
Abstract
The present work investigates the contribution of various second messenger systems to Ca2+-induced phosphatidylserine (PS) exposure in red blood cells (RBCs) from sickle cell disease (SCD) patients. The Ca2+ dependence of PS exposure was confirmed using the Ca2+ ionophore bromo-A23187 to clamp intracellular Ca2+ over 4 orders of magnitude in high or low potassium-containing (HK or LK) saline. The percentage of RBCs showing PS exposure was significantly increased in LK over HK saline. This effect was reduced by the Gardos channel inhibitors, clotrimazole and charybdotoxin. Nevertheless, although Ca2+ loading in the presence of an outwardly directed electrochemical gradient for K+ stimulated PS exposure, substantial exposure still occurred in HK saline. Under the conditions used inhibitors of other second messenger systems (ABT491, quinacrine, acetylsalicylic acid, 3,4-dichloroisocoumarin, GW4869 and zVAD-fmk) did not inhibit the relationship between [Ca2+] and PS exposure. Inhibitors of phospholipase A2, cyclooxygenase, platelet-activating factor, sphingomyelinase and caspases, therefore, were without effect on Ca2+-induced PS exposure in RBCs, incubated in either HK or LK saline.
Similar content being viewed by others


Introduction
Aminophospholipids including phosphatidylserine (PS) are normally confined to the inner leaflet of the plasma membrane [18]. Exposure is a pathophysiological process associated with apoptosis as PS is recognised by macrophages and other targets. A similar occurrence happens in red blood cells (RBCs) although in this case the term eryptosis is sometimes used, with that of apoptosis more appropriate for nucleated cells [18, 21]. Thus, in healthy individuals, only a small percentage of circulating RBCs show externalisation of PS. This is important because PS is prothrombotic and exposure causes RBC removal and thrombus formation [2, 11]. Increased PS exposure does occur in various disease states including sickle cell disease (SCD), in which an increased, but variable, percentage of RBCs show externalised PS, associated with both the anaemic and ischaemic complications of the condition [5, 18, 19, 26]. As such, the mechanism of exposure has received considerable attention.
In normal RBCs, asymmetrical distribution of PS is maintained by an active aminophospholipid translocase (or flippase) which transports aminophospholipids including PS from outer to inner leaflet using energy from ATP hydrolysis [10]. In addition, there is low activity of a Ca2+-dependent scrambling process (or scramblase), of which the transporter responsible remains unclear. In RBCs from SCD patients, PS exposure is associated by a rise in the activity ratio of scramblase to flippase [1] This can be caused by a number of factors but elevation of intracellular Ca2+ ([Ca2+]i) has been proposed as a significant precipitant. High [Ca2+]i will both inhibit the flippase and activate the scramblase. Recently members of the TMEM16 protein family have been identified. They may function as Ca2+-dependent phospholipid scramblases as well as Ca2+-dependent Cl- channels [35, 36], and as such may be important in PS exposure.
RBCs from SCD patients contain the abnormal haemoglobin HbS, in which the normal amino acid at the sixth position of the β chain is replaced with valine [3]. This substitution allows neighbouring HbS molecules to aggregate on deoxygenation, forming long polymers which distort RBC shape (forming the eponymous sickles and other bizarre morphologies) and have other deleterious sequelae. A further important difference with normal RBCs is a high membrane permeability to cations [14, 27, 37]. One pathway that is activated on HbS polymerisation and sickling, and which is not seen in normal RBCs, is a deoxygenation-induced cation conductance sometimes termed Psickle [13, 27, 28]. Psickle activity has several consequences, one of which is entry of Ca2+. Psickle is therefore associated with deoxygenation-induced elevation of [Ca2+] [32]. It may therefore encourage PS exposure. Indeed, we have shown previously that deoxygenation-induced PS exposure requires extracellular Ca2+ at physiological concentrations, that Ca2+ entry and elevation of [Ca2+]i is required, and that inhibitors of the Psickle pathway also reduce deoxygenation-induced PS exposure [4, 39]. PS exposure in response to elevation of Ca2+ shows a biphasic response with high and low affinity components, requiring [Ca2+]i of about 1 and 100 μM, respectively [39].
