
Abstract
The phyllosphere is a challenging microbial habitat in which microorganisms can flourish on organic carbon released by plant leaves but are also exposed to harsh environmental conditions. Here, we assessed the relative importance of canopy position—top, mid, and bottom at a height between 31 and 20 m—and tree species identity for shaping the phyllosphere microbiome in a floodplain hardwood forest. Leaf material was sampled from three tree species—maple (Acer pseudoplatanus L.), oak (Quercus robur L.), and linden (Tilia cordata MILL.)—at the Leipzig canopy crane facility (Germany). Estimated bacterial species richness (Chao1) and bacterial abundances approximated by quantitative PCR of 16S rRNA genes exhibited clear vertical trends with a strong increase from the top to the mid and bottom position of the canopy. Thirty operational taxonomic units (OTUs) formed the core microbiome, which accounted for 77% of all sequence reads. These core OTUs showed contrasting trends in their vertical distribution within the canopy, pointing to different ecological preferences and tolerance to presumably more extreme conditions at the top position of the canopy. Co-occurrence analysis revealed distinct tree species-specific OTU networks, and 55–57% of the OTUs were unique to each tree species. Overall, the phyllosphere microbiome harbored surprisingly high fractions of Actinobacteria of up to 66%. Our results clearly demonstrate strong effects of the position in the canopy on phyllosphere bacterial communities in a floodplain hardwood forest and—in contrast to other temperate or tropical forests—a strong predominance of Actinobacteria.
Similar content being viewed by others

Introduction
The phyllosphere is an important microbial habitat which spans about 108 km2 on a global scale [1]. It is host to central biogeochemical processes as well as plant-microbe interactions that affect plant community dynamics, and ecosystem functioning and productivity [2, 3]. Microbiota on leaf surfaces contribute to biogeochemical processes such as N2 fixation [4, 5], nitrification [6], and transformation of C1 compounds or terpenes and monoterpenes released from the plants [7]. Especially in forest canopies, they may play a central role in the bioremediation of air pollutants [8] and are in exchange with atmospheric and cloud microbiota, suggesting important implications for climate regulation [7]. Beyond their role in biogeochemical processes, phyllosphere microbiota are not only passive inhabitants on surfaces of plants but interact with their host in multiple ways [9], resulting in plant-microbe relationships that range from loose associations to defined symbioses [7]. They produce phytohormones or affect the production of these hormones by the plant [10,11,12] and they improve host resistance against pathogens [13, 14].
However, the phyllosphere is also a harsh environment where microorganisms are exposed to extreme conditions such as high UV radiation, desiccation, rainfall, antimicrobial substances released by leaves, and strong nutrient limitation [7]. Altogether, these environmental parameters positively select for the bacterial taxa that are able to persist on leaves [15]. As a consequence, phyllosphere bacterial diversity has been shown to be much lower than diversity of the rhizosphere, soil, or marine ecosystems [16,17,18]. Especially in the canopy of large trees, selective environmental forces that restrict microbial diversity are likely to vary along vertical gradients [19] with the severest stress by abiotic parameters presumably acting at the top of the canopy. However, studies addressing intra-individual variability of phyllosphere microbial communities have so far mostly focused on a single tree species such as Gingko biloba or Magnolia grandiflora [20, 21] or on rather small trees at a maximum height of 6 m [19]. Ongoing colonization of leaves by microorganisms and their continuous removal, e.g., by rain fall, result in complex community dynamics in the forest canopy phyllosphere [7]. It is unclear if microbial taxa differ in their preference for a particular canopy position and how this translates into the spatial heterogeneity of canopy-associated biogeochemical processes. Despite their high relevance for ecosystem functioning, phyllosphere microbiota in forest canopies have so far received comparatively little attention. Factors such as host species identity, leaf age, location in the canopy, light incidence, and microclimate conditions have been identified as central factors shaping the phyllosphere environment in tropical and North American temperate forests [22–27; 3]. However, more insight is needed if the phyllosphere diversity patterns observed for tropical and North American forests, especially the strong effect of host species identity, also apply to European forests with a different tree species composition.
Here, we hypothesize that (i) forest trees harbor species-specific phyllosphere bacterial communities, and that (ii) microbial communities at the treetop are the most distinct, as they are the most exposed to abiotic stress factors. Taking advantage of the Leipzig canopy crane facility located in central Germany, allowing us to sample leave material from up to 33 m height, we compared phyllosphere microbial communities between three different tree species abundant in the Leipzig floodplain forest—Acer pseudoplatanus L., Quercus robur L., and Tilia cordata MILL.—and across three different height levels within the canopy—top, mid, and bottom. Our results revealed clear vertical trends of increasing bacterial diversity and abundances, and changes in community structure from the top of the canopy to mid and bottom canopy, which were further modulated by plant species identity.
Methods
Leipzig Floodplain Hardwood Forest and Canopy Crane Facility
Leaf samples were obtained from three tree species—Q. robur L. (oak; Qr), A. pseudoplatanus L. (maple; Ap), and T. cordata MILL. (linden; Tc)—in the Leipzig floodplain hardwood forest, located near the city of Leipzig in Germany (Supplementary Fig. 1a). Situated in the floodplain of the Elster, Pleiße, and Luppe rivers, the Leipzig floodplain forest is one of the largest floodplain forests in Central Europe [22]. Climatic conditions are characterized by warm summers and an annual mean temperature of 8.4 °C with an annual precipitation of 516 mm [23]. The forest consists of the ash-elm floodplain forest (Fraxino-Ulmetum) and is dominated by maple (A. pseudoplatanus L.), ash (Fraxinus excelsior L.), oak (Q. robur L.), and hornbeam (Carpinus betulus L.), with smaller contribution of linden (T. cordata MILL.) and elm (Ulmus minor MILL.) [24]. A crane facility (Leipzig Canopy Crane facility, LCC) for the investigation of forest tree canopies was established in this floodplain forest in 2001, allowing access to about 800 tree individuals in up to 33 m height, covering a total area of 1.65 ha (Supplementary Fig. 1a). The estimated age of the trees sampled in this study ranged from 102 to 370 years for oak, 97 to 146 years for maple, and 205 to 240 years for linden (Supplementary Table 1). Different positions within the tree canopy were accessed by using a gondola attached to the crane. We sampled leaf material from the top, mid, and bottom position of the tree canopy. Depending of the height of individual trees, these positions ranged from 27.0 to 30.7 m, 18.6 to 26.3 m, and 12.9 to 23.2 m, respectively (Supplementary Fig. 1b). Samples were obtained from three individuals per tree species. At each canopy position, we sampled leaves in three spatial replicates, that is, leaves were sampled from different twigs which were between 0.2 and 0.5 m apart on the same height level. This way, a total of 81 samples were collected. For each tree individual, canopy position, and spatial replicate, between five and ten leaves were sampled and pooled for further analysis.
