
Abstract
Background
Budigalimab is a humanized, recombinant immunoglobulin G1 monoclonal antibody targeting programmed cell death protein 1 (PD-1). We present the safety, efficacy, pharmacokinetic (PK), and pharmacodynamic data from patients enrolled in the head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancer (NSCLC) expansion cohorts of the phase 1 first-in-human study of budigalimab monotherapy (NCT03000257; registered 15 December 2016).
Patients and methods
Patients with recurrent/metastatic HNSCC or locally advanced/metastatic NSCLC naive to PD-1/PD-1-ligand inhibitors were enrolled; patients were not selected on the basis of oncogene driver mutations or PD-L1 status. Budigalimab was administered at 250 mg intravenously Q2W or 500 mg intravenously Q4W until disease progression/unacceptable toxicity. The primary endpoints were safety and PK; the secondary endpoint was efficacy. Exploratory endpoints included biomarker assessments.
Results
In total, 81 patients were enrolled (HNSCC: N = 41 [PD-L1 positive: n = 19]; NSCLC: N = 40 [PD-L1 positive: n = 16]); median treatment duration was 72 days (range, 1–617) and 71 days (range, 1–490) for the HNSCC and NSCLC cohorts, respectively. The most frequent grade ≥ 3 treatment-emergent adverse event was anemia (HNSCC: n = 9, 22%; NSCLC: n = 5, 13%). Both dosing regimens had comparable drug exposure and increased interferon gamma-induced chemokines, monokine induced by gamma interferon, and interferon-gamma-inducible protein 10. Objective response rates were 13% (90% CI, 5.1–24.5) in the HNSCC cohort and 19% (90% CI, 9.2–32.6) in the NSCLC cohort. Median progression-free survival was 3.6 months (95% CI, 1.7–4.7) and 1.9 months (95% CI, 1.7–3.7) in the HNSCC and NSCLC cohorts.
Conclusions
The safety, efficacy and biomarker profiles of budigalimab are similar to other PD-1 inhibitors. Development of budigalimab in combination with novel anticancer agents is ongoing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Programmed cell death protein 1 (PD-1), a cell surface protein predominantly expressed on activated T cells, is an inhibitory immune checkpoint receptor and important target for cancer therapy [1, 2]. Its ligands, PD-L1 and PD-L2, are expressed on antigen-presenting cells of the immune system and upregulated in various cancers [3, 4]. Dysregulation of the PD-L1/PD-1 pathway is a mechanism by which malignant cells within the tumor microenvironment subvert protective antitumor immune responses by the host [5, 6], and PD-1/PD-L1 blockade is a promising anticancer strategy. PD-1 inhibitors, such as nivolumab and pembrolizumab, have been evaluated in a number of cancer types, and several PD-1 inhibitors are now approved as monotherapy and in combination with other anticancer agents in multiple cancers, including head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancer (NSCLC) [7,8,9].
Budigalimab, formerly called ABBV-181, is a PD-1 inhibitor currently under development. Unlike nivolumab and pembrolizumab, which are both of the immunoglobulin (Ig)G4 subclass, budigalimab is a humanized, recombinant IgG1 anti-PD-1 monoclonal antibody. It has been modified by point mutations (L234A, L235A) to reduce Fc receptor interactions and limit effector function. Preclinical experiments have demonstrated that budigalimab exhibits potent PD-1–blocking activity with high specificity [10] and has an affinity similar to that of nivolumab [11] and pembrolizumab [12]. Dose-finding and preliminary safety data from this first-in-human phase 1 study of budigalimab in patients with solid tumors (NCT03000257) have been previously presented [13]. The recommended phase 2 dose was determined to be 250 mg every 2 weeks (Q2W), 375 mg Q3W, or 500 mg Q4W, on the basis of pharmacokinetic (PK) modeling and simulations and PK/pharmacodynamic (PD) assessments that indicated these dosing regimens would lead to comparable exposure ranges and produce similar PD activity and a consistent toxicity profile [14, 15].
This report describes safety, efficacy, biomarker, and PK data from the budigalimab monotherapy expansion HNSCC and NSCLC cohorts of study NCT03000257.
Patients and methods
Patient and public involvement
There was no patient or public involvement in design, planned recruitment, or planned dissemination of this study.
Patient eligibility
Eligible patients were at least 18 years old with advanced HNSCC (arising from the oral cavity, oropharynx, hypopharynx, or larynx) or squamous or nonsquamous NSCLC, Eastern Cooperative Oncology Group (ECOG) performance status of 2 or lower, and measurable disease by Response Evaluation Criteria In Solid Tumors (RECIST; version [v]1.1 [16]. Patients were also required to have adequate organ function (including absolute neutrophil count ≥ 1,500/mm3, platelets ≥ 100,000/mm3, hemoglobin ≥ 9.0 g/dL, and creatinine clearance ≥ 50 mL/min as assessed by the Cockcroft-Gault formula or 24-h creatinine clearance). Eligible patients in the NSCLC expansion cohort had locally advanced or metastatic NSCLC, had previously experienced platinum-based therapy failure, and were naive to PD-1/PD-L1-targeting agents; in the HNSCC expansion cohort, patients had recurrent or metastatic disease that was not amenable to curative treatment with local or systemic therapy and were naive to PD-1/PD-L1-targeting agents. For this first-in-human study, patients were not selected on the basis of the presence or absence of any particular driver oncogenic mutations nor on their PD-L1 status. Key exclusion criteria included a history of inflammatory bowel disease, immune-mediated pneumonitis, active autoimmune disease (with exceptions of vitiligo, type I diabetes mellitus, hypothyroidism, and psoriasis), primary immunodeficiency, bone marrow or solid organ transplantation, HIV-positive ( +) status, chronic active hepatitis B or C infection, uncontrolled central nervous system metastasis, or evidence of hemolysis on screening laboratory studies.
The study protocol and informed consent form were approved by the institutional review board at each participating site prior to initiation of any screening or study-specific procedures. Written informed consent was obtained from each individual participating in the study. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, as defined by the International Conference on Harmonization. This study is registered at ClinicalTrials.gov (NCT03000257).
