mirtazapine potently blocks histamine H1 receptors, an action related to a variety of central, cardiovascular and autonomic side effects including obesity and somnolence (Glassman 1998; Anttila and Leinonen 2001; Rogoz et al. 2002; Kroeze et al. 2003; Sommer et al. 2003), though its sedative effects are counteracted by the arousing effect of enhanced noradrenergic transmission (Kent 2000; Szegedi and Schwertfeger 2005; Schmid et al. 2006).
In light of these observations concerning the distinctive multi-receptorial profile of mirtazapine (Table 1), the present study examined the mechanisms underlying its interoceptive properties following training with a standard, two-lever, food-reinforced, drug versus saline discrimination procedure.
Materials and methods
Animals
Thirty male Wistar rats (Iffa-Credo, L’Arbresle, France), weighing 180–200 g upon arrival, were housed singly in sawdust-lined cages with free access to water and restricted access to food (10–11 g/day) in order to maintain their weight at 80% of free-feeding values. Laboratory temperature was 21 ± 1.0°C and humidity 60 ± 5%. There was a 12/12 h light–dark cycle with lights on at 07:00 hours. All animal use procedures conformed to international European ethical standards (86/609-EEC) and the French National Committee (décret 87/848) for the care and use of laboratory animals.
Drug discrimination procedure
Employing a procedure extensively described in our previous work (e.g. Millan and Dekeyne 2007), rats were trained to discriminate mirtazapine (2.5 mg/kg, i.p.) from saline, using a two-lever, fixed-ratio 10, food-reinforced procedure. Each 15-min daily session (5 days/week) commenced 15 min after injection. Drug (D) or saline (S) sessions alternated irregularly (DSSDS–SDDSS–SDSDD–DSDSD, etc). During each session, correct responding was defined as no more than 13 presses on both levers to obtain the first reinforcement. The discrimination criterion was ten consecutive sessions with correct responding, and animals failing to reach the criterion after 100 sessions were not used further. Thereafter, test sessions were undertaken every Wednesday and Friday, while training sessions were pursued on the other days. Only rats responding appropriately on the two most recent training days were tested, whereas the others underwent an additional training session. In the course of testing, responses on the selected lever—i.e., the lever for which ten responses were emitted first—were reinforced for the remainder of the session. Tested drugs were administered instead of mirtazapine, 15 min before the beginning of the session. The order of drug and dose testing was irregular, and rats were never retested with the same dose of a drug. The data recorded during the test sessions were lever selection (i.e., the percentage of rats selecting “mirtazapine” lever) and response rate (i.e., the total number of presses on both levers).
Data analysis
Lever selection data were expressed as the percentage of rats selecting the drug lever and were compared by Fisher exact probability tests to control values (0%). In line with previous studies (e.g., Millan and Dekeyne 2007), “full” substitution was defined as ≥80% “mirtazapine” lever selection. Response rates in the presence of drugs were compared by paired t tests to those acquired during the preceding saline sessions.
Drug dosing
The training dose of mirtazapine was based upon a broad-based pharmacological characterization in this laboratory, including dialysis studies of its influence upon release of NA and DA in frontal cortex, and behavioral observations of its actions in diverse rodent models of potential anxiolytic and antidepressant properties (Millan et al. 2000a; Dekeyne and Millan 2006; Millan MJ, Brocco M and Dekeyne A, unpublished observation). Inasmuch as no differences in active doses were observed between (acute) s.c. and i.p. administration, the latter route was preferred for this chronic work to avoid any potential problems of cutaneous toxicity upon repeated injection of mirtazapine. For all drugs tested, at least two doses were examined. Doses were not increased further in the event that response rates were significantly suppressed or that one or more of the tested animals failed to select a lever. In addition, dose ranges were not extended further when a biphasic dose–response curve for lever selection was obtained, even in the absence of a parallel decrease in response rate. Drug dose ranges were selected on the basis of our extensive internal in vivo data base and relevant literature studies (see “Introduction” and “Discussion” sections for citations).
Drug sources and structures
All drug doses are expressed in terms of the base. Owing to solubility limits, and the risk of cutaneous toxicity, mirtazapine and certain other drugs (SB204,741, SB242,084, SB243,213, pyrilamine, and WB4101) were prepared as suspensions in sterile water plus a few drops of Tween 80 and were injected i.p. in volumes of 1.0 ml/kg i.p. All other drugs were dissolved in sterile water, plus a few drops of lactic acid (85%) if necessary and the pH adjusted to >5.0. Drug structures, salts and sources were as follows. SR46349-B (1(Z)-[2-(dimethyl amino)ethoxyimino]-1-(2-fluorophenyl)-3-(4-hydroxyphenyl)-2(E)-propene) hemifumarate was a generous gift of Sanofi Winthrop (Montpellier, France). Bupropion HCl was obtained from Burroughs Wellcome (NC, USA). Fluvoxamine maleate was obtained from Tocris Cookson (Bristol, UK). GBR12935 (1-[2-(diphenylmethoxy)ethyl]-3-(phenylpropyl) piperazine) diHCl, 8-OH-DPAT (8-hydroxy-2-(di-n-propylamino)-tetralin) HBr, RX821,002 (2-[2-(2-methoxy-1,4-benzodioxanyl)]imidazoline) HCl and WB4101 (2-(2,6-dimethoxyphenoxyethyl) aminomethyl-1,4-benzodioxane) HCl were obtained from Research Biochemicals International (Natick, MA, USA). Buspirone HCl, prazosin HCl, pyrilamine maleate, and yohimbine HCl were puchased from Sigma (St Quentin-Fallavier, France). Atipamezole HCl, citalopram HBr, duloxetine camphosulfonate, MDL100,907 (R(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidine-methanol) base, mianserin HCl, mirtazapine, ondansetron HCl, paroxetine HCl, reboxetine methane sulfonate, S32006 (N-pyridin-3-yl-1,2-dihydro-3H-benzo[e]indole-3-carboxamide) base, S33005 ((−)1-(1-dimethylaminomethyl 5-methoxy benzocyclobutan-1-yl) cyclohexanol) HCl, SB204,741 (1-(1-methylindol-5-yl)-3-(3-methylisothiazol-5-yl)urea) base, SB242,084 (6-chloro-5-methyl-1-[6-(2-methyl pyridin-3-yloxy) pyridin-3-yl carbamoyl] indoline) HCl, and SB243,213 (5-methyl-N-[6-(2-methylpyridin-3-yloxy)pyridin-3-yl]-6-(trifluoromethyl) indoline-1-carboxamide) HCl were all synthesized by Servier chemists (G. Lavielle and J.-L. Péglion).