Sampling of Leaf Material and Detachment of Surface-Associated Microbes
Leaves were sampled by clipping off leaves with ethanol-cleaned scissors, followed by immediate transfer to autoclaved polyphenylene ether (PPE) containers. Leaves were stored at ambient temperature (approx. 15 °C) during transport and were immediately processed upon arrival at the laboratory within 2 h. Leaves were amended with 250 ml suspension buffer (0.15 M NaCl, 0.1% Tween 20; [9]) in the autoclaved containers in which leaves had been sampled, subjected to mild sonication (1 min at 10% intensity, turned and another 1 min at 10% intensity), followed by shaking for 20 min at 100 rpm at room temperature. Subsequently, suspensions were filtered through 0.2-μm polyethersulfone filters (Supor, Pall Corporation), and filters were stored at −80 °C until nucleic acid extractions were performed. The remaining leaf material was dried at 50 °C for 1 week for determination of dry weight.
Nucleic Acid Extraction, Illumina MiSeq Amplicon Sequencing, and Quantitative PCR
DNA was extracted from the filters using the DNeasy PowerSoil Extraction kit (Qiagen) following the manufacturer’s protocol. Filters were cut into smaller pieces to facilitate cell disruption during the bead-beating step. Amplicon sequencing of bacterial 16S rRNA genes was carried out targeting the V3-V4 region with the primer combination Bakt_0341F/Bakt_0785R [25]. PCR amplification, library preparation, and sequencing on an Illumina MiSeq platform using v3 chemistry was performed at LGC (Berlin) as previously described [26]. Abundances of bacterial 16S rRNA genes were determined by quantitative PCR using Brilliant SYBR Green II Mastermix (Agilent Technologies) on a Mx3000P system (Agilent Technologies) and the primer combination Bakt_0341F [25] and Bakt_0799R, which is the reverse complement version of the primer 799F [27] discriminating against chloroplast-derived 16S rRNA genes. For qPCR, we used the following cycling conditions: 10 min at 95 °C, followed by 45 cycles of 30 s at 95 °C, 30 s at 53 °C, and 40 s at 72 °C, and subsequent melting curve analysis. Due to loss of plant material of some samples before determination of leaf dry weight, abundance data are only available for a subset of all samples (see Supplementary Table 2).
Sequence Analysis
Sequence analysis was carried out using Mothur v1.39.1 [28] following the Schloss MiSeq SOP [29] as previously described [26] along with the SILVA taxonomy reference database v132 [30]. Chimera search was performed using the uchime algorithm implemented in Mothur. Operational taxonomic units (OTUs) were assigned on a 0.03 distance level using the vsearch algorithm. We obtained 6,037,303 high-quality sequence reads across 81 samples with read numbers per sample ranging from 1610 to 198,790. For further statistical analysis, read numbers were normalized to the same number for all samples (11,188 reads) using the sub.sample function implemented in Mothur, resulting in the exclusion of two samples with too low read numbers from the data set (Ap6 and Qr18). Sample Tc12 was also excluded from further analysis because a large part of the sample had been lost during filtration, making the bacterial community composition less comparable. Sequences obtained in this study have been submitted to the European Nucleotide Archive (ENA) under the study accession number PRJEB36420, sample accession numbers SAMEA6502636–SAMEA6502715.
Statistical Analysis
Variation of bacterial species richness (number of observed and estimated OTUs (Chao)) across canopy positions and tree species was analyzed using two-factorial ANOVA followed by Tukey’s post hoc test. Vertical changes in relative abundances of phylum-level taxa between top, mid, and bottom canopy position were analyzed by linear regression. Principal coordinate analysis (PCoA) and PERMANOVA analysis based on Bray-Curtis dissimilarities was employed to assess effects of tree species identity and position in the canopy on phyllosphere bacterial community composition. PERMANOVA analysis was run with 999 permutations. For PCoA analysis, only OTUs with at least 10 reads across all samples were included. To further investigate the effect of tree species on the phyllosphere bacterial communities, we merged all the OTUs observed in association with a given tree species in the different samples (tree individuals, canopy positions, and spatial replicates) to one OTU pool and compared the resulting three tree species-dependent OTU pools to each other using Venn diagrams in Mothur. To assess whether the fraction of OTUs shared between top, mid, and bottom canopy position was different, we first pooled all the OTUs found in association with one tree individual and canopy position and determined the shared OTUs between top/mid, top/bottom, and mid/bottom canopy for each tree individual. Values from all nine tree individuals were used to analyze if top/mid, top/bottom or mid/bottom shared fractions were significantly different from each other, which was done using the Mann-Whitney U test. Finally, we followed distribution patterns of the 20 most abundant OTUs across tree species and position in the canopy by subjecting relative abundances of these OTUs to hierarchical clustering based on Bray-Curtis dissimilarities, combined with visualization using a heatmap. Except for PERMANOVA analysis, which was done in R [31] using the Adonis functions from the R package vegan [32], all these calculations were carried out using the software PAST v3.11 [33]. This software was also used for the generation of box and whisker plots.