Study design and treatment
This was a multicenter, open-label, phase 1 study of budigalimab in adult patients with advanced solid tumors, consisting of two parts: dose escalation and dose expansion. The primary objectives were to examine the safety and PK of budigalimab monotherapy. The secondary objective was to evaluate preliminary activity of budigalimab, and exploratory objectives included (1) evaluation of PD and exploratory biomarkers for association with safety, PK, and clinical responses; and (2) evaluation of baseline PD-L1 expression and relationship with outcome.
The overall study schema is shown in supplementary Fig. 1. The dose-escalation portion of the study followed a standard 3 + 3 design to determine the safety, maximum tolerated dose, and PK profile of budigalimab. On the basis of previously reported safety, PK, and PD data from the dose-escalation portion of the study [13, 14], patients were then enrolled into two tumor-specific monotherapy dose-expansion cohorts, HNSCC and NSCLC, which are reported in this current analysis. Budigalimab was administered by intravenous infusion at either 250 mg Q2W or 500 mg Q4W until disease progression per RECIST v1.1 [16], confirmed disease progression per immune (i)RECIST [17], unacceptable toxicity, or other protocol-defined discontinuation criteria (supplementary Table 1). Patients experiencing radiographic progression per RECIST v1.1 could continue budigalimab treatment if they had no symptoms or signs of disease progression, no decline in ECOG performance status, and no evidence of rapid disease progression or progressive tumor at critical anatomic sites.
Budigalimab was administered as follows: the first infusion was delivered over 90 min; if the patient did not experience an infusion reaction, the second infusion was shortened to 60 min. Subsequent infusions could be administered over 30 min in the absence of infusion reactions following the first or second infusion. Dose reduction of budigalimab was not permitted.
Assessments
Safety evaluations were performed throughout the study and included assessment of treatment-emergent adverse events (TEAEs) and monitoring of additional clinical data (including vital signs, physical examination, electrocardiograms, echocardiograms, and laboratory test assessments). AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03. The criteria for permanent discontinuation of budigalimab following a TEAE are described in supplementary Table 1. Immune-related AEs were managed per published guidelines [18,19,20].
Intensive serial blood samples for measurement of budigalimab concentrations (PK) in serum were collected in cycles 1 and 3, and additional samples were collected during cycle 2 and cycles ≥ 4. PK parameters were estimated using noncompartmental analysis in Phoenix® WinNonlin® (Certara, Princeton, NJ) and included maximum observed concentration (Cmax), time to Cmax, area under the concentration–time curve, and half-life.
Biologic samples were collected from each patient to evaluate tumor-specific and systemic biomarkers. All patients consented to provide either archived formalin-fixed paraffin-embedded tumor tissue or a pretreatment, fresh tumor biopsy. Tumor tissue was analyzed for PD-L1 expression using the Dako 28–8 pharmDX IHC [immunohistochemistry] assay (Agilent Technologies, Santa Clara, CA); testing was performed at a single laboratory (Mosaic Laboratories, Lake Forest, CA). Blood samples for exploratory biomarker assessment were collected prior to infusion (0 h, predose), 2-h postinfusion, and on days 2, 3, 8, and 15 in cycles 1 and 3, and days 1 and 15 of cycle 2. Biomarkers evaluated included immune cell counts and PD-1 saturation on CD4 + central memory T cells by real-time flow cytometry, as well as soluble cytokine quantification in cryopreserved serum by Luminex® (Austin, TX).
Efficacy endpoints included objective response rate (ORR; defined as confirmed complete response [CR] or confirmed partial response [PR]), best overall response (CR, PR, or stable disease [SD]), progression-free survival (PFS), and duration of objective response (DOR). Tumor assessments by radiographic imaging (contrast-enhanced computed tomography or magnetic resonance imaging) were performed at baseline and repeated every two treatment cycles for the first 12 months and every three cycles thereafter; these were investigator assessed according to RECIST v1.1 and iRECIST.
Statistical analyses
Approximately 40 patients were enrolled in each of the HNSCC and NSCLC expansion cohorts to evaluate safety and tolerability of budigalimab. All patients who received any amount of budigalimab were included in the demographic, baseline, and safety analyses. All patients who received at least one dose of study drug and had at least one postdose tumor assessment were included in the efficacy analyses. The two-sided 90% CIs for ORR were provided on the basis of the Clopper–Pearson (exact) method. PFS was defined as time from first dose of study drug to radiographic progression or death, whichever occurred first. For each responder, DOR was defined as time from initial response to the study drug to radiographic progression or death. Both PFS and DOR were summarized using the Kaplan–Meier method.
Results
Patient demographics and baseline characteristics
Between November 2017 and January 2019, 81 patients were enrolled in the HNSCC (N = 41) and NSCLC (N = 40) expansion cohorts (data cutoff: October 31, 2019). For the HNSCC cohort, the first patient was screened on 4 January 2018, and the last patient on 22 January 2019; for the NSCLC cohort, the first patient was screened on 8 November 2017, and the last patient on 20 December 2018. Baseline demographics and clinical characteristics of both cohorts are summarized in Table 1. Sufficient tumor samples for IHC analysis were obtained from 38 patients with HNSCC and 33 patients with NSCLC; 19 patients in the HNSCC cohort and 16 in the NSCLC cohort were PD-L1 + . There was insufficient tumor tissue for analysis from three patients considered responders per RECIST v1.1: 1 patient with HNSCC, and two patients with NSCLC.
Patient disposition and safety
The median duration of exposure to budigalimab was 72 days (range, 1–617) for the HNSCC cohort and 71 days (range, 1–490) for the NSCLC cohort (supplementary Table 2). In total, 24% of patients (N = 10) and 33% of patients (N = 13) in the HNSCC and NSCLC cohorts, respectively, reported budigalimab dose interruption. As of the data cutoff, two patients in the HNSCC cohort and four patients in the NSCLC cohort continued to receive budigalimab; the reasons for budigalimab treatment discontinuation were progressive disease (HNSCC: 88%; NSCLC: 70%), AEs (HNSCC: 7%; NSCLC: 18%), and withdrawn consent (NSCLC: 2.5%).
