
Abstract
Purpose
Patients with recurrent glioblastoma (rGBM) have a poor prognosis. Epidermal growth factor receptor (EGFR) gene amplification is present in ~ 50% of glioblastomas (GBMs). Depatuxizumab mafodotin (depatux-m), formerly ABT-414, is an antibody–drug conjugate that preferentially binds cells with EGFR amplification, is internalized and releases a potent antimicrotubule agent, monomethyl auristatin F (MMAF). Here we report the safety, pharmacokinetics, and efficacy of depatux-m monotherapy at the recommended Phase 2 dose (RPTD) in patients with EGFR-amplified, rGBM.
Methods
M12-356 (NCT01800695) is an open-label study with three escalation and expansion cohorts. Sixty-six patients with EGFR-amplified, rGBM were treated with depatux-m monotherapy at 1.25 mg/kg intravenously every 2 weeks. Adults with measurable rGBM, who were bevacizumab-naïve, with EGFR amplification were eligible.
Results
Among 66 patients, median age was 58 years (range 35–80). All patients were previously treated with radiotherapy/temozolomide. The most common adverse events (AEs) were eye related (91%), including blurred vision (65%), dry eye (29%), keratitis, and photophobia (27% each). Grade 3/4 AEs occurred in 42% of all patients, and ocular Grade 3/4 AEs occurred in 33% of patients overall. One patient (2%) had a Grade 4 ocular AE. Ocular AEs were manageable and usually resolved once treatment with depatux-m ceased. The objective response rate was 6.8%, the 6-month progression-free survival rate was 28.8%, and the 6-month overall survival rate was 72.5%.
Conclusion
Depatux-m monotherapy displayed frequent but mostly Grade 1/2 ocular toxicities. A PFS6 of 28.8% was observed in this rGBM population, warranting further study.
Similar content being viewed by others
Introduction
Glioblastoma (GBM) is the most common malignant brain cancer with an incidence of 2–3 of every 100,000 adults per year. Patients afflicted with GBM have a poor prognosis, with a median survival of 14–16 months from original diagnosis [1, 2]. Many patients will experience recurrent disease (rGBM), and treatment options are limited, with survival under 12 months and rare responses [3]. Six-month progression-free survival rates exceeding 20–25% are considering promising in this setting [4].
Given the dismal survival rates in rGBM, there is an urgent need to develop effective novel therapies. Amplification of the Epidermal Growth Factor Receptor (EGFR) gene, observed in 50% of GBMs [5,6,7], creates a tumor-specific target for experimental treatment. About 50% of GBMs with EGFR amplification also harbor the EGFRvIII deletion variant [8]. Of note, EGFR amplification usually remains unchanged at the time of tumor recurrence [9]. Several types of targeted therapies have been used to target EGFR in GBM. Tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib have been found to increase PFS in non-small cell lung cancer [10] but have not proven efficacious in GBM, [11,12,13,14,15,16]. Antibodies that target and bind the extracellular domain of EGFR, such as cetuximab, have shown decreased tumor growth and increased survival in mouse xenograft models [17] but again, did not demonstrate a survival benefit in patients [18]. There are several explanations for the failures of these agents, in particular the absence of the EGFR exon 19 deletion and exon 21 mutations that are correlated with activity in NSCLC [19]. Immunotherapy has improved outcomes for many cancers, and the vaccine rindopepimut, which targets EGFRvIII, showed promising results in early stage testing in GBM. However, a Phase 3 trial was recently discontinued due to a lack of survival benefit [20]. Numerous ongoing studies are evaluating the potential activity of various immunotherapy agents, including vaccines and immune checkpoint inhibitors, in newly diagnosed and recurrent GBM.
Depatuxizumab mafodotin (depatux-m), formerly ABT-414, is an antibody–drug conjugate (ADC) composed of the EGFR-directed monoclonal antibody, depatuxizumab (depatux), formerly ABT-806, conjugated to the potent antimicrotubule agent monomethyl auristatin F (MMAF, now mafodotin) via a non-cleavable maleimidocaproyl linker [21, 22]. EGFR amplification leads to a unique conformation of the EGFR protein that exposes a tumor-specific binding site for depatux-m. This epitope is also exposed in the EGFRvIII deletion variant. Once depatux-m enters the cell, MMAF is released, leading to cell death. Depatux-m has limited binding to EGFR in normal tissues and thus does not lead to other toxicities typically associated with other EGFR-targeted therapies, usually dermatological [23]. Preclinical data suggest that depatux-m has potent anti-tumor activity in GBM cell lines and xenograft models [6].
