
Abstract
In this study, the potential of Corynebacterium glutamicum for reductive whole-cell biotransformation is shown. The NADPH-dependent reduction of the prochiral methyl acetoacetate (MAA) to the chiral (R)-methyl 3-hydroxybutyrate (MHB) by an alcohol dehydrogenase from Lactobacillus brevis (Lbadh) was used as model reaction and glucose served as substrate for the regeneration of NADPH. Since NADPH is mainly formed in the oxidative branch of the pentose phosphate pathway (PPP), C. glutamicum was engineered to redirect carbon flux towards the PPP. Mutants lacking the genes for 6-phosphofructokinase (pfkA) or glyceraldehyde 3-phosphate dehydrogenase (gapA) were constructed and analyzed with respect to growth, enzyme activities, and biotransformation performance. Both mutants showed strong growth defects in glucose minimal medium. For biotransformation of MAA to MHB using glucose as reductant, strains were transformed with an Lbadh expression plasmid. The wild type showed a specific MHB production rate of 3.1 mmolMHB h−1 g −1cdw and a yield of 2.7 molMHB mol −1glucose . The ∆pfkA mutant showed a similar MHB production rate, but reached a yield of 4.8 molMHB mol −1glucose , approaching the maximal value of 6 molNADPH mol −1glucose expected for a partially cyclized PPP. The specific biotransformation rate of the ΔgapA mutant was decreased by 62 % compared to the other strains, but the yield was increased to 7.9 molMHB mol −1glucose , which to our knowledge is the highest one reported so far for this mode of NADPH regeneration. As one fourth of the glucose was converted to glycerol, the experimental yield was close to the theoretically maximal yield of 9 molNADPH mol −1glucose .
Similar content being viewed by others

Introduction
Whole-cell biotransformation has become an important method in chemoenzymatic synthesis, e.g., for the production of amino acids and chiral alcohols (Ishige et al. 2005). Corynebacterium glutamicum is a Gram-positive, non-pathogenic soil bacterium which is predominantly used for the large-scale industrial production of the flavor enhancer l-glutamate and the food additive l-lysine (Pfefferle et al. 2003; Kimura 2003; Hermann 2003). Recent metabolic engineering studies have shown that C. glutamicum is also capable of producing a variety of other commercially interesting compounds, e.g., other l-amino acids (Wendisch et al. 2006), d-amino acids (Stäbler et al. 2011), organic acids such as succinate (Okino et al. 2008; Litsanov et al. 2012a, b), diamines such as cadaverine (Mimitsuka et al. 2007) or putrescine (Schneider and Wendisch 2010), biofuels such as ethanol or isobutanol (Inui et al. 2004; Smith et al. 2010; Blombach et al. 2011), or proteins (Meissner et al. 2007). An overview of the product spectrum of C. glutamicum can be found in a recent review (Becker and Wittmann 2011).
C. glutamicum was also shown to be a suitable host for whole-cell biotransformation with resting cells for production of mannitol (Bäumchen and Bringer-Meyer 2007) and cyclohexanone derivatives (Doo et al. 2009; Yun et al. 2012). These reactions are often NAD(P)H dependent and cofactor recycling is crucial for profitable processes. For example, formate dehydrogenase or glucose dehydrogenase are used, but only 1 mol NAD(P)H can be generated from 1 mol formate or 1 mol glucose (Kaup et al. 2004, 2005; Ernst et al. 2005; Eguchi et al. 1992; Tan 2006). Use of metabolically active cells gives the opportunity to regenerate reduced cofactors via sugar metabolism and to gain a higher reduced cofactor to glucose ratio (Chin and Cirino 2011).
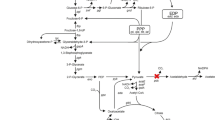
In Escherichia coli, several attempts were made for engineering cellular metabolism towards a higher NADPH per glucose yield (Fasan et al. 2011; Akinterinwa and Cirino 2011). NADPH is mainly generated in the oxidative branch of the pentose phosphate pathway (PPP), where glucose 6-phosphate dehydrogenase catalyzes the oxidation of glucose 6-phosphate to 6-phopshoglucono-δ-lactone and 6-phosphogluconate dehydrogenase, which catalyzes the oxidative decarboxylation of 6-phosphogluconate to ribulose 5-phosphate, yielding 2 mol NADPH (Fig. 1). Therefore, employment of the PPP is an interesting option for NADPH-dependent processes (Chin and Cirino 2011; Chemler et al. 2010). In a recent study with E. coli, we analyzed the NADPH-dependent reduction of the prochiral β-ketoester methyl acetoacetate (MAA) to the chiral hydroxy ester (R)-methyl 3-hydroxybutyrate (MHB) using glucose as substrate for the generation of NADPH (Siedler et al. 2011, 2012). The reduction was catalyzed by an R-specific alcohol dehydrogenase (ADH) from Lactobacillus brevis. MHB serves as a building block of statins (Panke and Wubbolts 2005). Deletion of pfkA and pfkB encoding phosphofructokinase I and II, respectively, resulted in a partial cyclization of the PPP and a yield of 5.4 molMHB mol −1glucose , which was near the theoretically maximal yield of 6 (Kruger and von Schaewen 2003).