All patients (100%) in the HNSCC (N = 41) and NSCLC (N = 40) expansion cohorts experienced ≥ 1 TEAE. In total, 25 patients (61%) in the HNSCC cohort and 27 patients (68%) in the NSCLC cohort reported grade ≥ 3 TEAEs; the most frequently reported was anemia (HNSCC: n = 9, 22%; NSCLC: n = 5, 13%). Patients were evaluated for the presence of hemolysis as a cause of anemia; no patients had this condition. TEAEs occurring in ≥ 20% of patients and the most common grade ≥ 3 TEAEs summarized by dose are provided in Table 2.
A total of 26 patients (63%) in the HNSCC cohort and 23 patients (58%) in the NSCLC cohort experienced an AE considered related to budigalimab by investigator assessment; the most common were hypothyroidism (n = 8; 20%), diarrhea (n = 6; 15%), and pruritus (n = 6; 15%) in the HNSCC cohort, and hypothyroidism (n = 6; 15%) and fatigue (n = 5; 13%) in the NSCLC cohort. Any-grade treatment-related AEs (TRAEs) that occurred in ≥ 10% of patients are summarized in supplementary Table 3. Four patients (10%) in the HNSCC cohort and five patients (13%) in the NSCLC cohort experienced a grade ≥ 3 AE related to budigalimab, with acute kidney injury (n = 2; 5%), anemia, diarrhea, and hypokalemia (n = 1; 2% each) in the HNSCC cohort, and reduced visual acuity, microscopic colitis, immune-mediated hepatitis, increased transaminase, hyponatremia, and hypophosphatemia (n = 1; 2% each) in the NSCLC cohort. Four patients (10%) in the HNSCC cohort and two patients (5%) in the NSCLC cohort experienced a serious TRAE, with acute kidney injury (n = 2; 5%), diarrhea, general physical health deterioration, and pyrexia (n = 1; 5%) in the HNSCC cohort, and immune-mediated hepatitis and acute kidney injury (n = 1; 3% each) in the NSCLC cohort.
The most common TEAEs considered immune-mediated reactions by the investigator, shown in supplementary Table 4, were hypothyroidism (n = 7; 17%), diarrhea (n = 5; 12%), and pruritus (n = 3; 7%) in the HNSCC cohort, and hypothyroidism (n = 6; 15%) and maculopapular rash (n = 3; 8%) in the NSCLC cohort. Overall, 17 (21%) patients experienced a TEAE that led to study drug discontinuation: 7 (17%) in the HNSCC cohort, and 10 (25%) in the NSCLC cohort (see supplementary Table 5). A single patient in the NSCLC cohort experienced grade ≥ 3 TRAEs leading to budigalimab discontinuation (immune-mediated hepatitis, grade 4). TEAEs leading to budigalimab dose interruption were reported by 14 patients (34%) in the HNSCC cohort and 15 patients (38%) in the NSCLC cohort (supplementary Table 6). The most common TEAEs leading to budigalimab dose interruption were acute kidney injury and dyspnea (n = 2; 5% each) in the HNSCC cohort, and upper respiratory tract infection and hypercalcemia (n = 2; 5% each) in the NSCLC cohort. No patients experienced a TRAE leading to death during the study; all TEAEs leading to death were considered unrelated to budigalimab (see supplementary Table 7). A single event of grade 5 cardiac arrest occurred. The patient was an 85-year-old male with an extensive history of cigarette smoking and left ventricular hypertrophy. The patient, who had no antecedent history of increasing dyspnea, chest pain, or any immune-related reactions to therapy, died in his sleep. The most likely causes for this event were coronary thrombosis, cardiac arrhythmia, or pulmonary embolism; myocarditis was not considered a likely cause.
Pharmacokinetics
Budigalimab PK results from dose-escalation and dose-expansion cohorts, across varying doses and regimens, have been reported previously [14, 15]. Budigalimab PK was approximately dose-proportional across the clinical doses evaluated. The two dosing regimens of 250 mg Q2W and 500 mg Q4W resulted in comparable dose-normalized exposures (supplementary Table 8) and maintained receptor saturation, as was previously predicted from population PK modeling and simulations and PK/PD assessments [14, 15].
Biomarkers
Budigalimab demonstrated complete sustained receptor saturation on circulating CD4 + central memory T cells and the expected PD effects at both the 250-mg Q2W and 500-mg Q4W doses (Fig. 1). Complete PD-1 saturation was observed within 2 h of dosing, followed by a transient drop in the number of circulating T cells at cycle (C)1 day (D)2, and increased proliferation of CD8 + T cells in 23 of 49 tested patients (47%), as measured by a ≥ twofold change in Ki67 from baseline (Fig. 1a). Increases in interferon gamma-induced chemokines, monokine induced by gamma interferon (MIG), and interferon gamma-induced protein 10 (IP-10) were observed within a day of dosing and increased through C2D1, with similar kinetics and magnitude of induction observed at 250-mg Q2W and 500-mg Q4W doses (Fig. 1b).

PD-1 receptor saturation and pharmacodynamic effects of budigalimab administration by dose level. Data shown for each patient with assay baseline and at least 1 postbaseline value. Individual patient data shown for patients with ≥ twofold change in CD8 Ki67 staining. Mean + /– 95% CI shown for PD-1 staining, T-cell counts, and cytokines (number of patients for each mean shown in graph). C, cycle; D, day; HNSCC, head and neck squamous cell carcinoma; hr, hour; IP-10, interferon gamma-induced protein 10; mAb, monoclonal antibody; MIG, monokine induced by gamma interferon; NSCLC, non-small cell lung cancer; PD-1, programmed cell death protein 1; Q, every; TCM, central memory T cells; W, weeks
Antitumor activity
A total of 77 patients were included in the efficacy-evaluable population (HNSCC: n = 40; NSCLC: n = 37). One patient in the HNSCC cohort discontinued budigalimab prior to week 8 secondary to grade 5 acute respiratory distress syndrome (unrelated to budigalimab), and three patients in the NSCLC cohort discontinued budigalimab (two secondary to clinical progression and one secondary to grade 5 upper respiratory infection, both unrelated to budigalimab).