Co-occurrence network analysis was done using the MENA platform [34]. Networks were constructed for the phyllosphere microbiome of each tree species, including samples from all three canopy positions, tree individuals, and spatial replicates per tree species. Only OTUs with more than 20 sequence reads across all samples per tree species were included. Network calculations were based on Spearman rank correlation coefficients without log transformation of the data. Similarity thresholds were 0.68, 0.65, and 0.67 for maple, oak, and linden, respectively. Networks were graphically refined using Cytoscape 3.7.2.
Results
Effect of Canopy Position and Tree Species on Phyllosphere Bacterial Communities
Amplicon sequencing of bacterial 16S rRNA genes revealed clear vertical trends in OTU richness and community composition from the top to the bottom canopy position, further modulated by the tree species. The number of observed and estimated (Chao estimator) species-level OTUs tended to increase from the top of the canopy towards the mid position (Fig. 1a). For the phyllosphere of linden, the increase in bacterial OTU numbers was already visible between the mid and the bottom position of the canopy while such a trend was less obvious for the other two tree species. Median values of observed (estimated) OTU numbers ranged from 264 to 331 (607 to 655) at the top of the canopy, from 332 to 400 (729 to 937) at the mid position, and from 393 to 429 (875 to 933) at the bottom of the canopy with the highest numbers observed in association with maple. Overall, canopy position had a significant effect on both observed (F = 19.66, p < 0.001) and estimated OTU numbers (F = 11.86, p < 0.001), while the effect of tree species was only significant for the observed OTUs (F = 3.64, p = 0.03). Interactions between these two factors were not significant.

a Estimated bacterial species richness and b abundance of bacterial 16S rRNA genes per g leaf (dry weight) in the canopy of Q. robur L. (left panel), A. pseudoplatanus L. (mid panel), and T. cordata MILL. (right panel) with positions in the canopy categorized as “top,” “mid,” and “bottom.” Data are means (± standard deviation) of results obtained from three tree individuals with three replicates per sampled canopy area. For gene abundances, a reduced number of samples is available (see Supplementary Table 2)
Abundances of bacterial 16S rRNA genes per g dry weight leaf showed a similar trend as bacterial OTU richness, with lower abundances at the top of the canopy compared to the mid or bottom position. Both canopy position (F = 8.717, p < 0.001) and tree species (F = 9.09, p < 0.001) had a significant effect on bacterial abundances. However, these data rather represent trends, as for some of the samples, especially from the top of the canopy, only a reduced number of replicates is available. Across all tree species, individuals, and position in the canopy, bacterial 16S rRNA gene abundances ranged from 1.9 × 106 to 1.3 × 109 g−1 (dry weight) (Fig. 1b). Abundances observed in association with linden were significantly higher than the numbers in association oak (Tukey’s post hoc test, p < 0.001) or maple (p < 0.001).
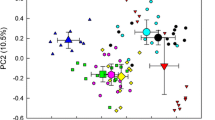
In line with these findings, PERMANOVA analysis based on Bray-Curtis dissimilarities revealed that position in the canopy as well as tree species had a significant effect on phyllosphere bacterial community structure (p < 0.001) with canopy position explaining 15.2% and tree species identity explaining 14.9% of the total community variation (Fig. 2b; Supplementary Table 3). In addition, interactions between these two factors also had a significant effect (p < 0.001). The observed height- and tree species-dependent trends in bacterial OTU richness were reflected by clustering patterns of samples using principal coordinate analysis. Here, especially for linden and maple, samples obtained from the top of the canopy clustered separate from samples obtained from mid or bottom positions with clear differences in OTU composition between these two tree species (Fig. 2a). In contrast, for the mid and bottom positions of the canopy, we observed minor clustering of communities according to tree species but also large overlaps. For each tree species, communities of the mid and bottom positions were more similar to each other than they were to the communities at the top of the canopy. In fact, for all three tree species together, the fraction of OTUs shared between the top and mid canopy positions (15–19.4%) or the top and bottom canopy positions (15.5–20.5%) was significantly lower than the fraction of OTUs shared between the mid and bottom positions (19.4–25.1%; Mann-Whitney U test, p = 0.00172) (Supplementary Fig. 2).

a Principal coordinate analysis of phyllosphere bacterial communities across three tree species and top, mid, and bottom positions of the canopy. b Variation partitioning resulting from PERMANOVA analysis. Analyses were based on distribution patterns of species-level OTUs using Bray-Curtis dissimilarities. Colors denote tree species, symbols denote position within the tree canopy
Composition of the Hardwood Forest Canopy Microbiome
The phyllosphere bacterial communities associated with all three tree species were largely dominated by Actinobacteria, Bacteroidetes, Alphaproteobacteria, and Gammaproteobacteria which together accounted for at least 95% of the sequence reads in each sample (Fig. 3). Members of Deinococcus-Thermus, candidate phylum FBP, representatives of the Candidate Phyla Radiation [35] such as Cand. Saccharimonadia, and Deltaproteobacteria were consistently present in the phyllosphere communities but rarely reached relative abundances of more than 3%. Taxa associated with chemolithoautotrophic lifestyles within the Gammaproteobacteria, such as Nitrosomonas, Nitrosospira, or Ferribacterium, were only represented by a few sequence reads across all samples.

Composition of the bacterial communities associated with the phyllosphere of oak (Q. robur L.), maple (A. pseudoplatanus L.), and linden (T. cordata MILL.) at the top, mid, and bottom positions of the canopy. Each bar represents mean values of three tree individuals and three spatial replicates per tree individual. Taxonomic affiliation is shown on the phylum level or class level for Proteobacteria and Patescibacteria
Notably, we observed major shifts in the relative fractions among the four dominant phyla in dependence of canopy position. For both maple and oak, the fraction of Actinobacteria increased strongly from the top of the canopy (18 and 24%, respectively) to 37 and 44% at the bottom of the canopy (linear regression, p < 0.006) while no such height-dependent trend was visible in association with linden trees (p = 0.55375) (Fig. 3; Supplementary Fig. 3). Moreover, relative abundances of Actinobacteria were lower in the linden phyllosphere compared to maple and oak for the bottom position of the canopy (pairwise Mann-Whitney U test; p = 0.008 and p = 0.006, respectively) and, compared to oak, also for the top position of the canopy (p = 0.004). In turn, the relative fraction of Gammaproteobacteria mostly represented by Burkholderiacaea, Enterobacteriacaea, Diplorickettsiacaea, and Pseudomonadaceae tended to decrease from the top towards the bottom of the canopy for all three tree species (Fig. 3); however, these trends were not significant (Supplementary Fig. 3). Bacteroidetes, represented mostly by the families of Hymenobacteraceae and Spirosomaceae, did not show any obvious changes in relative abundance with canopy position on the phylum level.