Results
Acquisition of a mirtazapine-induced discriminative stimulus
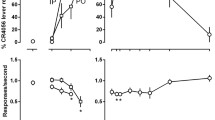
From the 30 rats initially recruited for this drug discrimination study, five rats failed to reach the discrimination criterion of ten consecutive sessions of correct responding (Fig. 1). A further 11 failed to show dose-dependent substitution for mirtazapine itself (indeed, four out of 11 selected saline lever at the training dose of 2.5 mg/kg, and the other seven chose the “mirtazapine” lever at a ∼64-lower dose). The total of 14 rats which fulfilled all criteria for successful training reached the discrimination criterion after a 63 ± 8 sessions (mean ± SEM). Throughout the studies, during training sessions, no changes in response rates (with absolute individual values ranging between 1,550 and 2,100) were observed in the presence of mirtazapine compared to saline.

Acquisition of the mirtazapine-mediated discriminative stimulus in rats. Data are percentage of correct responses (appropriate “mirtazapine” or “saline” lever selection) over blocks of five sessions during acquisition of a discriminative stimulus with mirtazapine at a training dose of 2.5 mg/kg, i.p.. Data are for rats (N = 14) which fulfilled the discrimination criteria and subsequently exhibited dose-dependent substitution for mirtazapine itself (Fig. 2)
Substitution with mirtazapine itself
The selected population of rats (N = 14) dose-dependently recognized the mirtazapine cue, with 0% and 100% selecting the mirtazapine lever at doses of 0.04 and 2.5 mg/kg, i.p., respectively (Fig. 2). As observed during “drug” training sessions, mirtazapine had no effect upon response rate.

Substitution for the discriminative stimulus elicited by mirtazapine with mirtazapine itself, and with selective 5-HT2C receptor antagonists. N = 14 per value for mirtazapine, and N = 5 per value for other compounds. Upper panel, lever selection. Data are percentage of animals selecting the “mirtazapine” lever. Asterisks indicate significance of differences (*P < 0.05; Fisher exact probability test) versus control values (0%). Lower panel, response rate. Data are means ± SEMs of percentage of control values obtained during the most recent saline-training session
Substitution with 5-HT2C receptor antagonists
The 5-HT2C receptor antagonists, SB242,084 and SB243,213, displayed dose-dependent and full (80%) substitution for mirtazapine over comparable dose ranges, without modifying response rate (Fig. 2). These effects were mimicked by S32006, a novel, selective 5-HT2C receptor antagonist (Dekeyne et al. 2008), which moreover expressed its actions more potently than mirtazapine itself and the two other compounds, as indicated by a leftward shift in the dose–response curve.
Lack of significant substitution with antagonists at 5-HT2A, 5-HT2B, and 5-HT3 receptors
The 5-HT2A antagonists, MDL100,907 and SR46349-B, displayed maximal observed substitution of 60% at doses of 0.16 and 2.5 mg/kg, s.c., respectively (Fig. 3). Both drugs displayed U-shaped dose–response curves. On the other hand, 0% “mirtazapine” lever selection was obtained with the selective 5-HT2B antagonist, SB204,741, at doses of 2.5 and 10.0 mg/kg, i.p. None of these compounds significantly modified response rates over the dose ranges tested. The prototypical 5-HT3 antagonist, ondansetron, produced a biphasic dose–response curve for “mirtazapine” lever selection with a maximum of 60% substitution attained at an intermediate dose of 0.63 mg/kg, s.c.

Lack of significant substitution for the discriminative stimulus elicited by mirtazapine with 5-HT3, 5-HT2A, and 5-HT2B receptor antagonists. Left panels, influence of the 5-HT2A receptor antagonists, MDL100,907 and SR46349-B. Middle panels, influence of the 5-HT2B receptor antagonist SB204,741. Right panels, influence of the 5-HT3 receptor antagonist, ondansetron. N = 5 per value. Upper panels, lever selection. Data are percentage of animals selecting the “mirtazapine” lever. Lower panels, response rates. Data are means ± SEMs of percentage of control values obtained during the most recent saline-training session
Lack of significant substitution with 5-HT1A agonists
8-OH-DPAT, which behaves as an agonist at 5-HT1A receptors, produced 60% substitution for mirtazapine at a dose of 0.01 mg/kg (Table 2). A further increase in doses to 0.04 and 0.16 mg/kg yielded less marked “mirtazapine” lever selection (40% and 0%), together with a decrease in response rates. At the dose of 0.16 mg/kg, half of the animals were unable to select a lever, and the other half revealed a significant decrease of response rates. With another 5-HT1A receptor agonist, buspirone, maximal observed substitution was 60% at the highest dose tested (2.5 mg/kg) which significantly decreased response rate.
Lack of significant substitution with antagonists and α2 at α1-adrenoceptors
The α2-AR antagonist, yohimbine (0.16–2.5 mg/kg) exhibited a “plateau” of 40% “mirtazapine” lever selection (Table 3). The more selective α2-AR antagonists, RX821,002 and atipamezole, displayed biphasic dose–response curve with maximal observed substitution of 60% and no more than 20% substitution, respectively. Over the dose ranges evaluated, these α2-AR antagonists did not significantly influence response rates. Concerning α1-AR antagonists, prazosin or WB4101 produced biphasic dose–response curves for lever selection with maximal observed substitution of 60% at doses of 0.16 and 2.5 mg/kg, s.c. for prazosin and of 40% for the dose of 2.5 mg/kg, i.p., of WB4101. A further increase of the dose of prazosin yielded 0% “mirtazapine” lever selection together with a marked—though not significant—decrease in response rate. Similarly, at 10.0 mg/kg, WB4101 yielded no substitution (0%) for the four out of seven animals able to select a lever, and there was a significant decrease in response rate.