PS exposure in normal RBCs has also been extensively investigated in the context of eryptosis [21]. Several other second messenger pathways besides high [Ca2+]i have been implicated including cyclooxygenase activity and prostaglandin E2 (PGE2) [23], phospholipase A2 (PLA2), platelet-activating factor (PAF) [23], sphingomyelinase activation and ceramide production [24, 25], caspases [29], and shrinkage [20, 22, 25, 31, 33]. These other pathways may interact with entry of Ca2+ via Psickle. For example, it has been proposed in normal RBCs that PGE2 may open non-specific cation channels to mediate Ca2+ entry [15–17]. In addition, there is evidence that Ca2+-induced PS exposure also involves PKC, likely PKCα [38]. It is therefore important to establish the extent to which Ca2+-induced PS scrambling is an indirect phenomenon involving any of these other pathways. In this study, we investigated this possibility using inhibitors of other second messenger pathways. Results suggest that neither cyclooxygenase, PLA2, PAF, sphingomyelinease, nor caspases are involved. Activation of the Ca2+-activated K+ channel (or Gardos channel [9]), with subsequent hyperpolarization or RBC shrinkage, however, do increase PS exposure.
Materials and methods
Solutions and chemicals
The main experimental saline comprised (in mM): NaCl 140, KCl 4, MgCl2 0.15, N-2-hydroxyethylpiperzine-N′-2-ethanesulphonic acid (HEPES) 10 and inosine 10 with a pH of 7.4 at 37 °C (low potassium-containing saline, LK saline). For washing RBCs, a high potassium-containing saline (HK saline) was usually used, comprising (in mM) NaCl 54, KCl 90, MgCl2 0.15, HEPES 10 and inosine 10 with a pH of 7.4 at room temperature. For Ca2+ titration curves, EGTA (2 mM) and appropriate concentrations of extracellular Ca2+ ([Ca2+]os) was used to clamp free [Ca2+]o at 0, 1, 10, 100 and 1,000 μM, with total [Ca2+]os of 0, 1.91, 2.00, 2.10 and 3.00 mM, respectively. RBCs were permeabilised to Ca2+ with the ionophore bromo-A23187 (2.5–6 μM) — control experiments were carried out to determine [A23187] for maximal effects for alteration of [Ca2+]i, PS exposure and also K+ flux. In control experiments, in the absence of bromo-A23187, PS exposure over 60 min incubation changed by <2 % (n = 3) with no systematic correlation between the extent of PS exposure and the extracellular calcium concentration. The ice-cold wash solution for K+ flux measurements comprised (in mM): MgCl2 107, 3-(N-morpholino)-propanesulphonic acid (MOPS) 10, pH 7.4 at 0 °C. Lactadherin (LA)-binding buffer consisted of LK or HK HBS with the appropriate [Ca2+]o, 1 mM vanadate and 16 nM LA-FITC. Osmolality of all solutions was 290 ± 5 mOsm kg−1. Lactadherin-FITC was obtained from Haematologic Technologies Inc. (Vermont, USA) supplied via Cambridge Bioscience (Cambridge, UK), anti-glycophorin A-PE from Becton Dickinson Biosciences (California, USA), HEPES and bromo-A23187 from Calbiochem (Merck, Darmstadt, Germany), the pan caspase inhibitor zVAD-fmk from Enzo Life Sciences Ltd (Essex, UK) and 86Rb+ from PerkinElmer (MA, USA). All other chemicals came from Sigma-Aldrich (Poole, Dorset, UK). Stock solutions of ABT491 and quinacrine were made in water; those of clotrimazole, charybdotoxin, diclofenac, acetylsalicylic acid, 3,4-dichloroisocoumarin, GW4869 and zVAD-fmk in DMSO (final concentration, 0.5 %). Where it was used as a solvent, control RBCs were treated with the same [DMSO]. Control experiments were carried out to investigate the effect of inhibitors on intracellular Ca2+. For these, RBCs were loaded with 5 μM fluo-4-AM for 45 min at 37 °C in the dark (1.5 % Hct) in HK saline, washed once into HK saline containing 1 μM Ca2+, pretreated with second messenger inhibitors prior to addition of ionophore. Mean fluorescence was then measured by FACS (see below) and did not vary by > 6 % compared to controls in the absence of second messenger inhibitors, indicative of the lack of effect of inhibitors on [Ca2+]i, with the exception of RBCs incubated with GW4869 in which mean fluorescence increased by 37 %; however, in this case RBCs showed some evidence of clumping.