Following changes in relative abundances of the most abundant 20 OTUs across the top, mid, and bottom positions of the canopy further demonstrated that the distribution patterns of individual OTUs were often linked to canopy position, which was further modulated by tree species identity. The strongest increase in relative abundance from the top towards the canopy mid and bottom was observed for OTU01 affiliated with Friedmaniella (Propionibacteriaceae), which constituted a dominant member of the phyllosphere community, accounting for up to 46% of the sequence reads in the individual samples at the canopy bottom and mid position but on average only for 4–11% at the top of the canopy (Supplementary Fig. 4). In turn, several OTUs decreased in relative abundance towards the mid and bottom position of the canopy for all three tree species, e.g., OTU09 (Massilia), OTU10 (Hymenobacter), OTU15 (Methylobacterium), and OTU17 (Kineococcus).
Hierarchical clustering based on Bray-Curtis dissimilarities of these 20 OTUs across all samples provided further insight into their preferential association with canopy position or a particular tree species. In general, clustering patterns according to tree species appeared to be less pronounced than those according to canopy position and confirmed the preferential association of OTU01 with bottom and mid canopy positions of oak and maple and its clear distinction from the distribution patterns of the other abundant OTUs (Fig. 4). OTU02, OTU03, OTU04, OTU05, OTU06, OTU07, and OTU09 affiliated with Beijerinckiaceae, Sphingomonadaceae, Hymenobacter, and Massilia showed distribution patterns complementary to those of OTU01. OTU05 (Sphingomonas), OTU07 (Hymenobacter), and OTU09 (Massilia) occurred primarily in association with the canopy top position across all three tree species. Clustering of bacterial communities according to tree species was more pronounced for the canopy top position compared to mid and bottom position.

Relative abundance of the 20 most abundant species-level OTUs across all samples. Affiliation with top, mid, or bottom positions of the canopy is depicted by triangles, circles, or squares, respectively. Names of samples refer to microbial communities in association with A. pseudoplatanus L. (Ap), Q. robur L. (Qr), and T. cordata MILL. (Tc). Two-way hierarchical clustering was performed based on Bray-Curtis dissimilarities
Phyllosphere Core Microbiome and Co-occurrence Networks
Integrating across tree individuals and positions in the canopy, we further compared the sets of OTUs associated with a given tree species to each other. For each tree species, about 55–57% of the OTUs were unique to that tree species, while a fraction of 26–29% was shared between all three tree species. Maple and oak shared a slightly higher fraction of OTUs between their phyllosphere microbiomes (36–37%) compared to the fraction shared with linden (33–34%) (Supplementary Fig. 5).
Thirty species-level OTUs from four different phyla and 13 different families were present across all tree species and individuals, canopy positions, and spatial replicates, forming the phyllosphere core microbiome. Altogether, these 30 OTUs accounted for 77% of the sequence reads but only for 0.3% of the observed phyllosphere bacterial diversity, indicating that the phyllosphere communities were strongly dominated by these core microbiome representatives. Among the core microbiome members, Hymenobacteraceae (Cytophagales, Bacteroidetes) contributed the largest number of OTUs, followed by Burkholderiaceae (Betaproteobacteriales, Gammaproteobacteria) and Beijerinckiacaea (Rhizobiales, Alphaproteobacteria) (Supplementary Table 4).
Bacterial communities associated with a given tree species were subjected to co-occurrence network analysis, which revealed substantially different networks for each tree species (Fig. 5). We obtained networks with 156 nodes and 332 links for maple, 216 nodes and 258 links for oak, and 161 nodes and 365 links for linden. Notably, co-occurrence networks of the oak phyllosphere microbiomes showed the largest fraction of negative interactions (44.2%), while negative interactions accounted for only 11.1 or 20.3%, respectively, of all interactions in the phyllosphere OTU network of maple and linden. Across samples, OTU01 was not only the OTU with the highest relative abundance but also among the top five OTUs with the highest number of links to other OTUs within the maple and oak canopy. In contrast, OTU01 was less strongly connected in the phyllosphere of linden, coinciding with its lower relative abundance in association with that tree species. Overall, OTU01 exhibited mostly positive links to other OTUs. However, these associated OTUs differed substantially across tree species. For the oak phyllosphere, 63% of the OTUs associated with OTU01 were also Actinobacteria, while Proteobacteria dominated the associated OTUs in the linden phyllosphere. In association with maple, OTU01 exhibited the most diverse connections to other OTUs, including an especially high contribution of Spirosomacea and Saccharimonadales compared to the other two tree species.

Co-occurrence network of bacterial species-level OTUs across height levels and individuals for each tree species. a Q. robur L., b A. pseudoplatanus L., c T. cordata MILL. Only network modules with more than 10 OTUs are shown. Color of circles denotes taxonomic affiliation on the family level. Numbers indicate core OTUs
Discussion
Phyllosphere microbiota in tree canopies play central roles in biogeochemical cycling and contribute to host plant fitness, protection and productivity [3]. Here, we hypothesized that both position in the canopy and tree species identity shape the phyllosphere microbial community in a floodplain hardwood forest in central Germany. In fact, we found evidence of an influence of both factors; however, canopy position-dependent effects were more pronounced and pointed to vertical gradients across the canopy. Bacterial abundance and OTU richness were lower at the top of the canopy compared to the mid canopy and bottom canopy positions across all three tree species—Q. robur L., A. pseudoplatanus L., T. cordata MILL.. While previous studies reported a large variation of phyllosphere bacterial community structure within a single tree canopy [19, 20], trends of increasing diversity from the top towards the mid and bottom canopies have rarely been described for bacterial [21] or fungal communities [36,37,38]. Here, we demonstrate that not only microbial diversity but also microbial abundances follow the same trends and are likely affected by the same canopy position-dependent factors.