Recently published results from this study show that depatux-m in combination with either chemoradiation or TMZ in both newly diagnosed and recurrent GBM has a tolerable safety and pharmacokinetics (PK) profile [24, 25]. Here, we present efficacy data, including objective response rate (ORR) and PFS6, for depatux-m monotherapy at the recommended Phase 2 dose (RPTD) in patients with recurrent, EGFR-amplified GBM.
Materials and methods
Study M12-356 (NCT01800695) was a multi-center, Phase 1, open-label study designed to evaluate the safety, preliminary efficacy and PK of depatux-m alone or in combination with other treatments in patients with GBM. The trial had three treatment arms: Arm A, depatux-m with radiation therapy (RT) and temozolomide (TMZ) in newly diagnosed GBM; Arm B, depatux-m with TMZ after RT in newly diagnosed or recurrent GBM; and Arm C, depatux-m monotherapy in rGBM. Each arm was composed of a dose escalation and dose expansion cohort [24]. This study was performed in accordance with the 1964 Declaration of Helsinki and its later amendments. All patients provided written informed consent prior to enrollment according to national regulation; the study design was approved by the Institutional Review Board/Ethics Committees of participating institutions.
Patients
This analysis encompassed 66 patients from Arm C who had EGFR-amplified, rGBM and received at least one dose of depatux-m at the RPTD of 1.25 mg/kg. Inclusion and exclusion criteria were as described previously [24]. Only patients with rGBM and centrally confirmed EGFR amplification were included. More specifically, patients had Response Assessment in Neuro-Oncology (RANO) defined [26] disease progression which included either: (1) measurable progressive or rGBM as seen by contrast-enhancing MRI and an interval of at least 12 weeks from completion of RT to study entry; (2) progression outside the radiation field; or (3) biopsy or surgically proven disease progression. An MRI with contrast was required within 14 days of Study Day 1, and patients were required to be on a stable or decreasing dose of corticosteroids for at least 5 days prior to the scan. Patients were ineligible if: they had received bevacizumab as prior treatment for rGBM, had a secondary GBM, or had been exposed to prior EGFR therapy for GBM, including EGFRvIII-specific immunotherapies.
Study design
Study design of M12-356 has been described previously [24, 25]. The primary objective was to determine the ORR [partial response (PR) + complete response (CR)]. The secondary objectives were to determine the PFS6, PFS, OS, and safety and tolerability of depatux-m.
Treatment regimen
The RPTD of depatux-m monotherapy was determined previously as 1.25 mg/kg via intravenous (IV) infusion every 2 weeks [25]. All patients received 1.25 mg/kg of depatux-m via intravenous infusion over 30–40 min on Days 1 and 15 of a 28-day cycle (Supplementary Fig. 1). Radiographic assessment of disease progression was performed before every other cycle. Treatment was intended to continue until either intolerable toxicity or disease progression as assessed locally by the investigator using RANO criteria [26]. Central review was not performed. Depatux-m dosing could be reduced to 1.0 or 0.5 mg/kg for Grade 3/4 toxicities. Re-escalation was permitted.
Pharmacokinetics
Serum samples for the determination of depatux-m concentrations were collected before and immediately after depatux-m infusions on Day 1 of Cycles 1 and 2, and before depatux-m infusions on Day 15 of Cycles 1 and 2. Serum samples for the determination for anti-drug antibody (ADA) were collected biweekly before each depatux-m infusion up to Day 1 of Cycle 3 and once every four weeks before depatux-m infusion in the subsequent cycles. For patients who were able to return to the clinic for the follow-up visit, ADA samples were also collected approximately 35 days after the last depatux-m infusion.
Depatux-m serum concentrations and ADA titers were determined using validated electrochemiluminscence immunoassays [24]. The depatux-m concentrations in the Arm C expanded cohort were compared to those in the Arm C dose escalation cohort only with intensive pharmacokinetic sampling [24].
Tumor molecular characterization
Molecular characterization of archival tumor tissue, including testing performed to determine EGFR expression, amplification, and EGFRvIII mutation status before protocol therapy was performed as described previously [24]. Briefly, fluorescence in situ hybridization (FISH) was used to detect locus-specific EGFR amplification. Two probes were employed: Vysis Locus Specific Identifier (LSI) EGFR SpectrumOrange Probe, and Vysis Chromosome Enumeration Probe (CEP) 7 SpectrumGreen Problem (Abbott Laboratories, Abbott Park, IL, USA). To call a tumor EGFR amplified, the sample should show ≥ 15% tumor cells with an EGFR/CEP 7 ratio ≥ 2.