RBCs preparation
Blood samples were collected from HbSS SCD patients, with consent and ethical permission (11/LO/0065), in the anticoagulant EDTA. RBCs were washed three times into HK HBS for 3 min at 600×g and subsequently twice into LK or HK HBS with 2 mM EGTA to remove contaminant Ca2+. RBC suspensions, final haematocrit (Hct) of 0.5 % (except in Fig. 7, where Hct was initially 5 %), were incubated at 37 °C for 30 or 60 min in the absence or presence of various second messenger inhibitors, followed by treatment with bromo-A23187 (nominally 2.5–6 μM with separate batches titrated to establish the optimal concentration, final [DMSO] <0.5 %) at the indicated free [Ca2+]o for 30 min at 37 °C. Vanadate (1 mM) was present in the last step to inhibit both the flippase and the plasma membrane calcium pump (PMCA).
Labelling of PS exposure
For PS labelling, 5-μl aliquots (105 RBCs) of each sample were placed in 250 μl of LA-FITC binding buffer and incubated in the dark at room temperature for 10 min. RBCs were then pelleted by centrifugation for 10 s at 16,100×g, washed once in LK or HK HBS to remove unbound LA-FITC and kept on ice until flow cytometry analysis. Unlike annexin-V, LA-FITC binds to PS in a Ca2+-independent manner and control experiments showed that binding was irreversible. Inhibitors were tested for self-fluorescence at their highest concentration with unlabelled RBCs.
FACS acquisition and analysis
Externalised PS was measured with LA-FITC using an excitation wavelength of 488 nm in the FL1 channel with an emission wavelength of 519 nm on a fluorescence-activated flow cytometer (FACSCalibur, Becton Dickinson, BD) and analysed with the BD CellQuest Pro software using a protocol published previously [4]. Measurements were taken using a logarithmic gain. Forward scatter (FSC, size) and side scatter (SSC, granularity) gates for RBCs were identified in control experiments using anti-glycophorin A-PE labelled RBCs. The positive fluorescent gate was set using RBCs unlabelled with LA-FITC. For each measurement 10,000 events were gated. PS positive cells were defined as all events falling within the preset FSC, SSC and positive fluorescent gates.
K+ flux measurements
Potassium fluxes were measured using 86Rb+ as a tracer for K+ [6, 34]. RBCs (Hct about 20 %) were first equilibrated in Eschweiler tonometers (Kiel, Germany) in air at 37 °C, pH 7.4, in HK saline with added CaCl2 (10 μM) in which glucose (5 mM) was substituted for inosine and MOPS for HEPES. Aliquots were then diluted 10-fold into test tubes containing the same saline with added A23187 (nominally 4 μM). Bumetanide (10 μM) and ouabain (100 μM) were present to inhibit K+ transport through the Na+–K+–2Cl− cotransport and Na+/K+ pump. Influx was started by adding 86Rb+ (final activity about 0.05 MBq ml−1) and was performed in the absence and presence of clotrimazole (5 μM) over 10 min, to give the activity of the Ca2+-activated K+ (or Gardos) channel after which RBCs were washed four times in ice-cold isotonic MgCl2 wash solution, the final cell pellet lysed with Triton X-100 (0.1 %), protein precipitated with 5 % trichloroacetic acid and activity was measured as Čerenkov radiation by liquid scintillation (Packard Tri-carb 2100TR). Microhaematocrit determination was used to measure Hct.
Statistics
With the exception of those shown in Figs. 1a and 7, all data are presented as means ± SEM for blood samples of n different SCD patients. Statistical significance was tested with Student's t-test, and a p value <0.05 was considered significant.