Leaf-associated bacteria might simply be washed off with rainwater, leading to continuous loss of biomass and OTUs to the mid and bottom positions of the canopy or eventually resulting in their transport to the soil via throughfall or stemflow [39]. In fact, Stone and Jackson [21] found that rainfall influenced compositional similarity of bacterial communities throughout the canopy of Magnolia trees, which could also be one the mechanisms underlying the higher similarity between mid canopy and bottom canopy communities versus communities at the top of the canopy observed in our study. Similarly, interior canopy and upper canopy communities were the most distinct in the Magnolia canopy [21]. However, these authors also reported that rain did not have any effect on bacterial species richness, questioning to which extent rainfall imposes physical disturbance on the phyllosphere-associated microbiota. Moreover, leaf-associated bacteria are protected against such physical forces by aggregates and biofilms and also by the surface structure of the leaves [40]. Consequently, rainfall is likely not the main driver of lower abundance and diversity at the top of the canopy in our study.
Alternatively, lower diversity at the top of the canopy could be a direct result of harsher environmental conditions acting on the microbial colonizers, such as higher exposure to UV radiation, weather extremes, desiccation, and depletion of nutrients due to rain-mediated wash off [21]. These conditions could lead to a selective enrichment of specialists at the top of the canopy, while the phyllosphere bacteria which are highly abundant at the mid and bottom canopy positions become less competitive under the conditions at the top of the canopy. Interestingly, Actinobacteria, in particular one OTU affiliated with the genus Friedmaniella, appeared to be the most responsive bacterial group to canopy position and showed a strong increase in its relative abundance from the top towards the mid and bottom canopy positions. In general, distribution patterns of the 20 most abundant OTUs appeared to be strongly linked to canopy position, suggesting contrasting ecological preferences for bacteria related to Friedmaniella versus those related to Hymenobacter, Methylobacterium, Kineococcus, or Massilia, whose relative abundance increased at the top of the canopy. Previous findings suggested that general stress response is an essential mechanism for plant colonization by Methylobacterium, including responses to heat shock and desiccation, and oxidative, UV, ethanol, and osmotic stresses [11, 41]. In addition, increased abundances of Methylobacterium in upper parts of the canopy of Magnolia trees have been explained by a positive response of this genus to changes in leaf physiology following higher light or higher temperature, or by a direct response to these environmental parameters [21]. In addition to a better adaption to harsh environmental conditions at the top of the canopy, increased relative abundances of Methylobacterium, Hymenobacter, Kineococcus, and Massilia may also have been supported by reduced abundances of Friedmaniella as a potentially very competitive inhabitant of the hardwood forest phyllosphere.
Plant species identity was identified as another key factor that influenced phyllosphere bacterial community composition in the floodplain forest, similar to tropical forests and temperate forest ecosystems in North America [42,43,44]. Bacterial communities associated with oak and maple were more similar to each other than to those associated with linden trees, suggesting that oak and maple provided more favorable and more similar conditions for certain taxa, e.g., for Actinobacteria related to Friedmaniella, than did linden. Plant host attributes such as plant taxonomic identity and phylogeny, wood density, leaf mass per area, seed mass, leaf water content, and leaf nitrogen and phosphorus concentrations have been suggested as key factors underlying the relationship between plant species and their microbiome in neotropical as well as in temperate forests [43, 45, 46]. Additional factors may include the rate of production of volatile organic compounds such as methanol, which can act as important substrate for the phyllosphere microbiota [7, 42, 47, 48].
Co-occurrence network analysis revealed that the three tree species did not only differ in their bacterial community structure and OTU composition but also in the patterns how these OTUs were connected to each other across tree individuals, canopy position, and spatial replicates. The observed larger fraction of negative interactions between OTUs in association with oak may point to stronger vertical gradients within the oak canopy or larger variation across individuals of the same tree species. Moreover, OTUs with a central position in the network, e.g., by multiple connections to other OTUs, differed between tree species. These findings suggest that plant host-related factors and the chemical environment that they shape select for specific microbial core consortia that are strongly tree species-dependent.
Since Actinobacteria made up to 65% of the phyllosphere bacterial community in some cases, they were by far more prominent in our study compared to neotropical or tropical forests [43, 48] but also to previous investigations of Canadian temperate forests or the phyllosphere of hornbeam (Carpinus betulus), where they only accounted for 5–9% of the total community [46, 49]. The most abundant OTU in our study was closely related to Friedmaniella okinawensis and F. sagamiharensis originally isolated from spider webs in a Japanese forest [50]. Bacteria related to Friedmaniella have been found in lower abundances in the phyllosphere of apple orchards or in urban environments [51, 52] and can also grow as endophytes [53, 54]. In fact, species within the genus Friedmaniella isolated from forest spider webs or the bark of mangrove plants have the capability to utilize a broader range of organic carbon compounds than other species of that genus [50, 53], suggesting that this broader substrate spectrum could be one the mechanisms underlying their success in the phyllosphere. Alternatively, the high relative abundance of Friedmaniella could also be linked to a stage of early senescence of the leaves, since Actinobacteria are generally known as major decomposers of leaf litter [55].
Most of the other genus-level taxa representing the hardwood forest core microbiome, such as Hymenobacter, Methylobacterium, Sphingomonas, and Pseudomonas have frequently been observed in association with other temperate forest tree species [19, 21] but also with herbaceous plants [16]. The genus Methylobacterium uses methanol as its carbon and energy source, a C1 compound typically released by plants [56]. Besides methanol, small amounts of nutrients, such as glucose, fructose, and sucrose [1], but also amino acids, methane, terpenes, and chloromethane [16, 49, 57, 58] can leach from the interior of the plant and be available for the phyllosphere microbiota. Overall, our findings suggest that microbial processes in the hardwood forest canopies are largely dominated by heterotrophic or C1-dependent metabolisms. Although nitrification has previously been proposed as an important process in tree canopies, stimulated by excess atmospheric deposition of ammonia [6, 59], we found only few sequence reads affiliated with chemolithoautotrophic Nitrosomonadaceae.