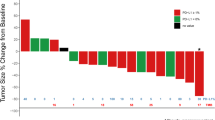
The best percentage change from baseline in size of target lesions for HNSCC patients is shown in Fig. 2a and for NSCLC patients in Fig. 2b. The percentage change over time in the size of target lesions is shown in Fig. 3a and Fig. 3b for HNSCC and NSCLC patients, respectively. A best overall response (defined as unconfirmed responses as per RECIST v1.1. or iRECIST) of PR or CR was achieved in 15% (90% CI, 6.7–27.5) of patients in the HNSCC and 19% (90% CI, 9.2–32.6) of patients in the NSCLC cohort. The ORR (defined as confirmed responses per RECIST v1.1. or iRECIST) for the HNSCC and NSCLC cohorts was 13% (90% CI, 5.1–24.5) and 19% (90% CI, 9.2–32.6), respectively (Table 3). The ORR for PD-L1 + (≥ 1%) patients in the HNSCC and NSCLC cohorts was 16% (90% CI, 4.5–35.9; 3 confirmed PRs in 19 PD-L1 + HNSCC patients) and 13% (90% CI, 2.3–34.4; 2 confirmed PRs in 16 PD-L1 + NSCLC patients), respectively (Table 3). The ORR for patients with NSCLC who had ≥ 50% PD-L1 expression was 29% (2 of 7 evaluable patients); when patients with epidermal growth factor receptor (EGFR) mutation or anaplastic lymphoma kinase (ALK) rearrangement are excluded from this group, the ORR was 40% (2 of 5 evaluable patients). Median PFS in HNSCC patients was 3.6 months (95% CI, 1.7–4.7 and 1.9 months (95% CI, 1.7–3.7) in NSCLC patients (Table 3; supplementary Fig. 2); median DOR was 9.4 months (95% CI, 1.9–not estimable) and 10.1 months (95% CI, 7.8–13.1) in the HNSCC and NSCLC cohorts, respectively (Table 3). The Kaplan–Meier estimate for the 6-month DOR rate was 80% in HNSCC and 100% in NSCLC, with only one responder exhibiting progressive disease within 6 months of response. Overall, responses were observed in both PD-L1 + and PD-L1–negative (PD-L1–) patients and were durable. In the NSCLC cohort, no responses were observed in patients with known EGFR mutation (n = 7), KRAS mutation (n = 7), or with ALK rearrangement (n = 1), regardless of PD-L1 expression.

Best percentage change in target lesions from baseline for a HNSCC and b NSCLC cohorts receiving budigalimab monotherapy. aPD-L1 + status; bPD-L1 status missing (i.e., unknown). ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; HNSCC, head and neck squamous cell carcinoma; NSCLC, non-small cell lung cancer; PD-L1 + , programmed cell death protein 1 ligand 1 positive; Q, every; W, weeks

Percentage change in target lesions from baseline for a HNSCC and b NSCLC cohorts receiving budigalimab monotherapy. ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; HNSCC, head and neck squamous cell carcinoma; NSCLC, non-small cell lung V; PD-1, programmed cell death protein 1; Q, every; W, weeks
Discussion
This first-in-human phase 1 study demonstrated that budigalimab administration at doses of 250 mg IV Q2W or 500 mg IV Q4W in patients with HNSCC and NSCLC was equally safe and well tolerated. Budigalimab showed dose-proportional PK and had comparable dose-normalized exposures at the evaluated dosing regimens of 250 mg Q2W and 500 mg Q4W.
The safety profile of budigalimab observed in the current study was comparable to that observed with other approved PD-1–targeted agents, including nivolumab and pembrolizumab. The incidents of anemia that was observed following budigalimab treatment was likely due to the prior chemotherapy and radiation therapy that the patients received. Careful monitoring for development of hemolysis during the study found no such events. In the CheckMate 017 study, the most common AEs in patients with advanced-stage squamous NSCLC treated with nivolumab were fatigue, decreased appetite, and asthenia [21]. In CheckMate 057, a study of nivolumab in nonsquamous NSCLC patients, the most common AEs were fatigue, decreased appetite, cough, constipation, and dyspnea [22]. In KEYNOTE-010, fatigue, pruritus, and decreased appetite were the most common AEs reported in NSCLC patients treated with pembrolizumab [23]. Accumulating evidence suggests that only a fraction of cancer patients benefit from immune checkpoint inhibitors, and severe immune-related AEs are associated with immune checkpoint inhibitor therapy [24]. The most commonly reported immune-related AEs reported in the HNSCC and NSCLC cohorts of the present study were hypothyroidism, diarrhea, hyperthyroidism, pruritus, and rash, which are similar to those reported in previous studies of immune checkpoint inhibitors [25].
Antitumor activity was observed following budigalimab treatment, with one patient in the NSCLC cohort achieving CR and six patients achieving PR. In the HNSCC cohort, six patients achieved PR, with one PR per iRECIST criteria after initial progressive disease per RECIST v1.1. Additionally, one patient in the HNSCC cohort and three in the NSCLC cohort achieved immune SD following previous progression per RECIST. The observation of pseudoprogression (disease progression per RECIST followed by subsequent reduction in tumor burden) [26] in several patients enrolled in this study is characteristic of immune checkpoint inhibitors, and similar observations have been reported in studies of other immune checkpoint inhibitors, including ipilimumab, nivolumab, and pembrolizumab [27,28,29,30]. Durable responses were observed both in PD-L1 + and PD-L1– patients in the current study, similar to responses observed in clinical studies of other PD-1-targeting agents [21, 22].