Scheme of the upper part of glycolysis and pentose phosphate pathway of C. glutamicum. Gene deletions and NADPH generating reactions are indicated. PTS phosphotransferase system, IolT1/IolT2 alternative glucose import system, Glk ATP-dependent glucokinase, PpgK polyphosphate/ATP-dependent glucokinase, Pgi phosphoglucose isomerase, PfkA phosphofructokinase, GapA glyceraldehyde-3-phosphate dehydrogenase, DHAP dihydroxyacetone phosphate, PEP phosphoenolpyruvate
To determine whether this metabolic engineering strategy can be generalized, is e.g. transferable to C. glutamicum, was one major goal of this study. It has to be kept in mind that differences exist in the repertoires of metabolic enzymes of E. coli and C. glutamicum. Of relevance for the present work is the occurrence of only one gene encoding a 6-phosphofructo1-kinase (pfkA) and the absence of genes encoding transhydrogenases and the key enzymes of the Entner–Doudoroff-pathway in C. glutamicum (Yokota and Lindley 2005). To further improve the NADPH per glucose yield, deletion of the glyceraldehyde 3-phosphate dehydrogenase (gapA) gene would be beneficial, as it should result in a complete cyclization of the PPP. Deletion of gapA theoretically enables a yield of 12 mol NADPH per mole of glucose 6-phosphate by complete recycling of fructose 6-phosphate and triose 3-phosphate through the oxidative PPP (Kruger and von Schaewen 2003). The gapB gene encoding a second glyceraldehyde 3-phosphate dehydrogenase in C. glutamicum should not be relevant in this context, as GapB does not function in the glycolytic direction (Omumasaba et al. 2004).
In this study, we analyzed C. glutamicum mutants lacking either pfkA or gapA for their behavior in reductive whole-cell biotransformation. The results supported the view that the PPP operates in cyclic manner, oxidizing glucose to CO2 with concomitant reduction of NADP+ to NADPH.
Materials and methods
Chemicals and enzymes
Chemicals were obtained from Sigma-Aldrich (Taufkirchen, Germany), Qiagen (Hilden, Germany), Merck (Darmstadt, Germany), and Roche Diagnostics (Mannheim, Germany).
Bacterial strains, plasmids, media, and growth conditions
Strains and plasmids used in this work are listed in Table 1. E. coli strains were transformed by the method described by Hanahan (1983) and cultivated in LB medium (Miller 1972). E. coli DH5α was used for cloning purposes and C. glutamicum ATCC 13032 and derivatives for gene expression and whole-cell biotransformation. When required, antibiotics were added to the medium at a final concentration of 50 μg kanamycin ml−1 (pEKEx2-LbADH) or 100 μg spectinomycin ml−1 (pEKEx3 derivatives).
For growth experiments with C. glutamicum, 50-ml LB overnight cultures were inoculated from LB plates, harvested by centrifugation (10 min, 3,220 × g), washed in CgXII medium (Eggeling and Bott 2005), and inoculated in CgXII medium containing 100 mM glucose to a final optical density at 600 nm (OD600) of 1. When appropriate, 1 mM isopropyl-β-d-thiogalactopyranosid (IPTG), 25 μg ml−1 kanamycin, and 100 μg ml−1 spectinomycin was added. For all growth experiments, 500 ml baffled shake flasks with 50 ml CgXII medium were used and incubated at 30 °C and 120 rpm. Growth was followed by OD600 determination using a UV-1650 PC photometer (Shimadzu, Duisburg, Germany). The biomass concentration was calculated from OD600 values using an experimentally determined correlation factor of 0.25 g (dry weight) of cells (cdw) per liter for an OD600 of 1 (Kabus et al. 2007). For the determination of enzyme activity in cell-free extracts, 50 ml LB medium containing 1 mM IPTG and 100 μg ml−1 spectinomycin was inoculated from LB overnight cultures to an OD600 of 0.5. At an OD600 of 4, cells were harvested by centrifugation (10 min, 3,220 × g, 4 °C) and stored at −20 °C until use.
Recombinant DNA work
Standard methods like polymerase chain reaction (PCR), restriction, or ligation were carried out according to established protocols (Sambrook and Russell 2001). E. coli cells were transformed by the CaCl2 method (Hanahan et al. 1991). DNA sequencing was performed by Eurofins MWG Operon (Germany). Oligonucleotides (listed in Table 2) were synthesized by Biolegio bv (Nijmegen, The Netherlands) and Eurofins MWG Operon (Germany).