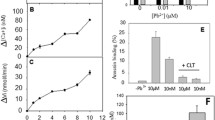
Ca2+-induced exposure of phosphatidylserine (PS) in red blood cells (RBCs) from patients with sickle cell disease (SCD). a Time course: RBCs were treated with the ionophore bromo-A23187 (2.5–6 μM) for up to 60 min in high potassium-containing (HK) saline with 100 μM extracellular [Ca2+] ([Ca2+]o). Symbols represent duplicate measurements from a single sample. b Ca2+ dependence: RBCs (0.5 % haematocrit, Hct; final [DMSO] <1 %) were treated with ionophore (2.5–6 μM) for 30 min, in HK or low potassium-containing (LK) saline, with [Ca2+]o varied between 0 μM and 1 mM. Symbols represent means ± SEM, n = 12. *p < 0.05 and **p < 0.002 between PS exposure in RBCs incubated in HK or LK saline
Results
Ca2+ dependence of PS exposure of RBCs from patients with SCD
The Ca2+ dependence of PS scrambling was confirmed using the ionophore bromo-A23187 to clamp intracellular [Ca2+]i. Due to the Donnan ratio, this method clamps [Ca2+]i at about double the value of [Ca2+]o [7, 30]. PS scrambling occurred within minutes with a plateau achieved by 20 min (Fig. 1a). In all subsequent experiments, therefore, RBCs were treated with ionophore for 30 min before labelling for PS. Elevation of [Ca2+]i, as well as causing PS scrambling, will also lead to activation of the Ca2+-activated K+ channel (or Gardos channel), and possibly a Ca2+-dependent Cl- channel [35]. In low potassium-containing (LK) salines, Gardos channel activation will result in loss of K+ and shrinkage and hyperpolarisation of the RBC membrane [12]. High potassium-containing salines (HK saline) were used to prevent these secondary effects. Typically, a biphasic response was apparent in both HK and LK salines with the major component of PS scrambling (accounting for about two-thirds of the total) occurring with high Ca2+ affinity (about 1 μM) and a minor component with low Ca2+ affinity (about 100 μM, Fig. 1b), as previously reported [39]. PS exposure was significantly increased in LK HBS. The Ca2+ affinities, however, appeared unaltered (Fig. 1b). These Ca2+ titration curves of PS exposure were subsequently used to investigate the effect of inhibitors of other second messengers, with RBCs left as untreated controls or treated with second messenger inhibitors for 30 min prior to and also during alteration of [Ca2+]i with ionophore.
The effect of inhibitors of second messengers in HK saline
The effect of diclofenac (10–500 μM) and acetylsalicylic acid (50–200 μM) as inhibitors of cyclooxygenase, ABT491 (1–50 μM) as an antagonist of PAF receptors, quinacrine (25 and 100 μM) as a non-specific PLA2 inhibitor, and two sphingomyelinase inhibitors, 3,4-dichloroisocoumarin (20 and 200 μM) — a serine proteinase inhibitor — and GW4869 (100 nM–10 μM), both active against neutral sphingomyelinase, were tested on PS scrambling in HK saline. Results are summarised in Table 1 for the highest inhibitor concentration used while Fig. 2 shows the effect of ABT491 and of acetylsalicylic acid over the full range of concentrations used. None of these reagents decreased the extent of PS exposure in the Ca2+ titration curves. Diclofenac and GW4896, however, showed a significant increase in PS exposure at some, but not all, concentrations (Table 1). For GW4896, this did not require the presence of Ca2+. In the presence of GW4896 at 10 μM there was some indication of RBC clumping which may have artefactually increased PS exposure. Such increases were not present for the alternative cyclooxygenase and sphingomyelinase inhibitors, acetylsalicylic acid and 3,4-dichloroisocoumarin.

Effect of platelet-activating factor (PAF) and cyclooxygenase inhibitors on Ca2+-induced PS exposure in RBCs from SCD patients. RBCs were incubated in HK saline in the presence or absence of inhibitor for 30 min prior to treatment with ionophore for 30 min (as in Fig. 1b). a Effect of treatment with the PAF inhibitor ABT491 (1–50 μM). b Effect of treatment with the cyclooxygenase inhibitor acetylsalicylic acid (50–200 μM). Symbols represent means ± SEM, n = 3, except for 5 μM ABT491, where n = 2. There was no significant difference between values in the presence or absence (controls) of the inhibitors
The effect of Gardos channel activity on PS exposure of RBCs from SCD patients
In the next series of experiments, the effect of Ca2+-induced K+ efflux via the Gardos channel was investigated. When RBCs were incubated with the Gardos channel inhibitor clotrimazole (5–20 μM, results shown for 20 μM only), PS exposure in LK saline was reduced (Fig. 3a). Clotrimazole, however, had no effect on PS exposure in RBCs incubated in HK saline (Fig. 3a). Charybdotoxin (600 nM), a spider venom which inhibits the Gardos channel with higher affinity than clotrimazole, was also tested. Similar to clotrimazole, PS exposure in LK saline was reduced to levels observed in HK saline (Fig. 3b). The inhibitory effect of these drugs on Ca2+-activated K+ fluxes across the RBC membrane was confirmed. With A23187-treated RBCs and [Ca2+]o of 10 μM Ca2+, K+ influx increased to 386 ± 79 mmol (l cells h)−1 being reduced to 18 ± 2 (means ± SEM, n = 9) in the presence of 5 μM clotrimazole, an inhibition of 95 %. These findings are consistent with activation of the Gardos channel with an outwardly directed K+ gradient responsible for elevation of PS exposure in LK saline in RBCs permeabilised to Ca2+, as has been observed in RBCs from normal individuals [22, 31].