Efficacy data from the current study indicate that the clinical activity of budigalimab is similar to that of approved anti-PD-1 agents in patients with NSCLC. Nivolumab exhibited ORRs of 20% [21] and 19% [22] in patients with squamous and nonsquamous NSCLC, respectively, while pembrolizumab exhibited an ORR of 18% in NSCLC patients with tumor proportion score ≥ 1% (KEYNOTE-010) [23]. Of note, most patients treated in these studies had 1 prior line of therapy (99% for CheckMate 017, 88% for CheckMate 057, and 68% for KEYNOTE-010) [21,22,23]. Such data may indicate that patients with ≥ 2 prior lines of therapy do not derive clinical benefit from these particular checkpoint inhibitors. In the current study, 53% of NSCLC patients treated with budigalimab had received 1 prior line of therapy, while 47% had ≥ 2 prior lines; among the seven responders, six patients had received one prior line of systemic therapy and one responder had received two prior lines of systemic therapy.
Although this study evaluated budigalimab in 40 patients with NSCLC, similar to other anti-PD-1 therapies a lower response rate was observed in NSCLC patients with tumors that harbor EGFR-activating mutations and ALK rearrangements [31]. The current study exhibited a higher proportion of patients with these genomic alterations (20%), compared with the proportion of patients with EGFR-activating mutations and ALK rearrangements in the CheckMate 057 (18%) and KEYNOTE-010 (9%) trials [22, 23]. Also, similar to other anti-PD-1 therapies, NSCLC patients treated with budigalimab with high (≥ 50%) tumor PD-L1 expression had higher ORR (29%) compared with the overall NSCLC cohort or with NSCLC patients with confirmed ≥ 1% tumor PD-L1 expression (19% and 13% ORR, respectively) [23]. It is worthy of mentioning that this trial was designed, and patients enrolled, at a time when the key oncogenic drivers in NSCLC were considered to be EGFR mutations and ALK rearrangements. In the intervening years since trial initiation, a number of other potential driver mutations have been identified in genes such as rearranged during transfection (RET), neurotrophic tyrosine receptor kinase (NTRK), human epidermal growth factor receptor 2 (HER2), and v-raf murine sarcoma viral oncogene homolog B1 (BRAF). However, as the majority of patients with NSCLC in this study had insufficient biopsy tissue, we were unable to perform an extended mutational analysis and determine the frequency of these mutations. Further studies may be warranted to evaluate the efficacy of budigalimab in patients with driver mutations other than EGFR and ALK.
Data from the HNSCC cohort are also consistent with response rates observed for nivolumab and pembrolizumab. In HNSCC, ORRs were 13% for nivolumab [22] and 15% for pembrolizumab (KEYNOTE-010) [23]. Budigalimab demonstrated a 13% ORR, with one responder meeting criteria for immune PR (on study day 101 after meeting criteria for immune unconfirmed progressive disease on study day 51).
PK assessments indicated that the 250-mg Q2W and 500-mg Q4W regimens resulted in similar dose-normalized exposures and PD activity, suggesting that either schedule is viable and thereby providing flexibility in potential combinations with other anticancer agents.
Biomarker assessment of the effect of budigalimab administration on PD-1 receptor occupancy showed complete saturation of PD-1 at 250 mg Q2W and 500 mg Q4W. PD-1 saturation resulted in expected biologic activities on T-cell proliferation and chemokines. These results are consistent with the activity of other anti-PD-1 agents, which enhance antitumor immune activity as detected by increases in peripheral CD8 T-cell proliferation [32, 33], interferon gamma-induced serum chemokines [34, 35], and therapeutic antitumor effects [36].
In conclusion, these data demonstrate that budigalimab has a manageable safety profile with evidence of biologic and clinical activity in patients with previously treated HNSCC and NSCLC that seems to be similar to approved PD-1 inhibitors. The data support the continued development of budigalimab in multiple oncology indications.
Availability of data and material
Availability of data and material AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Abbreviations
- + :
-
Positive
- –:
-
Negative
- AE:
-
Adverse event
- ALK:
-
Anaplastic lymphoma kinase
- C:
-
Cycle
- Cmax :
-
Maximum observed concentration
- CNS:
-
Central nervous system
- CR:
-
Complete response
- D:
-
Day
- DOR:
-
Duration of response
- ECOG:
-
Eastern cooperative oncology group
- EGFR:
-
Epidermal growth factor receptor
- HNSCC:
-
Head and neck squamous cell carcinoma
- IHC:
-
Immunohistochemistry
- IP-10:
-
Interferon gamma-induced protein 10
- iRECIST:
-
Immune response evaluation criteria in solid tumors
- IV:
-
Intravenous
- KM:
-
Kaplan–Meier
- mAb:
-
Monoclonal antibody
- MedDRA:
-
Medical dictionary for regulatory activities
- MIG:
-
Monokine induced by gamma interferon
- NE:
-
Not estimable
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- PD:
-
Pharmacodynamic
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death protein 1 ligand 1
- PD-L2:
-
Programmed cell death protein 1 ligand 2
- PFS:
-
Progression-free survival
- PK:
-
Pharmacokinetic
- PR:
-
Partial response
- Q:
-
Every
- RECIST:
-
Response evaluation criteria in solid tumors
- SD:
-
Stable disease
- TEAE:
-
Treatment-emergent adverse event
- TRAE:
-
Treatment-related adverse event
- v:
-
Version
- w:
-
Weeks
References
Herzberg B, Campo MJ, Gainor JF (2017) Immune checkpoint inhibitors in non-small cell lung cancer. Oncologist 22(1):81–88. https://doi.org/10.1634/theoncologist.2016-0189
Hamanishi J, Mandai M, Matsumura N, Abiko K, Baba T, Konishi I (2016) PD-1/PD-L1 blockade in cancer treatment: perspectives and issues. Int J Clin Oncol 21(3):462–473. https://doi.org/10.1007/s10147-016-0959-z
Keir ME, Butte MJ, Freeman GJ, Sharpe AH (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26:677–704. https://doi.org/10.1146/annurev.immunol.26.021607.090331
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L (2002) Tumor-associated B7–H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8(8):793–800. https://doi.org/10.1038/nm730
Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I (2016) Dual faces of IFNγ in cancer progression: a role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res 22(10):2329–2334. https://doi.org/10.1158/1078-0432.CCR-16-0224
Sanmamed MF, Chen L (2014) Inducible expression of B7–H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J 20:256–261. https://doi.org/10.1097/PPO.0000000000000061
KEYTRUDA® (pembrolizumab) injection [prescribing information]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2020. https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf. Accessed October 2, 2020.