Construction of deletion mutants and plasmids
C. glutamicum deletion mutants were constructed using pK19mobsacB (Schäfer et al. 1994) using the procedure described by Niebisch and Bott (2001). Upstream and downstream flanking regions of pfkA (cg1409), and gapA (cg1791) were amplified by PCR using the oligonucleotide pairs pfkA-Del-A/pfkA-Del-B and pfkA-Del-C/pfkA-Del-D for deletion of pfkA, and gapA-Del-A/gapA-Del-B and gapA-Del-C/gapA-Del-D for deletion of gapA (see Table 2 for primer sequences). The upstream and downstream flanking regions of each gene were fused by overlap extension PCR, resulting in a DNA fragment of about 1 kb. The resulting PCR products were cloned into SmaI-restricted vector pK19mobsacB resulting in pK19mobsacB∆pfkA, and pK19mobsacB∆gapA. The correctness of the cloned PCR fragments was confirmed by DNA sequencing. Transformation of C. glutamicum wild type with these plasmids and selection for the first and second homologous recombination was performed as described (Niebisch and Bott 2001; Rittmann et al. 2003). Kanamycin-sensitive and sucrose-resistant clones were analyzed by PCR using oligonucleotide pairs pfkA-Del-Ver-fw/pfkA-Del-Ver-rv or gapA-Del-Ver-fw/gapA-Del-Ver-rv.
For the complementation of deletion mutants, the genes pfkA (cg1409), and gapA (cg1791) from C. glutamicum and the genes pfkA (b3916) and pfkB (b3916) from E. coli were amplified via PCR from genomic DNA of C. glutamicum WT, which was prepared as described previously (Eikmanns et al. 1995), and E. coli MG1655 genomic DNA, which was prepared by using the DNA isolation kit (Roche, Mannheim, Germany). PCR was performed using the following oligonucleotide pairs: pfkA-cgl-fw/pfkA-cgl-rv, gapA-cgl-fw/gapA-cgl-rv, pfkA-eco-fw/pfkA-eco-rv, and pfkB-eco-fw/pfkB-eco-rv (see Table 2). To allow IPTG-inducible expression of pfkA, and gapA from C. glutamicum and pfkA, and pfkB from E. coli the corresponding PCR products were ligated into the SmaI-restricted vector pEKEx3 resulting in pEKEx3-pfkA Cgl, pEKEx3-gapA Cgl, pEKEx3-pfkA Eco, and pEKEx3-pfkB Eco.
For the construction of the expression plasmid pEKEx2-Lbadh, the adh gene of L. brevis was amplified together with a 9-bp linker and an artificial ribosome binding site (AAGGAG) using the oligonucleotides Lbadh_for and Lbadh_rev and the plasmid pBtacLbadh as template (Ernst et al. 2005). The PCR product was digested with BamHI and EcoRI and cloned into the vector pEKEx2. The correctness of the cloned PCR fragments in the plasmids was confirmed by DNA sequencing.
Enzyme activity assays
For the determination of alcohol dehydrogenase activity, cells were harvested by centrifugation (10,000 × g, 4 °C, 5 min) 30 min after start of biotransformation and stored at −20 °C until use. The cells were resuspended in 100 mM potassium phosphate buffer, pH 6.5, with 1 mM dithiothreitol and 1 mM MgCl2. Cells were disrupted at 4 °C by 3 × 15 s bead-beating with 0.1-mm-diameter glass beads using a Silamat S5 (Ivoclar Vivadent GmbH, Germany) and crude extracts were centrifuged at 16,000 × g (4 °C, 20 min) to remove intact cells and cell debris. The supernatants were used as cell-free extracts. Alcohol dehydrogenase activity was determined photometrically at 340 nm using a mixture of 10 mM methyl acetoacetate, 250 μM NADPH, and 1 mM MgCl2 in 100 mM potassium phosphate buffer, pH 6.5. The reactions were started by adding different dilutions of the cell-free extract. For rate calculation, an extinction coefficient for NADPH at 340 nm of 6.22 mM−1 cm−1 was used. One unit of enzyme activity corresponds to 1 μmol NADPH consumed per minute.
For the determination of the specific activity of phosphofructokinase and glyceraldehyde 3-phosphate dehydrogenase, cells were harvested by centrifugation (3,220 × g, 4 °C, 10 min) and washed in the appropriate buffer (see below) and stored at −20 °C until use. Cells were resuspended in 1 ml of the buffer and cell-free extracts were prepared by sonification as described previously (Stansen et al. 2005). All enzyme activity measurements were carried out at 30 °C. Protein concentrations were determined with bovine serum albumin as standard using Bradford reagents (Sigma, Taufkirchen, Germany).
6-Phosphofructokinase activity was measured spectrophotometrically at 340 nm according to Babul (1978) by a coupled enzymatic assay with pyruvate kinase and lactate dehydrogenase. ADP formed in kinase reaction was used to convert phosphoenolpyruvate to pyruvate, which was subsequently reduced to lactate with concomitant oxidation of NADH to NAD+. The assay solution contained 100 mM Tris–HCl pH 7.5, 0.2 mM NADH, 1 mM ATP, 10 mM MgCl2, and 0.2 mM phosphoenolpyruvate. One unit of enzyme activity corresponds to 1 μmol NADH oxidized per minute.
Glyceraldehyde 3-phosphate dehydrogenase activity was measured according to Omumasaba et al. (2004). The assay contained 1 mM NAD+, 50 mM Na2HPO4, 0.2 mM EDTA, and 0.5 mM glyceraldehyde 3-phosphate in 50 mM triethanolamine hydrochloride (TEA) buffer pH 8.5. One unit of enzyme activity corresponds to 1 μmol NADH formed per minute.