Involvement of the Ca2+-activated K+ channel (Gardos channel) in Ca2+-induced PS exposure in RBCs from SCD patients. RBCs were treated with ionophore for 30 min (as in Fig. 1b). a RBCs were incubated in either HK or LK HEPES-buffered saline (HBS) in the absence and presence of 20 μM clotrimazole (CLT). Symbols represent means ± SEM, n = 5; black circles LK HBS, black squares LK HBS with CLT (20 μM), grey triangles HK HBS and grey diamonds HK HBS with CLT (20 μM). *p < 0.03 between PS exposure in RBC incubated in LK saline in the presence and absence of CLT, # p < 0.05 between LK and HK saline. b RBCs were incubated in HK or LK saline, in the absence and presence of charybdotoxin (600 nM). Histograms represent means ± SEM, n = 3. *p < 0.05
The effect of inhibitors of second messengers in LK saline
Although Gardos channel activity likely accounts for the higher PS exposure in LK saline (compared to HK saline), other second messenger pathways may also be involved. The various inhibitors were therefore tested on the augmented PS exposure observed in LK saline. PS exposure was measured in LK saline in ionophore-treated RBCs at 1, 10 and 100 μM [Ca2+]o at the highest concentration of inhibitors used in HK saline (Figs. 4, 5 and 6). In all, there was a significant increase in PS exposure when comparing RBCs incubated in LK with those in HK saline. Again, however, there was no significant difference in PS exposure in the absence or presence of diclofenac (500 μM), acetylsalicylic acid (200 μM), quinacrine (100 μM) or 3,4-dichloroisocoumarin (200 μM). While there was an increase in PS exposure (p < 0.05) with ABT491 (50 μM) at a free [Ca2+]i of 10 μM and GW4869 (10 μM) at a free [Ca2+]i of 100 μM PS exposure at the other [Ca2+]is was unchanged. However, none of the drugs used caused an inhibition of PS exposure. Finally, the effect of the pan caspase inhibitor zVAD-fmk (60 μM) was investigated (Fig. 7) in LK saline. Again, PS exposure was unaltered.

Effect of cyclooxygenase inhibitors on Ca2+-induced PS exposure in RBCs from SCD patients. RBCs were incubated in HK saline, LK saline or LK saline plus inhibitor for 30 min before treatment with ionophore for 30 min (as in Fig. 1b). a Effect of diclofenac (500 μM). b Effect of acetylsalicylic acid (200 μM). Histograms represent means ± SEM, n = 3. *p < 0.05

Effect of platelet-activating factor (PAF) and phospholipase A2 (PLA2) inhibitors on Ca2+-induced PS exposure in RBCs from SCD patients. RBCs were incubated in HK saline, LK saline or LK saline plus inhibitor for 30 min before treatment with ionophore for 30 min (as in Fig. 1b). a Effect of the PAF inhibitor ABT491 (50 μM). Histograms represent means ± SEM, n = 7. # p < 0.05, *p < 0.005. b Effect of the PLA2 inhibitor quinacrine (100 μM). Histograms represent means ± SEM, n = 3. *p < 0.03

Effect of sphingomyelinase (SMase) inhibitors on Ca2+-induced PS exposure in RBCs from SCD patients. RBCs were incubated in HK saline, LK saline or LK saline plus inhibitors for 30 min before treatment with ionophore for 30 min (as in Fig. 1b). a Effect of the Mg2+-dependent neutral SMase inhibitor GW4869 (10 μM). Histograms represent means ± SEM, n = 6. b Effect of the SMase inhibitor 3,4-dicloroisocoumarin (200 μM). Histograms represent means ± SEM, n = 6. *p < 0.03, # p < 0.05

Effect of the caspase inhibitor zVAD-fmk on Ca2+-induced PS exposure in RBCs from SCD patients. RBCs (5 % Hct) were incubated in LK saline for 60 min in the absence or presence of zVAD-fmk (60 μM) prior to treatment with ionophore (30 min, at 0.5 % Hct, as in Fig. 1b). Results are from a single experiment representative of four different SCD patients
Discussion
The present work investigates the contribution of various other second messenger systems to Ca2+-induced PS exposure in RBCs from SCD patients. Inhibitors of cyclooxygenase, PAF, PLA2, sphingomyelinase and caspases were without effect. By contrast, the percentage of RBCs showing PS exposure was significantly increased under circumstances which allowed K+ exit through the Ca2+-activated K+ channel (or Gardos channel). This effect was inhibited by the Gardos channel inhibitors, clotrimazole and charybdotoxin.