Statistical analysis
Descriptive statistics were provided for patient demographic variables. Safety/toxicity summaries were provided for all patients who received at least one dose of depatux-m. Frequencies of adverse events (AEs) were tabulated by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, version 4.1) and listed by MedDRA (version 19) system organ class and preferred term. Responses were assessed per RANO criteria. The primary efficacy endpoint was objective response rate (ORR, complete response (CR) and partial response (PR)) and was determined for patients with measurable disease at baseline. The secondary endpoints included PFS6, PFS, OS, safety and tolerability. PFS was defined as the time period from the first dose of depatux-m to RANO-defined disease progression or date of death, if disease progression did not occur. OS was determined from the time of first dose of depatux-m to death from any cause. Ninety-five percent confidence interval (CI) was constructed for the estimated ORR (determined from the exact binomial distribution), PFS, and OS. The Greenwood formula was used to calculate the confidence limits for the quartiles of survival distribution (PFS and OS).
Results
Patient characteristics
As of 15 March 2017, enrollment was completed with 66 patients. The median age was 58 years. Forty-one percent were women and 59% were men. All patients had EGFR-amplified rGBM, and were previously treated with RT/TMZ (Table 1). Thirty-one patients (47%) received depatux-m as the first treatment after initial RT/TMZ. Thirty-one (47%) had tumors which harbored an EGFRvIII mutation (Table 1), which is similar to previously reported mutation rates of 50% in patients with EGFR-amplified GBM [8].
Safety of depatux-m
All patients received depatux-m at the RPTD of 1.25 mg/kg [25]. Sixty-four of 66 patients experienced at least one AE (Table 2). Nearly, all patients (91%) experienced at least one ocular AE. The most frequent included blurred vision (65%) and dry eye (29%). The most common non-ocular event was fatigue (33%).
Forty-two percent of patients experienced a Grade 3/4 AE, with ocular Grade 3/4 adverse events (AEs) due to microcystic keratopathy being the most common (35%, Table 3). Ocular-related Grade 3/4 AEs included keratitis (17%), corneal epithelial microcysts (8%), blurred vision (5%), and reduced visual acuity (5%). Non-ocular Grade 3/4 AEs occurred in 15% of patients. A further breakdown of all ocular AEs by grade (1/2 vs. 3/4, Supplementary Table 1) showed that the majority were Grade 1/2. Only 1 Grade 4 AE of reduced visual acuity was observed. A serious AE was observed in 36% of patients (Supplementary Table 2), with seizure (9%) occurring most frequently. Two serious AEs were assessed by the investigator as having a reasonable possibility as being attributable to depatux-m. These included one case of seizure and one case of cerebrovascular accident, which are not uncommon in patients with rGBM.
All patients had discontinued depatux-m at the time of analysis, the majority for disease progression (85%). Two patients discontinued for an AE related to progression, and eight patients (12%) discontinued for an AE unrelated to disease progression. These included four patients with ocular side effects, two with thrombocytopenia, one with proteinuria, and one with left-sided muscle weakness. Interruption of depatux-m dosing occurred in 33/66 patients (50%), with the most common reason for interruption due to ocular side effects in 25/66 patients (38%). Finally, 21 patients (32%) underwent a dose reduction of depatux-m due to an AE, with ocular AEs again the most common reason for reduction in 19/66 patients (29%). Fifty-six patients (85%) died during the course of the study.
Resolution of ocular side effects
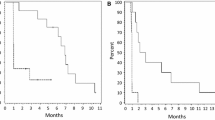
As mentioned, ocular side effects were very common in patients. The type and severity of ocular AEs varied, but all were attributed to generalized microcystic keratopathy, which is observed with some types of ADCs (see Discussion). Although the ocular side effects were common, they resulted in treatment discontinuation in only 4 patients (6%). The median time to onset of any ocular side effect was 3.2 weeks (95% CI 2.6, 3.6), as determined from all 66 patients. There was a trend toward reversibility of ocular side effects (Fig. 1); however, a precise definition of a median time to resolution could not be established, due to confounding factors including time on study.