Lack of significant substitution with a histamine H1 antagonist
Blockade of H1 sites with pyrilamine yielded a dose-dependent increase in substitution for mirtazapine, with an effect of 40% reached at the highest dose tested (20.0 mg/kg, i.p.). This dose significantly reduced response rates, precluding a further increase in doses (Table 3).
Substitution patterns with selective NA, mixed 5-HT/NA, and selective 5-HT reuptake inhibitors
The NARI, reboxetine, dose-dependently and fully substituted at a dose of 0.16 mg/kg, s.c. (Fig. 4). By contrast, the tricyclic antidepressant, desipramine, which inhibits NA reuptake among other activities, showed only 40% substitution at a dose that markedly, though not significantly, decreased response rate. A higher dose could not be tested owing to pronounced sedation leading to an inability to respond. The SNRI, duloxetine, displayed a maximal observed substitution of only 40% at 0.63 mg/kg, s.c. A further increase of dose yielded 20% “mirtazapine” lever selection in parallel with a decrease in response rate, which reached statistical significance at the highest dose tested (10.0). A further SNRI, S33005, likewise showed no substitution for mirtazapine, exerted no significant influence upon response rate when evaluated at high doses relative to those active in preclinical models of potential antidepressant activity (Millan et al. 2001a). The SSRIs, paroxetine and fluvoxamine, both failed to substitute for mirtazapine, up to the doses of 2.5 and 10.0 mg/kg, s.c., and citalopram displayed only 40% substitution at the highest dose tested (10.0 mg/kg). These SSRIs did not significantly affect response rates at the doses evaluated.

Substitution patterns for the discriminative stimulus elicited by mirtazapine with mixed serotonin/noradrenaline reuptake inhibitors (SNRIs) compared to selective noradrenaline (NA) and serotonin reuptake inhibitors (SSRIs). Left panels, NA reuptake inhibitors: significant substitution with reboxetine compared to desipramine. Middle panels, SNRIs: lack of significant substitution with duloxetine and S33005. Right panels, SSRIs: lack of significant substitution with paroxetine, citalopram and fluvoxamine. N = 5 per value. Upper panels, lever selection. Data are percentage of animals selecting the “mirtazapine” lever. Lower panels, response rates. Data are means ± SEMs of percentage of control values obtained during the most recent saline-training session. The asterisk indicates significant decrease in response rate (*P < 0.05; paired t test) compared to the control saline session
Lack of significant substitution with DARIs
Bupropion displayed only 40% substitution for mirtazapine at the highest dose tested (40.0 i.p.), and another DARI, GBR12,935, completely failed to substitute up to a dose of 10.0 mg/kg, i.p. (Table 4). Over the dose ranges evaluated, these compounds did not affect response rates.
Discussion
Establishment of a specific DS with mirtazapine
The present study demonstrated that the atypical, multi-receptorial antidepressant, mirtazapine, elicits a DS in rats employing a two-lever, food-reinforced, drug versus saline discrimination procedure. By comparison to other classes of psychotropic drugs like antipsychotics (Goudie et al. 2004; Prus et al. 2006; Cole et al. 2007), the mirtazapine DS proved difficult to train in terms of the long acquisition duration (almost 5 months), the particularly high number of animals which failed to show dose-dependent substitution for mirtazapine itself (11 out of 25), and the prolonged testing period (almost 18 months) required before DS properties of mirtazapine could be characterized using substitution tests. This challenging procedure is reminiscent of difficulties confronted in training animals to recognize other antidepressants like SSRIs and NARIs employing a similar training procedure (Olivier et al. 1993; Dekeyne et al. 2001b; Dekeyne and Millan 2003). Nonetheless, a sufficient number of subjects did reliably achieve the criterion requisite for thorough testing, and the finding that mirtazapine provoked a “salient” interoceptive stimulus herein is consistent with its ability to elicit a CTA in mice, albeit over a far shorter exposure period of 5 days (Berendsen and Broekkamp 1997). Moreover, underpinning the specificity of its DS properties, mirtazapine dose-dependently and fully substituted for itself over a dose range corresponding to those active in other procedures, such as expression of potential antidepressant and anxiolytic properties, and elevations in extracellular levels of DA and NA in the frontal cortex of freely moving rats (O’Connor and Leonard 1986; Andrews et al. 1994; Millan et al. 2000a; Millan 2003, 2006; Rauggi et al. 2005).
Substitution with antagonists at 5-HT2C versus 5-HT2B and 5-HT2A receptors
Mirtazapine possesses marked antagonist properties at 5-HT2C receptors, which contribute to its aforementioned functional actions (Millan et al. 2000a; Millan 2005). Accordingly, blockade of 5-HT2C sites is a strong candidate for implication in its interoceptive properties. Indeed, three 5-HT2C receptor antagonists—including the recently described urea derivative, S32006 (Dekeyne et al. 2008)—substituted for mirtazapine at doses corresponding to those which elevate DA and NA levels in frontal cortex, and that display antidepressant and anxiolytic properties in rodents (Kennett et al. 1997; Gobert et al. 2000; Wood et al. 2001; Dekeyne et al. 2008). A role of 5-HT2C receptors is supported by the observation that mirtazapine blocks the DS properties of the selective 5-HT2C agonist, Ro60,0175. Furthermore, it also abrogates the DS elicited by the SSRI, citalopram, which is mediated via 5-HT2C receptors (Millan et al. 2000a; Dekeyne et al. 2001b). Interestingly, mirtazapine and S32006 both behave as inverse agonists at constitutively active 5-HT2C receptors, and SB243,213 is an inverse agonist at 5-HT2C receptors coupled to phospholipase A2, though not phospholipase C (Wood et al. 2001; Berg et al. 2006; Chanrion et al. 2008; Dekeyne et al. 2008). However, since the neutral antagonist, SB242,084, likewise substituted for mirtazapine, the significance of inverse agonist properties of mirtazapine to its DS (and other in vivo) actions remains to be clarified. In this context, however, it is worth noting that attempts to train rats with SB242,084 (2.5 mg/kg, i.p.) were unsuccessful, so further efforts with an inverse agonist like S32006 may be justified (Dekeyne and Millan 2003; Dekeyne et al. 2008).