Given the temporal development of forest tree canopies throughout the growing season, our sampling provides only one snapshot, and the extent to which the September phyllosphere communities differ from earlier stages in spring and summer remains currently unclear. A previous study in a temperate mixed forest showed that temporal effects were smaller than those associated with host species identity [46]. Successional changes in phyllosphere communities can be associated with changes in the physiology of the host plant but can also be shaped by the constant import of microbes from various sources such as air, soils, rainwater, and animal and plant dispersal vectors [43, 60, 61]. Representatives of the genera Hymenobacter, Methylobacterium, and Massilia have been reported from air samples [62] or from aerosols originating from agricultural practices [7, 63], suggesting that airborne microbes play a major role in the early colonization of the surfaces of young leaves in spring [7] and could continuously be introduced to the phyllosphere communities throughout the season. Consequently, the September phyllosphere represents a stage that integrates the results of different mechanisms of colonization and competitive interactions between phyllosphere microbiota throughout the growing season.
Conclusions
Our findings clearly demonstrate that both position in the canopy and tree species have a strong effect on the structure of phyllosphere bacterial communities in a floodplain hardwood forest. Consistently lower bacterial diversity at the top of the canopy compared to the canopy mid and bottom positions pointed to a stronger selective pressure on phyllosphere bacteria given presumably harsher environmental conditions at the treetop. Across all three tree species, we observed a striking predominance of Actinobacteria related to Friedmaniella sp., which could be a typical feature of floodplain hardwood forests or linked to the early senescent state of leaves sampled in mid September.
References
Lindow SE, Brandl MT (2003) Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69:1875–1883. https://doi.org/10.1128/aem.69.4.1875-1883.2003
Vorholt JA (2012) Microbial life in the phyllosphere. Nature Rev Microbiol 10:828–840. https://doi.org/10.1038/nrmicro2910
Laforest-Lapointe I, Whitaker BK (2019) Decrypting the phyllosphere microbiota: progress and challenges. Am. J. Bot. 106:171–173. https://doi.org/10.1002/ajb2.1229
Freiberg E (1998) Microclimatic parameters influencing nitrogen fixation in the phyllosphere in a Costa Rican premontane rain forest. Oecologia 117:9–18 https://www.jstor.org/stable/4222127
Moyes AB, Kueppers LM, Pett-Ridge J, Carper DL, Vandehey N, O'Neil J, Frank AC (2016) Evidence for foliar endophytic nitrogen fixation in a widely distributed subalpine conifer. New Phytol. 210:657–668. https://doi.org/10.1111/nph.13850
Guerrieri R, Vanguelova EI, Michalski G, Heaton THE, Mencuccini M (2015) Isotopic evidence for the occurrence of biological nitrification and nitrogen deposition processing in forest canopies. Glob. Chang. Biol. 21:4613–4626. https://doi.org/10.1111/gcb.13018
Bringel F, Couée I (2015) Pivotal roles of phyllosphere microorganisms at the interface between plant functioning and atmospheric trace gas dynamics. Frontiers Microbiol 6:article 486. https://doi.org/10.3389/fmicb.2015.00486
Wei X, Lyu S, Yu Y, Wang Z, Liu H, Pan D, Chen J (2017) Phylloremediation of air pollutants: exploiting the potential of plant leaves and leaf-associated microbes. Frontiers Plant Sci 8:1318. https://doi.org/10.3389/fpls.2017.01318
Dees MW, Lysøe E, Nordskop B, Brurberg MB (2015) Bacterial communities associated with surfaces of leafy greens: shift in composition and decrease in richness over time. Appl. Environ. Microbiol. 81:1530–1539. https://doi.org/10.1128/AEM.03470-14
Brandl MT, Lindow SE (1998) Contribution of indole-3-acetic acid production to the epiphytic fitness of Erwinia herbicola. Appl. Environ. Microbiol. 64:3256–3263
Gourion B, Rossignol M, Vorholt JA (2006) A proteomic study of Methylobacterium extorquens reveals a response regulator essential for epiphytic growth. PNAS 103:13186–13191. https://doi.org/10.1073/pnas.0603530103
Reed SC, Townsend AR, Cleveland CC, Nemergut DR (2010) Microbial community shifts influence patterns in tropical forest nitrogen fixation. Oecologia 164:521–531. https://doi.org/10.1007/s00442-010-1649-6
Innerebner G, Knief C, Vorholt JA (2011) Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 77:3202–3210. https://doi.org/10.1128/AEM.00133-11
Balint-Kurti P, Simmons SJ, Blum JE, Ballaré CL, Stapleton AE (2010) Maize leaf epiphytic bacteria diversity patterns are genetically correlated with resistance to fungal pathogen infection. Mol. Plant-Microbe Interact. 23:473–484. https://doi.org/10.1094/MPMI-23-4-0473
Knief C, Ramette A, Frances L, Alonso-Blanco C, Vorholt JA (2010) Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J 4:719–728. https://doi.org/10.1038/ismej.2010.9
Delmotte N, Knief C, Chaffron S, Innerebner G, Roschitzki B, Schlapbach R, von Mering C, Vorholt JA (2009) Community proteogenomics reveals insight into the physiology of phyllosphere bacteria. PNAS 106:16428–16433. https://doi.org/10.1073/pnas.0905240106
Knief C, Delmotte N, Chaffron S, Stark M, Innerebner G, Wassmann R, von Mering C, Vorholt JA (2012) Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J 6:1378–1390. https://doi.org/10.1038/ismej.2011.192
Haas JC, Street NR, Sjödin A, Lee NM, Högberg MN, Näsholm T, Hurry V (2018) Microbial community response to growing season and plant nutrient optimisation in a boreal Norway spruce forest. Soil Biol. Biochem. 125:197–209. https://doi.org/10.1016/j.soilbio.2018.07.005
Laforest-Lapointe I, Messier C, Kembel SW (2016a) Tree phyllosphere bacerial communities: exploring the magnitude of intra- and inter-individual variation among host species. Peer J 4:e2367. https://doi.org/10.7717/peerj.2367
Leff JW, Del Tredici P, Friedman WE, Fierer N (2015) Spatial structuring of bacterial communities within individual Ginkgo biloba trees. Environ. Microbiol. 17:2352–2361. https://doi.org/10.1111/1462-2920.12695
Stone BWG, Jackson CR (2019) Canopy position is a stronger determinant of bacterial community composition and diversity than environmental disturbance in the phyllosphere. FEMS Microbiol Ecol 95:fiz032. https://doi.org/10.1093/femsec/fiz032
Müller GK (1995) Die Leipziger Auen - Bestandsaufnahme und Vorschläge für eine Gebietsentwicklung. Sächsisches Staatsministerium für Umwelt- und Landesentwicklung (SMU), Dresden. 102 pp.