Kaplan–Meier curve of time to resolution of ongoing ocular AEs (all grades) in patients after discontinuation of depatux-m. Time to resolution was defined as the number of days from the last dose date to the last end date of all ocular AEs
Pharmacokinetics
The PK of depatux-m in the Arm C expanded cohort was consistent with that in the Arm C dose escalation cohort [25] for both Cycle 1 and Cycle 2 (Supplementary Fig. 2). No ADA was detected or confirmed in any sample during therapy (n = 60 patients with at least one post-treatment ADA result) or at final follow-up visit (n = 13 patients with ADA results at final follow-up, which was not mandated).
Efficacy of depatux-m
Sixty of 66 patients had at least one post-baseline assessment allowing determination of change in tumor size (Fig. 2). Per RANO criteria, a best response of stable disease (SD) was observed in 27/66 patients (41%) and 34/66 patients (52%) had a best response of progressive disease (PD, Fig. 3). Of patients with measurable disease at baseline, the ORR was 6.8% (1/59 CR, 3/59 PR, 95% CI 1.9%, 16.5%). The median duration of response in 66 patients was 6.7 months (95% CI 1.6, 8.1).

The percent change in target lesion from baseline are shown for 60/66 patients who had at least one post-baseline measurement. Best tumor percent change is defined as the maximum reduction/minimum increase from baseline in tumor size. Values were determined per investigator measurements

The best responses as determined by the investigator using RANO criteria and time on depatux-m therapy are shown for 65/66 patients with available data. One patient had a baseline assessment but discontinued before the first follow-up, and is not included in this analysis
The PFS6 was 28.8% (95% CI 18.5, 39.9%) and median PFS was 1.7 months (95% CI 1.4, 3.3). The OS6 was 72.5% (95% CI 60.0, 81.7%) and median OS was 9.3 months (95% CI 6.6, 11.7). For patients with EGFRvIII mutation (n = 29 patients with available response data), the PFS6 was 17.2% (95% CI 6.3%, 32.7%) and median PFS was 1.6 months (95% CI 1.4, 3.3).
Discussion
Depatux-m monotherapy administered at the RPTD of 1.25 mg/kg in patients with EGFR-amplified rGBM demonstrated a PFS6 of 28.8% and OS6 of 72.5%, benchmarks [27] that suggest this drug could show benefits above current standard of care agents. These results, combined with a similar PFS6 of 27.1% in patients with EGFR-amplified rGBM treated with depatux-m alone or with TMZ (n = 126, which includes patients from this analysis and patients from Arm B) [28], suggest that further investigation of depatux-m in this population is warranted. Ten patients remained on treatment for more than 9 months (Fig. 3), suggesting that despite the ocular side effects, treatment was tolerated for a prolonged period of time. All 10 patients experienced typical ocular AEs, which were mainly Grade 1/2. Six of the ten had Grade 3 keratitis, one had Grade 3 corneal microcysts, and one had Grade 3 reduced visual acuity. One patient had proteinuria and one patient had significant neutropenia, both of which were managed by depatux-m dose interruption and the latter also by dose reduction.
The occurrence of microcystic keratopathy is a very predictable side effect in treatment with ADCs, particularly those with the MMAF toxin [29, 30]. It is not clear why certain ADC payloads induce such specific eye sensitivity, but it could be related to drug accumulation within various ocular tissues. Ocular side effects have been observed in the other arms of this study as well [24, 25]. Ocular side effects were generally manageable with dexamethasone eye drops, corneal bandages, dose reductions, and delays. Other prophylactic measures are being investigated, but have not yet been fully evaluated. Although ocular side effects were frequent, the majority of patients (56%, Supplementary Table 1) experienced only Grade 1/2 side effects, and only 6% of patients discontinued due to an ocular AE. Based on the details of the ten patients who had SD for more than 9 months mentioned above, the severity of ocular side effects did not correlate with time on therapy. Side effects did improve upon treatment discontinuation; however, the median time to resolution was unreliable based on data for a limited number of patients and is thus confounded by competing risks. These include patients who discontinued follow-up for progressive disease, initiated alternative therapies, or died, all of which led to a high censoring rate. Of note, six patients experienced complete resolution of ocular side effects before completion of study treatment.
Additionally, in this EGFR-amplified population, EGFRvIII mutation did not seem to further differentiate responders from non-responders, or patients likely to experience a PFS event within 6 months. Increased patient numbers are required to further understand the impact that depatux-m may have on the EGFRvIII vs. EGFR wild-type-amplified populations.