In fact, S32006 also possesses high affinity for 5-HT2B receptors, but their blockade is unlikely to be involved in the interoceptive effects of mirtazapine since SB242,084 and SB243,213 are both highly selective antagonists at 5-HT2C versus 5-HT2B receptors—and all other sites tested. Moreover, the selective 5-HT2B receptor antagonist, SB204,741 (Cussac et al. 2002), showed no substitution to mirtazapine at doses active in other behavioral procedures like modulation of sleep architecture (Kantor et al. 2004). In addition, though 5-HT2B receptor blockade reduces the locomotor actions of 3,4-methylenedioxymethamphetamine (ecstasy; MDMA; Doly et al. 2008), and 5-HT2B receptor stimulation is associated with anxiolytic properties (Kennett et al. 1996), there is no evidence that antagonism of central 5-HT2B receptors greatly affects mood. A role of 5-HT2A receptor blockade in the DS properties of mirtazapine also appears unlikely inasmuch as SB242,084, SB243,213, and S32006 all possess low affinity for 5-HT2A receptors, and the highly selective antagonist at 5-HT2A receptors, MDL100,907 and SR46349-B, failed to significantly substitute for mirtazapine. Nonetheless, it would be interesting to evaluate whether mirtazapine substitutes for a DS elicited by MDL100,907 itself and whether it blocks the interoceptive actions of the agonist, DOI, which are elicited via stimulation of 5-HT2A receptors (Schreiber et al. 1994; Dekeyne et al. 2002).
Lack of a major role for 5-HT3 and 5-HT1A receptors in the DS properties of mirtazapine
Inasmuch as ondansetron did not substitute for mirtazapine, antagonist properties at 5-HT3 receptors do not appear to be a salient component of its interoceptive effects. Notably, DS properties of 5-HT3 receptor inactivation do not appear to be robust, since it proved impossible to generate a DS with ondansetron itself (Olivier et al. 2002). It has been argued that indirect recruitment of 5-HT1A receptors participates in the functional and clinical profile of mirtazapine (see Introduction). Notably, pre-exposure to 8-OH-DPAT blunted a CTA provoked by mirtazapine while, reciprocally, pre-exposure to mirtazapine attenuated a CTA to 8-OH-DPAT (Berendsen and Broekkamp 1997). Nonetheless, it remains controversial whether mirtazapine elevates extracellular levels of 5-HT, and in the present study, neither 8-OH-DPAT nor a further 5-HT1A agonist, buspirone, showed significant substitution to mirtazapine at doses active in other models (Millan 2003). Notably, higher doses of 8-OH-DPAT preferentially activate post- versus presynaptic 5-HT1A receptors, yet they were not associated with greater substitution.
Lack of implication of α2- and α1-adrenoceptors, and of H1 receptors, in the DS properties of mirtazapine
We recently characterized the DS properties of potent and highly efficacious α2-AR agonist, S18616, which were blocked by mirtazapine in line with its antagonist properties at α2-ARs (Millan et al. 2000a; Dekeyne and Millan 2006). However, in the present study, the α2-AR antagonists, yohimbine, RX821,002, and atipamezole, all failed to fully substitute for mirtazapine suggesting that α2-AR blockade is not a predominant component of its interoceptive profile. The lack of substitution with yohimbine is of particular note in view of its modest 5-HT1A agonist properties (Winter and Rabin 1993; Millan et al. 2000b). Blockade of inhibitory α2-autoreceptors/heteroceptors facilitates noradrenergic (and dopaminergic) transmission, contributing to antidepressant actions and favoring arousal and motor activation (Millan et al. 2000a; Devoto et al. 2004; Invernizzi and Garattini 2004). Accordingly, α2-AR blockade may explain the lack of decrease in response rates during “mirtazapine” training and testing sessions in countering its histamine H1 receptor-mediated sedative actions (Szegedi and Schwertfeger 2005; Schmid et al. 2006). Nonetheless, the lack of substitution with pyrilamine suggests that H1 receptor blockade does not fulfill a pronounced role in the DS properties of mirtazapine. By contrast to α2-ARs, the affinity of mirtazapine for α1-ARs is less pronounced and, though involved in its cardiovascular–autonomic (and sedative) effects, blockade of α1-ARs does not play a major role in the influence of mirtazapine upon mood (Snoddy and Tessel 1985; Rogoz et al. 2002; Dekeyne and Millan 2006; Millan 2006). Not surprisingly, then, selective antagonists at α1-ARs, prazosin, and WB4101, did not significantly substitute for mirtazapine. This is an interesting distinction to the NARIs, reboxetine and nisoxetine, of which the DS effects were blocked by prazosin and WB4101 demonstrating a prominent role of α1-ARs in their interoceptive effects (Snoddy and Tessel 1985; Dekeyne and Millan 2006).