OPDIVO (nivolumab) injection [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company; 2020. https://packageinserts.bms.com/pi/pi_opdivo.pdf. Accessed October 2, 2020.
LIBTAYO® (cemiplimab-rwlc) injection [prescribing information]. Tarrytown, NY: Regeneron Pharmaceuticals, Inc.; 2020. https://www.regeneron.com/sites/default/files/Libtayo_FPI.pdf. Accessed October 2, 2020.
Lambert SL, Vosganian G, Harding F, Sheridan J, Englert S, Siggelkow S, Parikh A, Wang B, Hew K, Bankoti J, Stickler M, Lee E, Mcclellan M, Afar D, Reddy A, Lambert S (2018) Biomarker and preliminary pharmacodynamic evaluations of the PD-1 inhibitor ABBV-181 from an ongoing phase 1 clinical trial in patients with advanced solid tumors. J Immunother Cancer. https://doi.org/10.1186/s40425-018-0422-y
European Medicines Agency (2015) Assessment report. OPDIVO. International non-proprietary name: nivolumab. https://www.ema.europa.eu/en/documents/assessment-report/opdivo-epar-public-assessment-report_en.pdf. Accessed March 17, 2021.
European Medicines Agency (2015) Assessment report. Keytruda. International non-proprietary name: pembrolizumab. https://www.ema.europa.eu/en/documents/assessment-report/keytruda-epar-public-assessment-report_en.pdf. Accessed March 17, 2021.
Powderly J, Cassier PA, Cervantes A, Gao B, Gazzah A, Italiano A, Lin C, Luke JJ, Moreno V, Peltola K, Rasco D, Spira AI, Tanner MME, Tosi D, Afar D, Englert S, Parikh A, Reddy A, Vosganian G, Tolcher AW (2018) Safety and efficacy of the PD-1 inhibitor ABBV-181 in patients with advanced solid tumors: preliminary phase 1 results from study M15–891. Ann Oncol. https://doi.org/10.1093/annonc/mdy279
Powderly J, Spira A, Kondo S, Doi T, Luke JJ, Rasco D, Gao B, Tanner M, Cassier PA, Gazzah A, Italiano A, Tosi D, Afar DE, Parikh A, Engelhardt B, Englert S, Lambert SL, Kasichayanula S, Mensing S, Menon R, Vosganian G, Tolcher A (2020) Model informed dosing regimen and phase I results of the anti-PD1 antibody budigalimab (ABBV-181). Clin Transl Sci. https://doi.org/10.1111/cts.12855
Parikh A, Kasichayanula S, Menon R, Afar D, Lambert S, Engelhardt B, Mensing S (2018) Pharmacokinetics (PK) and dosing regimen selection of the PD-1 inhibitor ABBV-181 in patients with advanced solid tumors: preliminary phase 1 results from study M15–891. J Immunother Cancer. https://doi.org/10.1186/s40425-018-0423-x
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, Lin NU, Litière S, Dancey J, Chen A, Hodi FS, Therasse P, Hoekstra OS, Shankar LK, Wolchok JD, Ballinger M, Caramella C, de Vries EGE, RECIST working group, (2017) iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 18(3):e143–e152. https://doi.org/10.1016/S1470-2045(17)30074-8
Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, Jordan K (2017) ESMO Guidelines committee (2017) Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 28:iv119–iv142. https://doi.org/10.1093/annonc/mdx225
Puzanov I, Diab A, Abdallah K, Bingham CO, Brogdon C, Dadu R, Hamad L, Kim S, Lacouture ME, LeBoeuf NR, Lenihan D, Onofrei C, Shannon V, Sharma R, Silk AW, Skondra D, Suarez-Almazor ME, Wang Y, Wiley K, Kaufman HL, Ernstoff MS (2017) Society for Immunotherapy of Cancer Toxicity Management Working Group Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J immunother Cancer 5(1):95. https://doi.org/10.1186/s40425-017-0300-z
Thompson JA, Schneider BJ, Brahmer J, Andrews S, Armand P, Bhatia S, Budde LE, Costa L, Davies M, Dunnington D, Ernstoff MS, Frigault M, Hoffner B, Hoimes CJ, Lacouture M, Locke F, Lunning M, Mohindra NA, Naidoo J, Olszanski AJ, Oluwole O, Patel SP, Reddy S, Ryder M, Santomasso B, Shofer S, Sosman JA, Wahidi M, Wang Y, Johnson-Chilla A, Scavone JL (2019) Management of immunotherapy-related toxicities version 1. J Natl Compr Canc Network 17(3):255–289. https://doi.org/10.6004/jnccn.2019.0013
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Frontera OA, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR (2015) Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 373(2):123–135
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhäufl M, Arrieta O, Burgio MA, Fayette J, Lena H, Poddubskaya E, Gerber DE, Gettinger SN, Rudin CM, Rizvi N, Crinò L, Blumenschein GR Jr, Antonia SJ, Dorange C, Harbison CT, Finckenstein FG, Brahmer JR (2015) Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 373(17):1627–1639. https://doi.org/10.1056/NEJMoa1507643
Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G, Garrido M, Lubiniecki GM, Shentu Y, Im E, Dolled-Filhart M, Garon EB (2016) Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387(10027):1540–1550. https://doi.org/10.1016/S0140-6736(15)01281-7