Jansen E (1999) Das Naturschutzgebiet Burgaue. Staatliches Umweltfachamt, Leipzig, 43 pp.
Otto S, Floren A (2010) The canopy spiders (Araneae) of the floodplain forest in Leipzig. Arachnologische Mitteilungen 39:25–38. https://doi.org/10.5431/aramit3904
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2012) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nuc Acids Res 41:e1. https://doi.org/10.1093/nar/gks808
Rughöft S, Herrmann M, Lazar CS, Cesarz S, Levick SR, Trumbore SE, Küsel K (2016) Community composition and abundance of bacterial, archaeal and nitrifying populations in savanna soils on contrasting bedrock material in Kruger National Park, South Africa. Frontiers Microbiol 7:1638. https://doi.org/10.3389/fmicb.2016.01638
Chelius MK, Triplett EW (2001) The diversity of archaea and bacteria in association with the roots of Zea mays L. Microbial Ecol 41:252–263. https://doi.org/10.1007/s002480000087
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. https://doi.org/10.1128/AEM.01541-09
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79:5112–5120. https://doi.org/10.1128/AEM.01043-13
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Core Team R (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson GL, Solymos PM, Stevens HH, Wagner H (2008) The vegan package. Community Ecology Package 190
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontologica Electronica 4:1–9 http://palaeo-electronica.org/2001_1/past/issue1_01.htm
Deng Y, Jiang Y-H, Yan Y, He Z, Luo F, Zhou J (2012) Molecular ecological network analyses. BMC Bioinformatics 13:113 http://www.biomedcentral.com/1471-2105/13/113. Accessed Oct 2019
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle C et al (2016) A new view of the tree of life. Nat Microbiol 1:article 16048. https://doi.org/10.1038/nmicrobiol.2016.48
Harrison JG, Forister ML, Parchman TL, Koch GW (2016) Vertical stratification of the folia fungal community in the world's tallest trees. Am. J. Bot. 103:2087–2095. https://doi.org/10.3732/ajb.1600277
Izuno A, Kanzaki M, Artchawakom T, Wachrinrat C, Isagi Y (2016) Vertical structure of phyllosphere fungal communities in a tropical forest in Thailand uncovered by high-throughput sequencing. PLoS One 11:e0166669. https://doi.org/10.1371/journal.pone.0166669
Christian N, Herre EA, Mejia LC, Clay K (2017) Exposure to the leaf litter microbiome of healthy adults protects seedlings from pathogen damage. Proc. R. Soc. B 284:20170641. https://doi.org/10.1098/rspb.2017.0641
Bittar TB, Pound P, Whitetree A, Moore LD, van Stan II JT (2018) Estimation of throughfall and stemflow bacterial flux in a subtropical oak-cedar forest. Geophysical Res Letters 45:1410–1418. https://doi.org/10.1002/2017GL075827
Huber L, McCartney HA, Fitt BDL (1997) Influence of target characteristics on the amount of water splashed by impacting drops. Agric. For. Meteorol. 97:201–211. https://doi.org/10.1016/S0168-1923(97)00016-6
Gourion B, Francez-Charlot A, Vorholt JA (2008) PhyR is involved in the general stress response of Methylobacterium extorquens AM1. J. Bacteriol. 190:1027–1035. https://doi.org/10.1128/JB.01483-07
Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N (2010) The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 12:2885–2893. https://doi.org/10.1111/j.1462-2920.2010.02258.x
Kembel SW, O’Connor TK, Arnold HK, Hubbell SP, Wright SJ, Green JL (2014) Relationship between phyllosphere bacterial communities and plant functional traits in a neotropical forest. PNAS 111:13715–13720. https://doi.org/10.1073/pnas.1216057111
Laforest-Lapointe I, Paquette A, Messier C, Kembel SW (2017) Leaf bacterial diversity mediates plant diversity and ecosystem function relationships. Nature 546:145–147. https://doi.org/10.1038/nature22399
Yadav RKP, Karamanoli K, Vokou D (2005) Bacterial colonization of the phyllosphere of Mediterranean perennial species as influenced by leaf structural and chemical features. Microb. Ecol. 50:185–196. https://doi.org/10.1007/s00248-004-0171-y
Laforest-Lapointe I, Messier C, Kembel SW (2016b) Host species identity, site and time drive temperate tree phyllosphere bacterial community structure. Microbiome 4:27. https://doi.org/10.1186/s40168-016-0174-1
Westoby M, Falster DS, Moles AT, Vesk PA, Wright IJ (2002) Plant ecological strategies: some leading dimensions of variation between species. Annu. Rev. Ecol. Syst. 33:125–159. https://doi.org/10.1146/annurev.ecolsys.33.010802.150452
Kim M, Singh D, Lai-Hoe A, Go R, Abdul Rahim R, Ainuddin AN et al (2012) Distinctive phyllosphere bacterial communities in tropical trees. Microb. Ecol. 63:674–681. https://doi.org/10.1007/s00248-011-9953-1
Imperato V, Kowalkowski L, Portillo-Estrada M, Gawronski SW, Vangronsveld J, Thijs S (2019) Characterization of the Carpinus betulus phyllomicrobiome in urban and forest areas. Frontiers Microbiol 10:article 1110. https://doi.org/10.3389/fmicb.2019.01110
Iwai K, Aisaka K, Suzuki M (2010) Friedmaniella luteola sp. nov., Friedmaniella lucida sp. nov., Friedmaniella okinawensis sp. nov., and Friedmaniella sagamiharensis sp. nov., isolated from spiders. International J Syst Evol Microbiol 60:113–120. https://doi.org/10.1099/ijs.0.007815-0