Substitution by the NARI, reboxetine, in comparison to other antidepressant agents
In view of the aforementioned observation that prazosin blocks DS properties of reboxetine, but not those of mirtazapine, it is of special interest that reboxetine substituted for mirtazapine. One possible neurochemical substrate would be their common elevation in levels of NA, an action exerted by reboxetine at doses similar to those active herein (Dekeyne et al. 2001a; Millan et al. 2001b; Dekeyne and Millan 2003). Obviously, downstream of NA, postsynaptic ARs must be involved in mediating DS properties. One possibility would be β-ARs in view of the intriguing observation of Crissman and O’Donnell (2002) that mirtazapine generalizes to a DS elicited by intracerebral administration of the β1-AR agonist, isoproterenol. Unfortunately, systemically active agonists at β1 and β2-ARs are not currently available for substitution studies with mirtazapine under the current conditions (Millan and Dekeyne 2007). Despite substitution with reboxetine to the mirtazapine DS, no substitution was seen for mirtazapine to a reboxetine DS suggesting, in line with aforementioned findings with prazosin, that their interoceptive properties are not identical (Millan and Dekeyne 2007). In addition, by contrast to reboxetine, desipramine, which displays high affinity for NA transporters (Tatsumi et al. 1997; Millan et al. 2001a, 2001b), did not substitute for mirtazapine. One likely reason for this difference to reboxetine is the broad profile of interactions of desipramine with other receptors (Millan 2006). This may interfere with interoceptive properties mediated by suppression of NA reuptake. Similarly, as argued previously (Dekeyne and Millan 2003; Millan and Dekeyne 2007), the potent actions of the SNRIs, duloxetine and S33005, at 5-HT transporters may compromise any substitution to mirtazapine via reduced NA reuptake. Thus, together with substitution of mirtazapine to the β1-AR agonist, isoproterenol, generalization of reboxetine to mirtazapine, supports a role of adrenergic mechanisms in its DS properties. In this context, it is of note that increased NA levels is an action shared with 5-HT2C receptor antagonists. However, since other agents that elevate NA levels do not substitute for reboxetine, this issue justifies additional exploration.
The interoceptive actions of mirtazapine can unambiguously be differentiated from antidepressants increasing extracellular 5-HT, since no substitution was observed with three SSRIs, and mirtazapine itself did not substitute to DS induced by citalopram (Dekeyne et al. 2001b). Moreover, inasmuch as the DARIs, bupropion and GBR12,935, failed to substitute for the mirtazapine DS, a role of increased DA availability does not contribute to its interoceptive effects.
Comparison of the interoceptive effects of mirtazapine versus mianserin
Though this is the first study to examine DS properties of mirtazapine, previous work has focused on the interoceptive profile of its chemically related predecessor, mianserin, which potently blocks muscarinic receptors (Kelley et al. 1995): indeed, mianserin elicited a DS in rats for which the selective muscarinic antagonist, scopolamine, substituted. It is instructive to consider substitution/antagonism profiles of mianserin compared to mirtazapine versus various training drugs (Table 5). First, despite its affinity for NA transporters, mianserin did not substitute for reboxetine, and neither mianserin nor mirtazapine generalize to citalopram (Dekeyne et al. 2001b; Millan and Dekeyne 2007). Second, in line with a CTA study of Walker et al. (2005), mirtazapine and mianserin block DS actions of 5-HT2C receptor agonists (Gommans et al. 1998; Bourson et al. 1996; Dekeyne et al. 1999). Third, both mianserin and mirtazapine abolished a DS elicited by the α2-AR agonist, S18616 (Dekeyne and Millan 2006). Fourth, mianserin antagonized a DS elicited by the 5-HT2A agonist, DOI (Yamamoto et al. 1991; Smith et al. 1990, 2002; Schreiber et al. 1994) and substituted for the 5-HT2A receptor antagonist, MDL100,907 (Dekeyne et al. 2002; Dekeyne A and Millan MJ, unpublished observation). Finally, mianserin shared the DS properties of the 5-HT1A agonist, flesinoxan (Herremans et al. 1999). Thus, while mirtazapine and mianserin reveal similarities and differences in interoceptive properties, 5-HT2C receptor antagonism is a common component of their DS effects.
Concluding comments
The present study demonstrates that mirtazapine can induce stimulus control of behavior in rats. In line with its distinctive mechanism of action and its lack of cross-substitution with SSRIs, DS properties of mirtazapine do not simply reflect “antidepressant-like” effects per se. However, substitution of the NARI, reboxetine, to mirtazapine indicates a possible role for elevated extracellular levels of NA, an effect likewise elicited by 5-HT2C receptor antagonists, which generalized to mirtazapine. Indeed, 5-HT2C receptor blockade plays a prominent role in the interoceptive properties of mirtazapine. Nonetheless, it remains possible that mirtazapine elicits a “compound” DS—that is, composed of multiple stimulus elements—by analogy to atypical agents like the antipsychotics, clozapine and quetiapine, that interact with an array of targets (Hoenicke et al. 1992; Kelley and Porter 1997; Goudie et al. 2004; Prus et al. 2006; Cole et al. 2007). Accordingly, as previously proposed for antipsychotics, further insights into the DS actions of mirtazapine may be acquired in substitution studies with mirtazapine to selective ligands acting at sites potentially implicated in its interoceptive profile (Prus et al. 2006; Goudie et al. 2004; Cole et al. 2007). Furthermore, as for other classes of ligand that interact with multiple receptors and display complex mechanisms of action, it is conceivable that other (higher or lower) doses of mirtazapine may yield differing patterns of substitution (see above citations). Thus, despite the primary role of 5-HT2C blockade, additional investigation would be of interest to further characterize the precise mechanisms underlying DS effects of mirtazapine.