To conclude, we observed in this multicenter, dose expansion study that depatux-m monotherapy administered at the RPTD in patients with EGFR-amplified, rGBM demonstrated promising efficacy and manageable toxicity, indicating that further study of this novel targeted therapy in GBM is justified. Two other global, randomized trials are ongoing: depatux-m or placebo + RT/TMZ in EGFR-amplified, newly diagnosed GBM (INTELLANCE 1, NCT02573324); and depatux-m vs. depatux-m + TMZ vs. TMZ/lomustine in EGFR-amplified, rGBM has completed accrual with results expected in late 2017 (EORTC 1410-BTG, INTELLANCE 2, M14-483, NCT02343406).
References
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO, European Organisation for R, Treatment of Cancer Brain T, Radiation Oncology G, National Cancer Institute of Canada Clinical Trials G (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10(5):459–466. doi:10.1016/S1470-2045(09)70025-7
Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J (2014) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 16(Suppl 4):iv1-63. doi:10.1093/neuonc/nou223
Seystahl K, Wick W, Weller M (2016) Therapeutic options in recurrent glioblastoma–An update. Crit Rev Oncol Hematol 99:389–408. doi:10.1016/j.critrevonc.2016.01.018
Lamborn KR, Yung WK, Chang SM, Wen PY, Cloughesy TF, DeAngelis LM, Robins HI, Lieberman FS, Fine HA, Fink KL, Junck L, Abrey L, Gilbert MR, Mehta M, Kuhn JG, Aldape KD, Hibberts J, Peterson PM, Prados MD, North American Brain Tumor C (2008) Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol 10 (2):162–170. doi:10.1215/15228517-2007-062
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, Network TR (2013) The somatic genomic landscape of glioblastoma. Cell 155(2):462–477. doi:10.1016/j.cell.2013.09.034
Gan HK, Cvrljevic AN, Johns TG (2013) The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J 280(21):5350–5370. doi:10.1111/febs.12393
Yoshimoto K, Dang J, Zhu S, Nathanson D, Huang T, Dumont R, Seligson DB, Yong WH, Xiong Z, Rao N, Winther H, Chakravarti A, Bigner DD, Mellinghoff IK, Horvath S, Cavenee WK, Cloughesy TF, Mischel PS (2008) Development of a real-time RT-PCR assay for detecting EGFRvIII in glioblastoma samples. Clin Cancer Res 14(2):488–493. doi:10.1158/1078-0432.CCR-07-1966
Padfield E, Ellis HP, Kurian KM (2015) Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol 5:5. doi:10.3389/fonc.2015.00005
van den Bent MJ, Gao Y, Kerkhof M, Kros JM, Gorlia T, van Zwieten K, Prince J, van Duinen S, Sillevis Smitt PA, Taphoorn M, French PJ (2015) Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol 17(7):935–941. doi:10.1093/neuonc/nov013
Liang W, Wu X, Fang W, Zhao Y, Yang Y, Hu Z, Xue C, Zhang J, Zhang J, Ma Y, Zhou T, Yan Y, Hou X, Qin T, Dinglin X, Tian Y, Huang P, Huang Y, Zhao H, Zhang L (2014) Network meta-analysis of erlotinib, gefitinib, afatinib and icotinib in patients with advanced non-small-cell lung cancer harboring EGFR mutations. PLoS One 9(2):e85245. doi:10.1371/journal.pone.0085245
van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T (2009) Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol 27(8):1268–1274. doi:10.1200/JCO.2008.17.5984
Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, Fine HA, Mehta M, Deangelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD, North American Brain Tumor C (2010) A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol 12 (1):95–103. doi:10.1093/neuonc/nop015
Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, Schultz CJ, Choucair A, Dolled-Filhart M, Christiansen J, Gustavson M, Molinaro A, Mischel P, Dicker AP, Bredel M, Mehta M (2013) RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys 85(5):1206–1211. doi:10.1016/j.ijrobp.2012.10.008
Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, Shaffer D, Lis E, Abrey LE (2009) A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J Neuro Oncol 92(1):99–105. doi:10.1007/s11060-008-9741-z
Raizer JJ, Giglio P, Hu J, Groves M, Merrell R, Conrad C, Phuphanich S, Puduvalli VK, Loghin M, Paleologos N, Yuan Y, Liu D, Rademaker A, Yung WK, Vaillant B, Rudnick J, Chamberlain M, Vick N, Grimm S, Tremont-Lukats IW, De Groot J, Aldape K, Gilbert MR, Brain Tumor Trials C (2016) A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J Neuro oncol 126(1):185–192. doi:10.1007/s11060-015-1958-z
Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, Suh JH, Toms SA, Vogelbaum MA, Weil RJ, Elson P, Barnett GH (2010) Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neuro oncol 98(1):93–99. doi:10.1007/s11060-009-0067-2
Eller JL, Longo SL, Hicklin DJ, Canute GW (2002) Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery 51(4):1005–1013 (discussion 1013–1004)
Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, D’Hondt L, Strauven T, Chaskis C, In’t Veld P, Michotte A, De Greve J (2009) Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol 20(9):1596–1603. doi:10.1093/annonc/mdp032
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350(21):2129–2139. doi:10.1056/NEJMoa040938
Weller M, Butowski N, Tran D, Recht L, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust S, Iwamoto F, Drappatz J, O’Rourke D, Wong M, Finocchiaro G, Perry J, Wick W, He Y, Davis T, Stupp R, Sampson J (2016) ACT IV: an international double-blind, phase 3 trial of rindopepimut in newly diagnosed, EGFRvIII-expression glioblastoma. Neuro Oncol. doi:10.1093/neuonc/now212.068
Reilly EB, Phillips AC, Buchanan FG, Kingsbury G, Zhang Y, Meulbroek JA, Cole TB, DeVries PJ, Falls HD, Beam C, Gu J, Digiammarino EL, Palma JP, Donawho CK, Goodwin NC, Scott AM (2015) Characterization of ABT-806, a humanized tumor-specific anti-EGFR monoclonal antibody. Mol Cancer Ther 14(5):1141–1151. doi:10.1158/1535-7163.MCT-14-0820
Phillips AC, Boghaert ER, Vaidya KS, Mitten MJ, Norvell S, Falls HD, DeVries PJ, Cheng D, Meulbroek JA, Buchanan FG, McKay LM, Goodwin NC, Reilly EB (2016) ABT-414, an antibody-drug conjugate targeting a tumor-selective EGFR epitope. Mol Cancer Ther 15(4):661–669. doi:10.1158/1535-7163.MCT-15-0901
Fakih M, Vincent M (2010) Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr Oncol 17(Suppl 1):S18-30
Reardon DA, Lassman AB, van den Bent M, Kumthekar P, Merrell R, Scott AM, Fichtel L, Sulman EP, Gomez E, Fischer J, Lee HJ, Munasinghe W, Xiong H, Mandich H, Roberts-Rapp L, Ansell P, Holen KD, Gan HK (2016) Efficacy and safety results of ABT-414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro Oncol. doi:10.1093/neuonc/now257
Gan HK, Reardon DA, Lassman AB, Merrell R, van den Bent M, Butowski N, Lwin Z, Wheeler H, Fichtel L, Scott AM, Gomez EJ, Fischer J, Mandich H, Xiong H, Lee H-J, Munasinghe WP, Roberts-Rapp LA, Ansell PJ, Holen KD, Kumthekar P (2017) Safety, pharmacokinetics and antitumor response of depatuxizumab mafodotin as monotherapy or in combination with temozolomide in patients with glioblastoma. Neuro-Oncology (in press)
Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB, Tsien C, Mikkelsen T, Wong ET, Chamberlain MC, Stupp R, Lamborn KR, Vogelbaum MA, van den Bent MJ, Chang SM (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28(11):1963–1972. doi:10.1200/JCO.2009.26.3541
Han K, Ren M, Wick W, Abrey L, Das A, Jin J, Reardon DA (2014) Progression-free survival as a surrogate endpoint for overall survival in glioblastoma: a literature-based meta-analysis from 91 trials. Neuro Oncol 16(5):696–706. doi:10.1093/neuonc/not236
Lassman AB, Bent MJVD, Gan HK, Reardon DA, Kumthekar P, Butowski NA, Lwin Z, Mikkelsen T, Nabors LB, Papadopoulos KP, Penas-Prado M, Simes J, Wheeler H, Gomez EJ, Lee H-J, Roberts-Rapp L, Xiong H, Bain EE, Maag D, Merrell RT (2017) Efficacy analysis of ABT-414 with or without temozolomide (TMZ) in patients (pts) with EGFR-amplified, recurrent glioblastoma (rGBM) from a multicenter, international phase I clinical trial. J Clin Oncol 35(15_suppl):2003–2003. doi:10.1200/JCO.2017.35.15_suppl.2003
Donaghy H (2016) Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 8(4):659–671. doi:10.1080/19420862.2016.1156829
Eaton JS, Miller PE, Mannis MJ, Murphy CJ (2015) Ocular Adverse Events Associated with Antibody-Drug Conjugates in Human Clinical Trials. J Ocul Pharmacol Ther 31(10):589–604. doi:10.1089/jop.2015.0064
Acknowledgements
We thank patients and their families; study investigators and staff; Mrinal Y. Shah, PhD, for medical writing support; and Jingjing Gao, PhD, for statistical support (employees of AbbVie).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
AbbVie provided financial support for this study (NCT01800695) and participated in the design, study conduct, analysis and interpretation of the data, as well as the writing, review, and approval of the manuscript. All authors were involved in the data gathering, analysis, review, interpretation and manuscript preparation and approval.