Whole-cell biotransformation
For cultivation of the different recombinant C. glutamicum strains carrying the pEKEx2-Lbadh plasmid, a single colony of each strain was inoculated into 10 ml BHIS medium (37 g l−1 brain heart infusion, 91 g l−1 sorbitol) containing the appropriate selection marker as described above and grown overnight at 30 °C and 120 rpm. These pre-cultures were used for inoculation of the main cultures to an optical density at 600 nm (OD600) of 0.4. Main cultures were grown in 100 ml BHIS medium in shake flasks in the presence of the appropriate selection marker and 0.5 mM IPTG at 30 °C and 120 rpm. The cells were harvested at an OD600 between 2.5 and 5 by centrifugation (4,000 × g, 4 °C, 7 min) and resuspended in a solution containing 111 mM glucose, 2 mM MgSO4, and 250 mM potassium phosphate buffer, pH 6.5, to a cell density of 3 gcdw l−1. The biotransformation was started by adding 50 mM MAA and conducted in shake flasks at 30 °C and 120 rpm to prevent cell sedimentation. Specific productivities (mmolMHB h−1 g −1cdw ) were determined by taking samples at 30–60-min time intervals over a period of 3 h. MHB and glucose concentrations of the samples were determined (see below). Specific productivities were calculated by dividing the slope of graphs showing MHB concentration vs. time by the cell dry weight, which remained constant.
Analysis of substrates and products
Methyl acetoacetate (MAA), (R)-methyl 3-hydroxybutyrate (MHB), glucose, and extracellular metabolites were analyzed by HPLC as described previously (Siedler et al. 2011).
Results
Growth behavior and in vitro enzyme activities of C. glutamicum wild-type and mutant strains
In a C. glutamicum mutant lacking 6-phosphofructokinase, glucose catabolism is forced to proceed via the pentose phosphate pathway. Fructose 6-phosphate formed in the PPP by transaldolase or transketolase has to re-enter the oxidative part of the PPP again and only glyceraldehyde 3-phosphate can be catabolized further via the lower part of the glycolytic pathway. Thus, the initial part of glucose catabolism in a ∆pfkA mutant can be described by the following equation: Glucose 6-phosphate + 6 NADP+ ➔ Glyceraldehyde 3-phosphate + 3 CO2 + 6 NADPH + 6 H+. Thus, 6 mol NADPH are formed per mole of glucose.
The deletion of the pfkA gene prevented growth in CgXII medium with 100 mM glucose (Table 3). The growth defect of the ∆pfkA mutant was complemented to levels of the WT control (0.32 h−1) by plasmid-based overexpression of either the homologous pfkA gene from C. glutamicum (0.32 h−1) or of the heterologous pfkA gene from E. coli (0.33 h−1) and increased to 0.16 h−1 by heterologous expression of pfkB from E. coli. The slow growth of ∆pfkA/pEKEx3-pfkB Eco was accompanied by a significantly higher biomass yield of 10.8 g l−1 compared to 8.4 g l−1 of WT/pEKEx3 or 8.6 g l−1 of strain ΔpfkA/pEKEx3-pfkA Cgl.
6-Phosphofructokinase activity was absent in the pfkA deletion strain (Table 3). Plasmid-borne expression of C. glutamicum pfkA or of E. coli pfkA or pfkB increased phosphofructokinase activity in the WT background from 0.04 U mg−1 to 0.12, 0.11, and 0.19 U mg−1, respectively. In the ∆pfkA background, phosphofructokinase activities of 0.10 to 0.13 U mg−1 were determined when either C. glutamicum pfkA or E. coli pfkA or pfkB was overexpressed (Table 3).
C. glutamicum possesses two glyceraldehyde 3-phosphate dehydrogenases, GapA and GapB, but only GapA functions in the glycolytic direction as a ΔgapA deletion mutant was unable to grow in glucose minimal medium whereas a ΔgapB mutant showed no growth defect under these conditions (Omumasaba et al. 2004). A complete block of glyceraldehyde 3-phosphate conversion to 1,3-bisphosphoglycerate should lead to a complete oxidation of glucose in the PPP according to the equation: Glucose + 6 H2O + 12 NADP+ ➔ 6 CO2 + 12 NADPH + 12 H+.
In agreement with previous results (Omumasaba et al. 2004), a deletion of the gapA gene in strain ATCC 13032 resulted in an inability to grow in glucose minimal medium. This defect was complemented by plasmid-based overexpression of the gapA gene. NAD+-dependent glyceraldehyde-3-phosphate dehydrogenase activity of cell-free extracts was 0.15 U mg−1 in WT/pEKEx3 and absent in strain ∆gapA/pEKEx3. In strains WT/pEKEx3-gapA and ∆gapA/pEKEx3-gapA, the glyceraldehyde 3-phosphate dehydrogenase activity with NAD+ was found to be 0.26 and 0.13 U mg−1, respectively (Table 4).