References
Alici T, Kayir H, Aygoren MO, Saglam E, Uzbay IT (2006) Discriminative stimulus properties of tianeptine. Psychopharmacology 183:446–451
Andrews JS, Jansen JHM, Linders S, Princen A, Drinkenburg WHIM, Coenders CJH, Vossen JHM (1994) Effects of imipramine and mirtazapine on operant performance in rats. Drug Dev Res 32:58–66
Anttila SA, Leinonen EV (2001) A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev 7:249–264
Bengtsson HJ, Kele J, Johansson J, Hjorth S (2000) Interaction of the antidepressant mirtazapine with alpha2-adrenoceptors modulating the release of 5-HT in different rat brain regions in vivo. Naunyn Schmiedebergs Arch Pharmacol 362:406–412
Berendsen HHG, Broekkamp CLE (1997) Indirect in vivo 5-HT1A-agonistic effects of the new antidepressant mirtazapine. Psychopharmacology 133:275–282
Berg KA, Navailles S, Sanchez TA, Silva YM, Wood MD, Spampinato U, Clarke WP (2006) Differential effects of 5-methyl-1-[[2-[(2-methyl-3pyridyl)oxyl]-5-pyridyl]carbamoyl]-6-trifluoromethylindone (SB 243213) on 5-hydroxytryptamine2C receptor-mediated responses. J Pharmacol Exp Ther 319:260–268
Bourson A, Wanner D, Wyler R, Petit N, Zwingelstein RA, Sleight AJ (1996) Pharmacologic evaluation of the discriminative stimulus of metachlorophenylpiperazine. Pharmacol Biochem Behav 53:107–114
Browne RG (1981) Anxiolytics antagonize yohimbine’s discriminative stimulus properties. Psychopharmacology 74:245–249
Chanrion B, Mannoury la Cour C, Gavarini S, Seimandi M, Vincent L, Pujol JF, Bockaert J, Marin P, Millan MJ (2008) Inverse agonist and neutral antagonist actions of antidepressants at recombinant and native 5-HT2C receptors: differential modulation of cell surface expression and signal transduction. Mol Pharmacol 73:748–757
Cole JC, Field M, Sumnall HR, Goudie AJ (2007) Potentiation of olanzapine substitution in rats discriminating clozapine by the D2/3 agonist quinpirole. Behav Pharmacol 18:185–190
Costall B, Naylor RJ (2004) 5-HT3 receptors. Curr Drug Targets 3:27–37
Crissman AM, O’Donnell JM (2002) Effects of antidepressants in rats trained to discriminate centrally administered isoproterenol. J Pharmacol Exp Ther 302:606–611
Cussac D, Newman-Tancredi A, Quentric Y, Carpentier N, Poissonnet G, Parmentier JG, Goldstein S, Millan MJ (2002) Characterization of phospholipase C activity at h5-HT2C compared with h5-HT2B receptors: influence of novel ligands upon membrane-bound levels of [3H] phosphatidylinositols. Naunyn Schhmiedebergs Arch Pharmacol 365:242–252
Davis R, Wilde MI (1996) Mirtazapine: a review of its pharmacology and therapeutic potential in the management of major depression. CNS Drugs 5:389–402
De Boer (1996) The effects of mirtazapine on central noradrenergic and serotonergic neurotransmission. Int Clin Psychopharmacol 10:19–23
Dekeyne A, Millan MJ (2003) Discriminative stimulus properties of antidepressant agents: a review. Behav Pharmacol 14:391–407
Dekeyne A, Millan MJ (2006) Discriminative stimulus properties of the selective and highly potent a2-adrenoceptor agonist, S18616, in rats: mediation by the a2A subtype, and blockade by the atypical antidepressants, mirtazapine and mianserin. Neuropharmacology 51:718–726
Dekeyne A, Girardon S, Millan MJ (1999) Discriminative stimulus properties of the novel serotonin (5-HT)2C receptor agonist, Ro 60–0175: a pharmacological analysis. Neuropharmacology 38:415–423
Dekeyne A, Gobert A, Iob L, Cistarelli L, Melon C, Millan MJ (2001a) Discriminative stimulus properties of the selective norepinephrine reuptake inhibitor, reboxetine, in rats. Psychopharmacology 154:213–218
Dekeyne A, Iob L, Millan MJ (2001b) Following long-term training with citalopram, both mirtazapine and mianserin block its discriminative stimulus properties in rats. Psychopharmacology 153:389–392
Dekeyne A, Iob L, Hautefaye P, Millan MJ (2002) The selective serotonin2A receptor antagonist, MDL100,907, elicits a specific interoceptive cue in rats. Neuropsychopharmacology 26:552–556
Dekeyne A, Mannoury la Cour C, Gobert A, Brocco M, Lejeune F, Sharp T, Serres F, Daszuta A, Soumier A, Papp M, Rivet J-M, Cremers TI, Muller O, Lavielle G, Millan MJ (2008) S32006, a novel 5-HT2C receptor antagonist displaying broad-based antidepressant and anxiolytic properties in rodent models. Psychopharmacology. doi:10.1007/s00213-008-1177-9
Devoto P, Flore G, Pira L, Longu G, Gessa GL (2004) Mirtazapine-induced corelease of dopamine and noradrenaline from noradrenergic neurons in the medial prefrontal and occipital cortex. Eur J Pharmacol 487:105–111
Di Matteo V, Cacchio M, Di Giulio C, Esposito E (2002) Role of serotonin2C receptors in the control of brain dopaminergic function. Pharmacol Biochem Behav 71:727–734
Doly S, Valjent E, Setola V, Callebert J, Hervé D, Launay JM, Maroteaux L (2008) Serotonin 5-HT2B receptors are required for 3, 4-methylenedioxymethamphetamine-induced hyperlocomotion and 5-HT release in vivo and in vitro. J Neurosci 28:2933–2940
Eckler JR, Rabin RA, Winter JC (2003) Nefazodone in the rat: mimicry and antagonism of [-]-DOM-induced stimulus control. Pharmacol Biochem Behav 75:405–410
Evenden J, Ensler K, Jackson DM, Stephan-Dahlin C (1998) The discriminative stimulus effects of clomethiazole in the rat. Behav Pharmacol 9:379–388
Fabricio ASC, Tringali G, pozzoli G, Navarra P (2005) Mirtazapine acutely inhibits basal and K+ -stimulated release of corticotropin-releasing hormone from the rat hypothalamus via a non-genomic mechanism. Psychopharmacology 178:78–82