Conflict of interest
Martin van den Bent: received honoraria from Roche, AbbVie, Celldex, Merck Ag, Cavion, Actelion, BMS, Blue Earth Diagnostics and Novartis; received research funding from AbbVie. Hui K. Gan: has an investigator-initiated study with AbbVie; received travel support and research funding from AbbVie; received honoraria from Pfizer, BMS, and Merck Serono; affiliated with the Ludwig Institute for Cancer Research. Andrew B. Lassman: in the last 12 months received personal compensation from WebMD, Sapience Therapeutics, Genentech, Italian Association for Cancer Research, AbbVie, AstraZeneca, Novocure, and Kadmon; additional travel support from Karyopharm, AstraZeneca, Abbvie, Bioclinica, Genentech, and VBI Vaccines; and research support from RTOG Foundation, Genentech, Amgen, AbbVie, Novartis, Karyopharm, Celldex, NW Biotherapeutics, Plexxicon, Pfizer, Agenus, Medimmune, Boehringer Ingelheim, Angiochem, Novocure, Stemline, E-Therapeutics, Millennium. Priya Kumthekar: Consultant for AbbVie. Ryan Merrell: Serves on a Scientific Advisory Board for AbbVie. Nicholas Butowski: Received honoraria from and has a consulting or advisory role with, Roche/Genentech, Medicenna, VBL Theraputics, Omniox; is involved in speakers’ bureaus with Roche and Merck; received research funding from Insys. Zarnie Lwin: Has served on Abbvie Scientific Advisory Board and received honoraria. Tom Mikkelsen: No potential conflicts of interest to disclose. Louis B. Nabors: Serves on a Scientific Advisory Board for Cavion, Merck, and BMS; investigator for AbbVie. Kyriakos P. Papadopoulos: Received research funding from AbbVie, MedImmune, Daiichi Sankyo, GlaxoSmithKline, Onyx, Sanofi, Novartis. Marta Penas-Prado: No potential conflicts of interest to disclose. John Simes: Received research funding for an investigator-initiated trial from AbbVie. Helen Wheeler: Investigator for AbbVie. Tobias Walbert: Serves on a Scientific Advisory Board for Novocure. Andrew M. Scott: Received research funding and travel support from AbbVie; received research funding from Daiichi-Sankyo; is a consultant and has stock in Life Science Pharmaceuticals; is affiliated with the Ludwig Institute for Cancer Research. Erica Gomez, Ho-Jin Lee, Lisa Roberts-Rapp, Hao Xiong, Earle Bain, Peter J. Ansell, Kyle D. Holen, David Maag: Employees of AbbVie and may own stock. David A. Reardon: Received honoraria from and has a consulting or advisory role with Abbvie, Bristol Myers Squibb, Cavion, Celldex, Inovio, Merck, Novartis, Roche/Genentech, Amgen, Novocure, Oxigene, Regeneron and Stemline Therapeutics; is involved in speakers’ bureaus with Roche and Merck; received research funding from Incyte, Midatech and Celldex.
Ethical approval
This article does not contain any study with animals performed by any of the authors.
Human rights and animal statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van den Bent, M., Gan, H.K., Lassman, A.B. et al. Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: results from a multi-center, international study. Cancer Chemother Pharmacol 80, 1209–1217 (2017). https://doi.org/10.1007/s00280-017-3451-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3451-1