Biotransformation of MAA to MHB with the reference and the mutant strains
For biotransformation of MAA to MHB, the gene encoding the (R)-specific alcohol dehydrogenase of L. brevis (Lbadh) was overexpressed in C. glutamicum WT and in the deletion strains ∆pfkA and ∆gapA using plasmid pEKEx2-Lbadh. The specific NADPH-dependent MAA dehydrogenase activity in cell-free extracts of these strains was similar, ranging from 0.51 to 0.76 U mg−1 in independent experiments. Assuming that the in vivo activities are comparable, they are not limiting the biotransformation rate. The C. glutamicum wild type showed a MAA dehydrogenase activity below 0.01 U mg−1 with either NADPH or NADH as cofactor indicating that the biotransformation occurred only in the presence of the recombinant ADH from L. brevis. For the biotransformation, the strains were cultivated in BHIS medium to the exponential growth phase and then harvested and resuspended in 250 mM potassium phosphate buffer pH 6.5 containing 111 mM glucose and 2 mM MgSO4 to a cell density of 3 gcdw l−1. The resulting cell suspensions were incubated at 30 °C and 120 rpm and the biotransformation was started by adding 50 mM MAA.
The kinetics of MHB production and of glucose consumption of the wild-type and the two mutant strains carrying pEKEx2-Lbadh over a period of 180 min are shown in Fig. 2, and the rates and yields are listed in Table 5. It is evident from Fig. 2 that the rates of MHB production and glucose consumption were almost constant within the time period investigated and proportional to each other. The strain WT/pEKEx2-Lbadh showed an MHB production rate of 3.14 mmol h−1 g −1cdw and a glucose consumption rate of 1.17 mmol h−1 g −1cdw . This resulted in a MHB yield of 2.7 mol per mole of glucose, corresponding to an NADPH yield of 2.7 mol per mole of glucose. The strain ΔpfkA/pEKEx2-Lbadh had an 8 % reduced MHB production rate and a 49 % reduced glucose consumption rate, resulting in a 78 % increased MHB yield of 4.8 mol per mole of glucose. The strain ΔgapA/pEKEx2-Lbadh showed a 62 % decreased MHB production rate and an 87 % reduced glucose consumption rate, corresponding to a 193 % increase of the MHB yield of 7.9 mol per mole of glucose. As discussed below, the strongly reduced glucose uptake rate of the strain ΔgapA/pEKEx2-Lbadh is most likely a consequence of the fact that the strain does not form PEP.

Kinetics of MHB production (open squares) and glucose consumption (filled squares) during biotransformation of MAA to MHB using resting cells (3 gcdw l−1) of the indicated C. glutamicum strains carrying the plasmid pEKEx2-Lbadh. The cell suspensions were incubated at 30 °C and 120 rpm. Mean values and standard deviations from three independent experiments are shown
By-product formation of wild-type and mutant strains
During biotransformation, by-product formation was nearly constant and specific rates were calculated (Table 5). The strain WT/pEKEx2-Lbadh showed an acetate formation rate (1.19 mmol h−1 g −1cdw ) comparable to the glucose consumption rate (1.17 mmol h−1 g −1cdw ). In addition, WT/pEKEx2-Lbadh formed succinate as by-product with a rate of 0.19 mmol h−1 g −1cdw . A low acetate production rate of 0.05 mmol h−1 g −1cdw was shown by the strain ΔpfkA/pEKEx2-Lbadh, which corresponds to only 8 % of the glucose uptake rate. Succinate was not formed by ΔpfkA/pEKEx2-Lbadh. The strain ΔgapA/pEKEx2-Lbadh formed neither acetate nor succinate, but glycerol with a rate of 0.08 mmol h−1 g −1cdw , which corresponds to 53 % of the glucose consumption rate. As glyceraldehyde 3-phosphate cannot be catabolized to pyruvate in the ΔgapA mutant, reduction to glycerol presents an alternative pathway to oxidation in the cyclic PPP.
Discussion
For reductive whole-cell biotransformations requiring NADPH, attempts were made in this work to increase the NADPH yield per mole of glucose using C. glutamicum as host strain and the reduction of MAA to MHB as NADPH-requiring model reaction. Rerouting of glucose catabolism from glycolysis to the oxidative PPP was achieved by deletion of either the pfkA gene or the gapA gene.
C. glutamicum wild type carrying pEKEx2-Lbadh showed a 31 % lower specific MHB production rate compared to E. coli carrying pBtac-Lbadh, even when compared to an E. coli biotransformation conducted at 30 °C (unpublished data). This difference might be due to a lower glucose uptake capacity or to a generally lower metabolic flux capacity of C. glutamicum. Overexpression of the genes involved in glucose uptake and catabolism via glycolysis or PPP could improve the rate of glucose catabolism, as shown recently for oxygen-deprived conditions (Yamamoto et al. 2012; Jojima et al. 2010). The MHB per glucose yield found for C. glutamicum WT/pEKEx2-Lbadh (2.7 mol/mol) was 10 % higher than the corresponding value determined for E. coli BL21(DE3)/pBtac-Lbadh (2.44 mol/mol) (Siedler et al. 2011), which might be due to slight differences in the partition of glucose 6-phosphate between glycolysis and the PPP.