Friedman R, Barrett RJ, Sanders-Bush E (1983) Additional evidence that L-5-hydroxytryptophan discrimination models a unique receptor. Psychopharmacology 80:209–213
Glassman AH (1998) Cardiovascular effects of antidepressant drugs: updated. J Clin Psychiatry 59:13–18
Gobert A, Rivet JP, Lejeune F, Newman-Tancredi A, Adhumeau-Auclair A, Nicolas JP, Cistarelli L, Melon C, Millan MJ (2000) Serotonin2C receptors tonically suppress the activity of mesocortical dopaminergic and adrenergic, but not serotonergic, pathways: a combined dialysis and electrophysiological analysis in the rat. Synapse 36:205–221
Gommans J, Hijzen TH, Maes RAA, Olivier B (1997) Discriminative stimulus properties of Eltoprazine. Life Sci 61:11–19
Gommans J, Hijzen TH, Maes RAA, Olivier B (1998) Discriminative stimulus properties of mCPP: evidence for a 5-HT2C receptor mode of action. Psychopharmacology 137:292–302
Goudie AJ, Smith JA, Millan MJ (2004) Characterization of the effects of receptor-selective ligands in rats discriminating the novel antipsychotic quetiapine. Psychopharmacology 171:212–222
Haddjeri N, Blier P, de Montigny C (1998) Acute and long-term actions of the antidepressant drug mirtazapine on central 5-HT neurotransmission. J Affect Disord 51:255–266
Herremans AHJ, Van Der Heyden JAM, Van Drimmelen M, Olivier B (1999) The 5-HT1A receptor agonist flesinoxan shares discriminative stimulus properties with some 5-HT2 receptor antagonists. Pharmacol Biochem Behav 64:389–395
Hoenicke EM, Vanecek SA, Woods JH (1992) The discriminative stimulus effects of clozapine in pigeons: involvement of 5-Hydroxytryptamine1C and 5-hydroxytryptamine2 receptors. J Pharm Exp Ther 263:276–284
Invernizzi RW, Garattini S (2004) Role of presynaptic alpha2-adrenoceptors in antidepressant action: recent findings from microdialysis studies. Prog Neuropsychopharmacol Biol Psychiatry 28:819–827
Kantor S, Jakus R, Balogh B, Benko A, Bagdy G (2004) Increased wakefulness, motor activity and decreased theta activity after blockade of the 5-HT2B receptor by the subtype-selective antagonist SB-215505. Br J Pharmacol 142:1332–1342
Kelley BM, Porter JH (1997) The role of muscarinic cholinergic receptors in the discriminative stimulus properties of clozapine in rats. Pharmacol Biochem Behav 57:707–719
Kelley BM, Porter JH, Varvel SA (1995) Mianserin as a discriminative stimulus in rats: asymmetrical cross-generalization with scopolamine. Psychopharmacology 120:491–493
Kennett GA, Bright F, Trail B, Baxter GS, Blackburn TP (1996) Effects of the 5-HT2B receptor agonist, BW 723C86, on three rat models of anxiety. Br J Pharmacol 117:1443–1448
Kennett GA, Wood MD, Bright F, Trail B, Riley G, Holland V, Avenell KY, Stean T, Upton N, Bromidge S, Forbes IT, Brown AM, Middlemiss DN, Blackburn TP (1997) SB 242084, a selective and brain penetrant 5-HT2C receptor antagonist. Neuropharmacology 36:609–620
Kent JM (2000) SNaRIs, NaSSAs, and NaRIs: new agents for the treatment of depression. Lancet 355:911–918
Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, Jayathilake K, Meltzer HY, Roth BL (2003) H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 28:519–526
Millan MJ (2003) The neurobiology and control of anxious states. Prog Neurobiol 70:53–244
Millan MJ (2005) 5-HT2C receptors as a target for the treatment of depressive and anxious states: focus on novel therapeutic strategies. Therapie 60:441–460
Millan MJ (2006) Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther 110:135–370
Millan MJ, Dekeyne A (2007) Discriminative stimulus properties of the selective norepinephrine reuptake inhibitor, reboxetine, in rats: a characterisation with a/b-adrenoceptor subtype selective ligands, antidepressants, and antagonists at neuropeptide receptors. Int J Neuropsychopharmacol 10:579–593
Millan MJ, Girardon S, Dekeyne A (1999a) 5-HT2C receptors are involved in the discriminative stimulus effects of citalopram in rats. Psychopharmacology 142:432–434
Millan MJ, Gobert A, Girardon S, Dekeyne A (1999b) Citalopram elicits a discriminative stimulus in rats at a dose selectively increasing extracellular levels of serotonin vs. dopamine and noradrenaline. Eur J Pharmacol 364:147–150
Millan MJ, Gobert A, Rivet J-M, Adhumeau-Auclair A, Cussac D, Newman Tancredi A, Dekeyne A, Nicolas J-P, Lejeune F (2000a) Mirtazapine enhances frontocortical dopaminergic and corticolimbic adrenergic, but not serotonergic, transmission by blockade of a2 adrenergic and serotonin2C receptors: a comparison to citalopram. Eur J Neuroscience 12:1079–1095
Millan MJ, Newman-Tancredi A, Audinot V, Cussac D, Lejeune F, Nicolas J-P, Cogé F, Galizzi JP, Boutin JA, Rivet J-M, Dekeyne A, Gobert A (2000b) Agonist and antagonist actions of yohimbine as compared to fluparoxan at alpha2-adrenergic receptors (AR)s, serotonin (5-HT)1A, 5-HT1B, 5-HT1D, and dopamine D2 and D3 receptors. Significance for the modulation of frontocortical monoaminergic transmission and depressive states. Synapse 35:79–95
Millan MJ, Dekeyne A, Papp M, Drieu La Rochelle C, Macsweeny C, Peglion J-L, Brocco M (2001a) S33005, a novel ligand at both serotonin and norepinephrine transporters: II. Behavioral profile in comparison with venlafaxine, reboxetine, citalopram and clomipramine. J Pharm Exp Ther 298:581–591
Millan MJ, Gobert A, Lejeune F, Newman-Tancredi A, Rivet J-M, Auclair A, Peglion J-L (2001b) S33005, a novel ligand at both serotonin and norepinephrine transporters: I. Receptor binding, electrophysiological, and neurochemical profile in comparison to venlafaxine, reboxetine, citalopram and clomipramine. J Pharmacol Exp Ther 298:565–580