Jain P, Jain C, Velcheti V (2018) Role of immune-checkpoint inhibitors in lung cancer. Ther Adv Respir Dis 12:1753465817750075. https://doi.org/10.1177/1753465817750075
Khoja L, Day D, Chen TWW, Siu LL, Hansen AR (2017) Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol 28(10):2377–2385. https://doi.org/10.1093/annonc/mdx286
Martin-Romano P, Castanon E, Ammari S, Champiat S, Hollebecque A, Postel-Vinay S, Baldini C, Varga A, Michot JM, Vuagnat P, Marabelle A, Soria JC, Ferté C, Massard C (2020) Evidence of pseudoprogression in patients treated with PD1/PDL1 antibodies across tumor types. Cancer Med 9(8):2643–2652
Chanana R, Noronha V, Joshi A, Patil V, Prabhash K (2018) Evolving role of immunotherapy in head-and-neck cancers: a systemic review. J Head Neck Phys Surg 6(1):2–11. https://doi.org/10.4103/jhnps.jhnps_10_18
Tazdait M, Mezquita L, Lahmar J, Ferrara R, Bidault F, Ammari S, Balleyguier C, Planchard D, Gazzah A, Soria JC, Marabelle A, Besse B, Caramella C (2018) Patterns of responses in metastatic NSCLC during PD-1 or PDL-1 inhibitor therapy: comparison of RECIST 1.1, irRECIST and iRECIST criteria. Eur J Cancer 88:38–47. https://doi.org/10.1016/j.ejca.2017.10.017
Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, Sequist LV, Smith DC, Leming P, Carbone DP, Pinder-Schenck MC, Topalian SL, Hodi FS, Sosman JA, Sznol M, McDermott DF, Pardoll DM, Sankar V, Ahlers CM, Salvati M, Wigginton JM, Hellman MD, Kollia GD, Gupta AK, Brahmer JR (2015) Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol 33(18):2004–2012. https://doi.org/10.1200/JCO.2014.58.3708
Jia W, Gao Q, Han A, Zhu H, Yu J (2019) The potential mechanism, recognition and clinical significance of tumor pseudoprogression after immunotherapy. Cancer Biol Med 16(4):655–670. https://doi.org/10.20892/j.issn.2095-3941.2019.0144
Berghoff AS, Bellosillo B, Caux C, de Langen A, Mazieres J, Normanno N, Preusser M, Provencio M, Rojo F, Wolf J, Zielinski CC (2019) Immune checkpoint inhibitor treatment in patients with oncogene-addicted non-small cell lung cancer (NSCLC): summary of a multidisciplinary round-table discussion. ESMO Open 4(3):e000498. https://doi.org/10.1136/esmoopen-2019-000498
Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, Behera M, Wu H, McCausland M, Chen Z, Zhang C, Khuri FR, Owonikoko TK, Ahmed R, Ramalingam SS (2017) Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1–targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 114(19):4993–4998. https://doi.org/10.1073/pnas.1705327114
Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, Adamow M, Kuk D, Panageas KS, Carrera C, Wong P, Quagliarello F, Wubbenhorst B, D’Andrea K, Pauken KE, Herati RS, Staupe RP, Schenkel JM, McGettigan S, Kothari S, George SM, Vonderheide RH, Amaravadi RK, Karakousis GC, Schuchter LM, Xu X, Nathanson KL, Wolchok JD, Gangadhar TC, Wherry EJ (2017) T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545(7652):60–65. https://doi.org/10.1038/nature22079
Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, Dhodapkar KM (2015) Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol 194(3):950–959. https://doi.org/10.4049/jimmunol.1401686
Choueiri TK, Fishman MN, Escudier B, McDermott DF, Drake CG, Kluger H, Stadler WM, Perez-Gracia JL, McNeel DG, Curti B, Harrison MR, Plimack ER, Appleman L, Fong L, Albiges L, Cohen L, Young TC, Chasalow SD, Ross-Macdonald P, Srivastava S, Jure-Kunkel M, Kurland JF, Simon JS, Sznol M (2016) Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma. Clin Cancer Res 22(22):5461–5471. https://doi.org/10.1158/1078-0432.CCR-15-2839
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL (2010) Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 28:3167–3175. https://doi.org/10.1200/JCO.2009.26.7609
Acknowledgements
AbbVie and the authors thank the patients who participated in this clinical trial, the study coordinators, and support staff. We would like to acknowledge James P. Sheridan (Drug Metabolism Pharmacokinetics & Bioanalysis Department, AbbVie) for performing the preclinical and clinical pharmacokinetic studies, and Fiona Harding (Oncology Discovery, AbbVie) for creating, selecting, and validating the ABBV-181 antibody and filing the in vitro/in vivo IND supporting reports, including summaries of the preclinical activity of ABBV-181. Biomarker sample testing was performed by Covance, Princeton, NJ (flow cytometry), Mosaic Laboratories, Lake Forest, CA (immunohistochemistry), and Myriad RBM, Austin, TX (cytokine testing). This study was funded by AbbVie Inc., North Chicago, IL, USA. Medical writing support was provided by Joanne Franklin, PhD, CMPP, Aptitude Health, The Hague, the Netherlands, funded by AbbVie.