Biotransformation studies with E. coli ∆pfkA and ∆pfkA∆pfkB mutants expressing Lbadh showed yields of 4.8 and 5.4 molMHB mol −1glucose , respectively (Siedler et al. 2011). 13C metabolic flux analysis demonstrated a negative net flux through phosphoglucose isomerase in the ∆pfkA mutant, in compliance with the proposed partial cyclization of the PPP (Siedler et al. 2012). The MHB yield per glucose of the E. coli strain ∆pfkA/pBtac-Lbadh was comparable to that of the C. glutamicum strain ∆pfkA/pEKEx2-Lbadh (4.8 molMHB mol −1glucose ), indicating that a partial cyclization of the PPP occurred in the latter species, too. Furthermore, similarities were found when comparing by-product formation in E. coli and C. glutamicum. Less acetate and no succinate was produced in both ∆pfkA mutant strains compared to the reference strains within the experimental period, presumably as a consequence of a decreased carbon flux through the lower part of glycolysis and the TCA cycle in these mutants (Siedler et al. 2012).
C. glutamicum possesses two glyceraldehyde 3-phosphate dehydrogenases (GAPDH), but only GapA functions in the glycolytic direction (Omumasaba et al. 2004). Thus, a deletion of the corresponding gene theoretically should result in a cyclization of the PPP. The fact that the MHB per glucose yield of the strain ∆gapA/pEKEx2-Lbadh (7.9 mol/mol) was higher compared to the strain ∆pfkA/pEKEx2-Lbadh and corresponded to 66 % of the maximal value of 12 mol NADPH per mole of glucose indicated a more extended cyclic operation of the PPP in the ∆gapA mutant compared to the ∆pfkA mutant. The maximal value for a complete oxidation of glucose in the PPP was not reached because 25 % of the glucose carbon was lost by reduction of glyceraldehyde 3-phosphate to glycerol. Taking this loss into account, only 9 molMHB mol −1glucose could be achieved maximally. The experimental yield of 7.9 molMHB mol −1glucose corresponds to 88 % of this value and is 46 % above the best yields reported so far (Chin and Cirino 2011; Siedler et al. 2011, 2012). Future yield optimization could be achieved by deletion of the gene encoding glycerol 3-phosphatase. Such a deletion was recently shown to prevent glycerol formation, which predominantly occurs in fructose-utilizing C. glutamicum strains (Lindner et al. 2012).
The strongly reduced biotransformation rate of the strain ΔgapA/pEKEx2-Lbadh was probably a consequence of the diminished capability for glucose uptake. In a ΔgapA mutant, no PEP should be formed during glucose catabolism and consequently, glucose uptake via the PTS should be impossible. PTS-independent glucose uptake has recently been described for C. glutamicum. It involves the inositol transporters IolT1 and IolT2 which also function as low-affinity glucose permeases (Lindner et al. 2011). Subsequent phosphorylation of glucose to glucose 6-phosphate is catalyzed either by an ATP-dependent glucokinase encoded by glk (Park et al. 2000) or by the polyphosphate- or ATP-dependent glucose kinase PpgK (Lindner et al. 2010). It can be assumed that glucose uptake during biotransformation with the ΔgapA mutant occurs via this alternative pathway, as the observed glucose consumption rate of 2.5 nmol min−1 mg −1cdw (Table 5) at glucose concentrations >10-fold above the apparent K s values of IolT1 and IolT2 (2.8 and 1.9 mM, respectively) is in the range determined for PTS-independent glucose uptake at 1 mM glucose (0.7 nmol min−1 mg −1cdw ) (Lindner et al. 2011). Overexpression of either iolT1 or iolT2 together with ppgK was shown to allow almost wild-type growth rates in a PTS-negative mutant (Lindner et al. 2011) and thus would probably also allow higher biotransformation rates of a ΔgapA mutant. Alternatively, expression of the glucose facilitator gene glf from Zymomonas mobilis could help to increase glucose uptake (Weisser et al. 1995; Parker et al. 1995).
Overall, we could demonstrate the potential of C. glutamicum for NADPH-dependent reductive whole-cell biotransformation and show that deletion of either pfkA or gapA is beneficial to improve the NADPH per glucose yield, presumably by cyclization of the PPP.