Nakayama K, Sakurai T, Katsu H (2004) Mirtazapine increases dopamine release in prefrontal cortex by 5-HT1A receptor activation. Brain Res Bull 63:237–241
O’Connor WT, Leonard 1986 BE (1986) Effect of chronic administration of the 6-aza analogue of mianserin (Org 3770) and its enantiomers on behaviour and changes in noradrenaline metabolism of olfactory-bulbectomised rats in the “open field” apparatus. Neuropharmacology 25:267–270
Olivier B, Bosch L, Van Hest A, Van Der Heyden J, Mos J, Van Der Poel G, Schipper J, Tulp M (1993) Preclinical evidence on the psychotropic profile of fluvoxamine. Pharmacopsychiat 26:2–9
Olivier B, van Wijngaarden I, Soudijn W (2000) 5-HT3 receptor antagonists and anxiety; a preclinical and clinical review. J Eur Neuropsychopharm 10:77–95
Olivier B, Broersen LM, Slangen JL (2002) 5-HT3 receptor ligands lack discriminative stimulus properties. Prog Neuropsychopharmacol Biol Psychiatry 26:463–471
Pedersen L, Klysner R (1997) Antagonism of selective serotonin reuptake inhibitor–induced nausea by mirtazapine. Int Clin Psychopharmacol 12:59–60
Prus AJ, Philibin SD, Perhson AL, Porter JH (2006) Discriminative stimulus properties of the atypical antipsychotic drug clozapine in rats trained to discriminate 1.25 mg/kg clozapine vs. 5.0 mg/kg clozapine vs. Vehicle. Behav Pharm 17:185–194
Ramamoorthy R, Radhakrishnan M, Borah M (2008) Antidepressant-like effects of serotonin type-3 antagonist, ondansetron: an investigation in behaviour-based rodent models. Behav Pharmacol 19:29–40
Rauggi R, Cassanelli A, Raone A, Tagliamonte A, Gambarana C (2005) Study of mirtazapine antidepressant effects in rats. Int J Neuropsychopharmacol 15:1–11
Rogoz Z, Wrobel A, Dlaboga D, Maj J, Dziedzicka-Wasylewska M (2002) Effect of repeated treatment with mirtazapine on the central alpha1-adrenergic receptors. J Physiol Pharmacol 53:105–116
Schmid DA, Wichniak A, Uhr M, Ising M, Brunner H, Held K, Weikel JC, Sonntag A, Steiger A (2006) Changes of sleep architecture, spectral composition of sleep EEG, the nocturnal secretion of cortisol, ACTH, GH, prolactin, melatonin, ghrelin, and leptin, and the DEX-CRH test in depressed patients during treatment with mirtazapine. Neuropsychopharmacology 31:832–844
Schreiber R, Brocco M, Millan MJ (1994) Blockade of the discriminative stimulus effects of DOI by MDL100,907 and the ‘atypical’ antipsychotics, clozapine and risperidone. Eur J Pharmacol 264:99–102
Schule C, Baghai T, Bidlingmaier M, Strasburger C, Laakman G (2002) Endocrinological effects of mirtazapine in healthy volunteers. Prog Neuropsychopharmacol Biol Psychiatry 26:1253–1261
Smith RL, Barrett RJ, Sanders-Bush E (1990) Adaptation of brain 5-HT2 receptors after mianserin treatment: receptor sensitivity, not receptor binding, more accurately correlates with behavior. J Pharmacol Exp Ther 254:484–488
Smith RL, Gresch PJ, Barrett RJ, Sanders-Bush E (2002) Stimulus generalization by fenfluramine in a quipazine–ketanserin drug discrimination is not dependent on indirect serotonin release. Pharmacol Biochem Behav 72:77–85
Snoddy AM, Tessel RE (1985) Prazosin: effect on psychomotor-stimulant cues and locomotor activity in mice. Eur J Pharmacol 116:221–228
Sommer BR, Fenn H, Pompei P, DeBattista C, Lembke A, Wang P, Flores B (2003) Safety of antidepressants in the elderly. Expert Opin Drug Safety 2:367–383
Szegedi A, Schwertfeger N (2005) Mirtazapine: a review of its clinical efficacy and tolerability. Expert Opin Pharmacother 6:631–641
Tatsumi M, Groshan K, Blakely RD, Richelson E (1997) Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 340:249–258
Terry P, Katz JL (1997) Dopaminergic mediation of the discriminative stimulus effects of bupropion in rats. Psychopharmacology 134:201–212
Walker EA, Kohut Hass RW, Brown EK Jr, Prabandham A, Lefever T (2005) Selective and nonselective serotonin antagonists block the aversive stimulus properties of MK212 and m-chlorophenylpiperazine (mCPP) in mice. Neuropharmacology 49:1210–1219
Whale R, Clifford EM, Cowen PJ (2000) Does mirtazapine enhance serotonergic neurotransmission in depressed patients? Psychopharmacology 148:325–332
Winter JC, Rabin RA (1993) Antagonism of the stimulus effects of yohimbine and 8 hydroxydipropylaminotetralin. Pharmacol Biochem Behav 44:851–855
Wood MD, Reavill C, Trail B, Wilson A, Stean T, Kennett GA, Lightowler S, Blackburn TP, Thomas D, Gager TL, Riley G, Holland V, Bromidge SM, Forbes IT, Middlemiss DN (2001) SB-243213; a selective 5-HT2C receptor inverse agonist with improved anxiolytic profile: lack of tolerance and withdrawal anxiety. Neuropharmacology 41:186–199
Yamamoto T, Walker EA, Woods JH (1991) Agonist and antagonist properties of serotonergic compounds in pigeons trained to discriminate either quipazine or l-5-hydroxytryptophan. J Pharmacol Exp Ther 258:999–1007
Young R, Glennon RA (2002) Nicotine and bupropion share a similar discriminative stimulus effect. Eur J Pharmacol 443:113–118
Zhang L, Barrett JE (1991) Imipramine as a discriminative stimulus. J Pharmacol Exp Ther 259:1088–1093
Acknowledgements
We thank Loretta Iob for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dekeyne, A., Millan, M.J. Discriminative stimulus properties of the “atypical” antidepressant, mirtazapine, in rats: a pharmacological characterization. Psychopharmacology 203, 329–341 (2009). https://doi.org/10.1007/s00213-008-1259-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-008-1259-8