Funding
AbbVie Inc. provided financial support for the study (NCT03000257) and participated in the design, study conduct, analysis and interpretation of data, as well as the writing, review, and approval of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors were involved in the writing of this manuscript and read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict interest
Antoine Italiano: Consulting/advisory role: Roche, Daiichi Sankyo, Immune Design, Epizyme, Bayer, Lilly; honoraria: Bayer, Daiichi Sankyo, Lilly, Epizyme, Novartis, Roche; research funding: Roche, Bayer, AstraZeneca/MedImmune, PharmaMar, MSD Oncology, Merck Serono. Philippe A. Cassier: Honoraria: Novartis, Roche/Genentech, Blueprint Medicines, Amgen; research funding: Novartis, Roche/Genentech, Lilly, Blueprint Medicines, Bayer, AstraZeneca, Celgene, Plexxikon, AbbVie, Bristol-Myers Squibb, Merck Serono, Merck Sharp & Dohme; Consultancy/advisory role: Merck Serono, Roche/Genentech. Chia-Chi Lin: Consulting/advisory role: Novartis, Boehringer Ingelheim, Blueprint Medicines; travel/accommodations/expenses: Lilly, Daiichi Sankyo, BeiGene, Novartis; honoraria: Novartis, Roche, Daiichi Sankyo. Tuomo Alanko: Consulting/advisory role: Bayer, Baxalta/Shire, BMS, Celgene, Eli Lilly, MSD, Nordic Drugs, Roche, Kaiku Health; research funding: AbbVie, Bayer, Boehringer Ingelheim, BMS, Debiopharm, Eli Lilly, Incyte, MSD, Pfizer, Roche; travel/accommodations/expenses: Baxalta/Shire, BMS, MSD, Pfizer, Roche. Katriina J. Peltola: Consulting/advisory role: Orion Pharma, BMS, MSD, Novartis, Pfizer, Ipsen, Roche, Varian; stockholder: Faron Pharmaceuticals; speakers’ bureau: BMS, Pfizer, MSD; expert testimony: Ipsen; travel/accommodations/expenses: Roche, BMS; research funding: AbbVie, Bayer, BMS, MSD, Roche, Exelixis, Orion Pharma, Eisai, Novartis. Anas Gazzah: Travel, accommodations, congress registration expenses: Boehringer Ingelheim, Novartis, Pfizer, Roche; consultant/expert role: Novartis; principal/sub-investigator of clinical trials: Aduro Biotech, Agios Pharmaceuticals, Amgen, Argen-X BVBA, Arno Therapeutics, Astex Pharmaceuticals, AstraZeneca, Aveo, Bayer HealthCare Ag, BBB Technologies BV, BeiGene, BioAlliance Pharma, BioNTech AG, Blueprint Medicines, Boehringer Ingelheim, Bristol-Myers Squibb, Ca, Celgene Corporation, Chugai Pharmaceutical Co., Clovis Oncology, Daiichi Sankyo, Debiopharm SA, Eisai, Exelixis, Forma, GamaMabs, Genentech, Inc., Gilead Sciences, Inc, GlaxoSmithKline, Glenmark Pharmaceuticals, H3 Biomedicine, Inc, F. Hoffmann-La Roche AG, Incyte Corporation, Innate Pharma, Servier IRIS, Janssen, Kura Oncology, Kyowa Kirin Pharmaceutical Development, Lilly, Loxo Oncology, Lytix Biopharma AS, MedImmune, Menarini Ricerche, Merck Sharp & Dohme Chibret, Merrimack Pharmaceuticals, Merus, Millennium Pharmaceuticals, Nanobiotix, Nektar Therapeutics, Novartis Pharma, Octimet Oncology NV, OncoEthix, OncoMed, Oncopeptides, Onyx Therapeutics, Orion Pharma, Oryzon Genomics, Pfizer, PharmaMar, Pierre Fabre, Rigontec GmbH, Roche, Sanofi Aventis, Sierra Oncology, Taiho Pharma, Tesaro, Inc, Tioma Therapeutics, Inc., Xencor; research grants: AstraZeneca, BMS, Boehringer Ingelheim, Janssen Cilag, Merck, Novartis, Pfizer, Roche, Sanofi; nonfinancial support (drug supplied): AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Johnson & Johnson, Lilly, MedImmune, Merck, NH TherAGuix, Pfizer, Roche. Her-Shyong Shiah: The author declares no potential conflicts of interest. Emiliano Calvo: Consulting/advisory role: Novartis, Nanobiotix, Janssen-Cilag, PsiOxus, Seattle Genetics, EUSA Pharma, AbbVie, Celgene, AstraZeneca, Guidepoint Global, Roche/Genentech, GLG, Pfizer, Servier, amcure; speakers’ bureau: Novartis; research funding: AstraZeneca, BeiGene, Novartis, START; travel/accommodations/expenses reimbursement: Roche/Genentech; honoraria: HM Hospitales Group; stock/ownership interests: START, Oncoart Associated, International Cancer Consultants; president and founder of Foundation INTHEOS. Andrés Cervantes: Institutional research funding: AbbVie, Genentech, Merck Serono, BMS, MSD, Roche, BeiGene, Bayer, Servier, Lilly, Novartis, Takeda, Astellas, FibroGen; advisory board or speaker fees: Merck Serono, Roche, Bayer, Servier, Pierre Fabre. Desamparados Roda: The author declares no potential conflicts of interest. Diego Tosi: Consulting/advisory role: BioMarin (immediate family member); research funding: Novartis, Astellas, Janssen; patent pending on a new drug combination for prostate cancer treatment; travel/accommodations/expenses: Janssen, Pfizer, Astellas Pharma; immediate family member had travel/accommodations/expenses from Nutricia and Amicus. Bo Gao: Consulting/advisory role: MSD. Michael Millward: Consulting/advisory role: Merck Sharp & Dohme, Bristol-Myers Squibb, AstraZeneca, Roche, Pfizer, Takeda, Novartis; conference travel/support: Merck Sharp & Dohme, Bristol-Myers Squibb, AstraZeneca, Roche. Lydia Warburton: Travel/accommodations/expenses: MSD, Merck. Minna Tanner: Consulting/advisory role: Roche, Novartis, Pfizer; speakers’ bureau: Roche, Novartis, Pfizer, Amgen. Gregory Vosganian: Former employee of AbbVie and may own stock. Stefan Englert, Stacie Lambert, Apurvasena Parikh, Daniel E. Afar: AbbVie employees and may own stock. Victor Moreno: Consulting fees: Merck, BMS, Janssen, Pieris; travel/accommodations: Regeneron/Sanofi; presentations: Nanobiotix; educational grant: Medscape/Bayer.
Informed consents
This study was approved by the institutional review board at each participating site prior to initiation of any screening or study-specific procedures. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, as defined by the International Conference on Harmonization. Written informed consent was obtained from each individual participating in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Italiano, A., Cassier, P.A., Lin, CC. et al. First-in-human phase 1 study of budigalimab, an anti-PD-1 inhibitor, in patients with non-small cell lung cancer and head and neck squamous cell carcinoma. Cancer Immunol Immunother 71, 417–431 (2022). https://doi.org/10.1007/s00262-021-02973-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-021-02973-w