References
Abe S, Takayarna K, Kinoshita S (1967) Taxonomical studies on glutamic acid producing bacteria. J Gen Appl Microbiol 13(3):279–301
Akinterinwa O, Cirino PC (2011) Anaerobic obligatory xylitol production in Escherichia coli strains devoid of native fermentation pathways. Appl Environ Microbiol 77(2):706–709
Babul J (1978) Phosphofructokinases from Escherichia coli. Purification and characterization of the nonallosteric isozyme. J Biol Chem 253(12):4350–4355
Bäumchen C, Bringer-Meyer S (2007) Expression of glf Z.m. increases D-mannitol formation in whole cell biotransformation with resting cells of Corynebacterium glutamicum. Appl Microbiol Biotechnol 76(3):545–552
Becker J, Wittmann C (2011) Bio-based production of chemicals, materials and fuels—Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol. doi:10.1016/j.copbio.2011.11.012
Blombach B, Riester T, Wieschalka S, Ziert C, Youn JW, Wendisch VF, Eikmanns BJ (2011) Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol 77(10):3300–3310
Chemler JA, Fowler ZL, McHugh KP, Koffas MA (2010) Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab Eng 12(2):96–104
Chin JW, Cirino PC (2011) Improved NADPH supply for xylitol production by engineered Escherichia coli with glycolytic mutations. Biotechnol Prog 27(2):333–341
Doo EH, Lee WH, Seo HS, Seo JH, Park JB (2009) Productivity of cyclohexanone oxidation of the recombinant Corynebacterium glutamicum expressing chnB of Acinetobacter calcoaceticus. J Biotechnol 142(2):164–169
Eggeling L, Bott M (eds) (2005) Handbook of Corynebacterium glutamicum. CRC, Boca Raton, pp 535–568
Eguchi T, Kuge Y, Inoue K, Yoshikawa N, Mochida K, Uwajima T (1992) NADPH regeneration by glucose dehydrogenase from Gluconobacter scleroides for l-leucovorin synthesis. Biosci Biotechnol Biochem 56(5):701–703
Eikmanns BJ, Kleinertz E, Liebl W, Sahm H (1991) A family of Corynebacterium glutamicum/Escherichia coli shuttle vectors for cloning, controlled gene expression, and promoter probing. Gene 102(1):93–98
Eikmanns BJ, Rittmann D, Sahm H (1995) Cloning, sequence analysis, expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J Bacteriol 177(3):774–782
Ernst M, Kaup B, Müller M, Bringer-Meyer S, Sahm H (2005) Enantioselective reduction of carbonyl compounds by whole-cell biotransformation, combining a formate dehydrogenase and a (R)-specific alcohol dehydrogenase. Appl Microbiol Biotechnol 66(6):629–634
Fasan R, Crook NC, Peters MW, Meinhold P, Buelter T, Landwehr M, Cirino PC, Arnold FH (2011) Improved product-per-glucose yields in P450-dependent propane biotransformations using engineered Escherichia coli. Biotechnol Bioeng 108(3):500–510
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166(4):557–580
Hanahan D, Jessee J, Bloom FR (1991) Plasmid transformation of Escherichia coli and other bacteria. Methods Enzymol 204:63–113
Hermann T (2003) Industrial production of amino acids by coryneform bacteria. J Biotechnol 104(1–3):155–172
Inui M, Kawaguchi H, Murakami S, Vertes AA, Yukawa H (2004) Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J Mol Microbiol Biotechnol 8(4):243–254
Ishige T, Honda K, Shimizu S (2005) Whole organism biocatalysis. Curr Opin Chem Biol 9(2):174–180
Jojima T, Fujii M, Mori E, Inui M, Yukawa H (2010) Engineering of sugar metabolism of Corynebacterium glutamicum for production of amino acid L-alanine under oxygen deprivation. Appl Microbiol Biotechnol 87(1):159–165
Kabus A, Niebisch A, Bott M (2007) Role of cytochrome bd oxidase from Corynebacterium glutamicum in growth and lysine production. Appl Environ Microbiol 73(3):861–868
Kaup B, Bringer-Meyer S, Sahm H (2004) Metabolic engineering of Escherichia coli: construction of an efficient biocatalyst for D-mannitol formation in a whole-cell biotransformation. Appl Microbiol Biotechnol 64(3):333–339
Kaup B, Bringer-Meyer S, Sahm H (2005) D-Mannitol formation from D-glucose in a whole-cell biotransformation with recombinant Escherichia coli. Appl Microbiol Biotechnol 69(4):397–403
Kimura E (2003) Metabolic engineering of glutamate production. Adv Biochem Eng Biotechnol 79:37–57
Kruger NJ, von Schaewen A (2003) The oxidative pentose phosphate pathway: structure and organisation. Curr Opin Plant Biol 6(3):236–246
Lindner SN, Knebel S, Pallerla SR, Schoberth SM, Wendisch VF (2010) Cg2091 encodes a polyphosphate/ATP-dependent glucokinase of Corynebacterium glutamicum. Appl Microbiol Biotechnol 87(2):703–713
Lindner SN, Seibold GM, Henrich A, Krämer R, Wendisch VF (2011) Phosphotransferase system-independent glucose utilization in Corynebacterium glutamicum by inositol permeases and glucokinases. Appl Environ Microbiol 77(11):3571–3581
Lindner SN, Meiswinkel TM, Panhorst M, Youn JW, Wiefel L, Wendisch VF (2012) Glycerol-3-phosphatase of Corynebacterium glutamicum. J Biotechnol. doi:10.1016/j.jbiotec.2012.02.003
Litsanov B, Brocker M, Bott M (2012a) Towards homosuccinate fermentation: metabolic engineering of Corynebacterium glutamicum for anaerobic succinate production from glucose and formate. Appl Environ Microbiol. doi:10.1128/AEM.07790-11