Yashiro E, Spear RN, McManus PS (2011) Culture-dependent and culture-independent assessment of bacteria in the apple phyllosphere. J. Appl. Microbiol. 110:1284–1296. https://doi.org/10.1111/j.1365-2672.2011.04975.x
Espenshade J, Thijs S, Gawronski S, Bové H, Weyens N, Vangronsveld J (2019) Influence of urbanization on epiphytic bacterial communities of the Platanus x hispanica tree leaves in a biennieal study. Frontiers Microbiol 10:675. https://doi.org/10.3389/fmicb.2019.00675
Tuo L, Pan Z, Li F-N, Lou I, Guo M, Lee SM-Y, Chen L, Hu L, Sun C-H (2016) Friedmaniella endophytica sp. nov., an endophytic actinobacterium isolated from bark of Kandelia candel. Int. J. Syst. Evol. Microbiol. 66:3057–3062. https://doi.org/10.1099/ijsem.0.001146
Pirttilä AM (2018) Endophytic bacteria in tree shoot tissues and their effects on host. In: Pirttilä AM, Frank AC (eds) Endophytes of forest trees. Springer International Publishing AG, Cham, pp 177–190. https://doi.org/10.1007/978-3-319-89833-9_8
Buresova A, Kopecky J, Hrdinkova V, Kamenik Z, Omelka M, Sagova-Mareckova M (2019) Succession of microbial decomposers is determined by litter type, but site conditions drive decomposition rates. Appl. Environ. Microbiol. 85:e01760–e01719. https://doi.org/10.1128/AEM.01760-19
Sy A, Timmers AC, Knief C, Vorholt JA (2005) Methylotrophic metabolism is advantageous for Methylobacterium extorquens during colonization of Medicago trunculata under competitive conditions. Appl. Environ. Microbiol. 71:7245–7252. https://doi.org/10.1128/AEM.71.11.7245-7252.2005
Nadalig T, Haque MFU, Roselli S, Schaller H, Bringel F, Vuilleumier S (2011) Detection and isolation of chloromethane-degrading bacteria from the Arabidopsis thaliana phyllosphere, and characterization of chloromethane utilization genes. FEMS Microbiol. Ecol. 77:438–448. https://doi.org/10.1111/j.1574-6941.2011.01125.x
Iguchi H, Sato I, Sakakibara M, Yurimoto H, Sakai Y (2012) Distribution of methanotrophs in the phyllosphere. Biosci. Biotechnol. Biochem. 76:1580–1583. https://doi.org/10.1271/bbb.120281
Papen H, Gessler A, Zumbusch E, Rennenberg H (2002) Chemolithoautotrophic nitrifiers in the phyllosphere of a spruce ecosystem receiving high atmospheric nitrogen input. Curr. Microbiol. 44:56–60. https://doi.org/10.1007/s00284-001-0074-9
Bai Y, Muller DB, Srinivas G, Garrido-Oter R, Potthoff E, Rott M et al (2015) Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528:364–369. https://doi.org/10.1038/nature16192
Sanchez-Canizares C, Jorrin B, Poole PS, Tkacz A (2017) Understanding the holobiont: the interdependence of plants and their microbiome. Curr. Opin. Microbiol. 38:188–196. https://doi.org/10.1016/j.mib.2017.07.001
Zhen H, Krumins V, Fennell DE, Mainelis G (2018) Analysis of airborne microbial communities using 16S ribosomal RNA: potential bias due to air sampling stress. Sci. Total Environ. 621:939–947. https://doi.org/10.1016/j.scitotenv.2017.10.154
Rastogi G, Sbodio A, Tech JJ, Suslow TV, Coaker GL, Leveau JH (2012) Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition of field-grown lettuce. ISME J 6:1812–1822. https://doi.org/10.1038/ismej.2012.32
Acknowledgements
We thank Rolf Engelmann for technical support during sampling and Christian Wirth for providing access to the Leipzig canopy crane facility. Julia Rosenberger is acknowledged for help with sample processing.
Availability of Data and Material
Sequence data generated in this study have been submitted to the European Nucleotide Archive (ENA) under the study accession number PRJEB36420, sample accession numbers SAMEA6502636–SAMEA6502715. Results of quantitative PCR for each sample are provided as Supplementary Material (Supplementary Table 2).
Code Availability
Not applicable.
Funding
Open Access funding provided by Projekt DEAL. Sequencing was financially supported by the German Center for Integrative Biodiversity Research (iDiv) – Halle, Jena, Leipzig funded by the Deutsche Forschungsgemeinschaft (FZT 118). Additional support was provided by the Collaborative Research Centre AquaDiva (CRC 1076 AquaDiva) of the Friedrich Schiller University Jena, funded by the Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Contributions
This study was designed by MH, PG, and KK. RR supported sampling of leaves using the canopy crane facility and provided information on site characteristics, location of tree individuals, tree age, and sampling height. MH and PG performed most of the laboratory work. MH analyzed the data and wrote the manuscript with contributions from all other authors.
Corresponding author
Ethics declarations
Conflict of Interest/Competing Interests
The authors declare that they have no conflict of interest.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Electronic supplementary material
ESM 1
(PDF 813 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herrmann, M., Geesink, P., Richter, R. et al. Canopy Position Has a Stronger Effect than Tree Species Identity on Phyllosphere Bacterial Diversity in a Floodplain Hardwood Forest. Microb Ecol 81, 157–168 (2021). https://doi.org/10.1007/s00248-020-01565-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-020-01565-y