Litsanov B, Kabus A, Brocker M, Bott M (2012b) Efficient aerobic succinate production from glucose in minimal medium with Corynebacterium glutamicum. Microb Biotechnol 5(1):116–128
Meissner D, Vollstedt A, van Dijl JM, Freudl R (2007) Comparative analysis of twin-arginine (Tat)-dependent protein secretion of a heterologous model protein (GFP) in three different Gram-positive bacteria. Appl Microbiol Biotechnol 76(3):633–642
Miller JH (ed) (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory, New York
Mimitsuka T, Sawai H, Hatsu M, Yamada K (2007) Metabolic engineering of Corynebacterium glutamicum for cadaverine fermentation. Biosci Biotechnol Biochem 71(9):2130–2135
Niebisch A, Bott M (2001) Molecular analysis of the cytochrome bc 1-aa 3 branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c 1. Arch Microbiol 175(4):282–294
Okino S, Noburyu R, Suda M, Jojima T, Inui M, Yukawa H (2008) An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl Microbiol Biotechnol 81(3):459–464
Omumasaba CA, Okai N, Inui M, Yukawa H (2004) Corynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation. J Mol Microbiol Biotechnol 8(2):91–103
Panke S, Wubbolts M (2005) Advances in biocatalytic synthesis of pharmaceutical intermediates. Curr Opin Chem Biol 9(2):188–194
Park SY, Kim HK, Yoo SK, Oh TK, Lee JK (2000) Characterization of glk, a gene coding for glucose kinase of Corynebacterium glutamicum. FEMS Microbiol Lett 188(2):209–215
Parker C, Barnell WO, Snoep JL, Ingram LO, Conway T (1995) Characterization of the Zymomonas mobilis glucose facilitator gene product (glf) in recombinant Escherichia coli: examination of transport mechanism, kinetics and the role of glucokinase in glucose transport. Mol Microbiol 15(5):795–802
Pfefferle W, Möckel B, Bathe B, Marx A (2003) Biotechnological manufacture of lysine. Adv Biochem Eng Biotechnol 79:59–112
Rittmann D, Schaffer S, Wendisch VF, Sahm H (2003) Fructose-1,6-bisphosphatase from Corynebacterium glutamicum: expression and deletion of the fbp gene and biochemical characterization of the enzyme. Arch Microbiol 180(4):285–292
Sambrook J, Russell DW (eds) (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145(1):69–73
Schneider J, Wendisch VF (2010) Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol 88(4):859–868
Siedler S, Bringer S, Bott M (2011) Increased NADPH availability in Escherichia coli: improvement of the product per glucose ratio in reductive whole-cell biotransformation. Appl Microbiol Biotechnol 92(5):929–937
Siedler S, Bringer S, Blank LM, Bott M (2012) Engineering yield and rate of reductive biotransformation in Escherichia coli by partial cyclization of the pentose phosphate pathway and PTS-independent glucose transport. Appl Microbiol Biotechnol 93(4):1459–1467
Smith KM, Cho KM, Liao JC (2010) Engineering Corynebacterium glutamicum for isobutanol production. Appl Microbiol Biotechnol 87(3):1045–1055
Stäbler N, Oikawa T, Bott M, Eggeling L (2011) Corynebacterium glutamicum as a host for synthesis and export of D-amino acids. J Bacteriol 193(7):1702–1709
Stansen C, Uy D, Delaunay S, Eggeling L, Goergen JL, Wendisch VF (2005) Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Environ Microbiol 71(10):5920–5928
Tan I (2006) Applications of whole cell biotransformations for the production of chiral alcohols. Rheinische Friedrich-Wilhelms University of Bonn, Bonn, Dissertation
Weisser P, Krämer R, Sahm H, Sprenger GA (1995) Functional expression of the glucose transporter of Zymomonas mobilis leads to restoration of glucose and fructose uptake in Escherichia coli mutants and provides evidence for its facilitator action. J Bacteriol 177(11):3351–3354
Wendisch VF, Bott M, Eikmanns BJ (2006) Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr Opin Microbiol 9(3):268–274
Yamamoto S, Gunji W, Suzuki H, Toda H, Suda M, Jojima T, Inui M, Yukawa H (2012) Overexpression of glycolytic genes enhances Corynebacterium glutamicum glucose metabolism and alanine production under oxygen-deprived conditions. Appl Environ Microbiol. doi:10.1128/AEM.07998-11
Yokota A, Lindley ND (2005) Central metabolism: sugar uptake and conversion. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC, Boca Raton, pp 215–240
Yun JY, Lee JE, Yang KM, Cho S, Kim A, Kwon YE, Park JB (2012) Ethambutol-mediated cell wall modification in recombinant Corynebacterium glutamicum increases the biotransformation rates of cyclohexanone derivatives. Bioprocess Biosyst Eng 35(1–2):211–216
Acknowledgments
This work was supported by the Ministry of Innovation, Science, Research and Technology of North Rhine-Westphalia (BioNRW, Technology Platform Biocatalysis, RedoxCell support code—W0805wb001b).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Solvej Siedler and Steffen N. Lindner contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Siedler, S., Lindner, S.N., Bringer, S. et al. Reductive whole-cell biotransformation with Corynebacterium glutamicum: improvement of NADPH generation from glucose by a cyclized pentose phosphate pathway using pfkA and gapA deletion mutants. Appl Microbiol Biotechnol 97, 143–152 (2013). https://doi.org/10.1007/s00253-012-4314-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4314-7