The assay chosen for the current investigation was a Ca2+ titration curve in which RBCs were treated with bromo-A23187 and different levels of extracellular Ca2+ to alter the free [Ca2+]i between 0.1 μM and 1 mM. This allows the visualisation of both high and low Ca2+ affinities of PS scrambling [4, 39], a process that begins within 5 min and is complete within 20 min. The assay used enables any effects of second messenger inhibitors to be observed as either a change in Ca2+ affinity of the scrambling process or as a change of the percentage of RBCs showing PS exposure. Under the conditions chosen, however, Ca2+-induced scrambling did not appear to involve cyclooxygenase, PAF, PLA2, sphingomyelinase and caspases in either HK saline or in LK saline. Increases in PS exposure with diclofenac and GW4869 did occur but were variable, inconsistent between HK and LK salines, and not observed with alternative inhibitors of the same pathways (acetylsalicylic acid and 3,4-dichloroisocoumarin), perhaps indicative of a non-specific effect. The time taken for RBCs to traverse the microvasculature in vivo will usually be seconds to minutes, so these findings suggest that relatively brief elevations of intracellular [Ca2+] would probably need to act directly if they are to stimulate the scrambling process.
PS exposure involved in the process of RBC eryptosis has received considerable attention and has emerged as a complex event with many potential stimuli. Cyclooxygenase and PGE2 [8, 23], PAF and PLA2 [23], sphingomyelinase and ceramide [24, 25] and caspases [29] have all been implicated in PS exposure using inhibitor studies and reagents similar to those described here. These previous reports differ in the use of normal human (HbAA) RBCs, with one exception [26]. They also usually involve relatively prolonged incubation of RBCs with reagents tested on PS exposure over several hours to days (e.g., [25, 26]). The shorter term effects described in the current work may be more relevant to the initial trapping of PS-exposing RBCs via interaction with phagocytes or activated endothelial cells. The Ca2+ signal alone would appear to contribute to the initial PS exposure under these conditions, with other important pathways investigated in the present work becoming involved more slowly and playing an additional role in stimulating PS exposure.
Gardos channels have previously been shown to increase PS exposure in response to Ca2+ loading in normal (HbAA) human RBCs [22, 31]. In addition, treatment with the K+ ionophore valinomycin to increase K+ permeability directly also increases PS exposure [33]. We observed similar effects in RBCs from SCD patients. Thus in contrast to cyclooxygenase, PAF, PLA2, sphingomyelinase and caspase activity, activation of the Gardos channel in the presence of an outwardly directed electrochemical gradient for K+ was associated with a rise in the percentage of RBCs showing PS exposure. The rise of PS was inhibited by removing the gradient for K+ loss (HK saline) or by inhibition of the channel directly with either clotrimazole or charybdotoxin, as shown previously in RBCs from normal individuals [22, 31]. When activated, K+ channels will cause hyperpolarization of the RBC membrane as well as solute loss and shrinkage [12]. It has been suggested that the increased negative potential difference acts to increase Ca2+ entry and hence elicit PS exposure [22]. The lack of effect on Ca2+ affinity in the titration curves (e.g., Fig. 1b), however, suggests that this is not the case. It is also important to realise, nevertheless, that the considerable PS exposure in HK saline implies that K+ efflux and its sequelae are not prerequisites of Ca2+-induced PS exposure. Similarly, although high K+ may directly inhibit scrambling [40], this effect was not sufficient to prevent considerable PS exposure in RBCs from SCD patients.
In conclusion, Ca2+-induced PS exposure in RBCs from SCD patients does not appear to involve a number of second messengers in the time scale of the present experiments. K+ efflux, probably via shrinkage, however, does increase externalization of PS.
References
Barber LA, Palascak MB, Joiner CH, Franco RS (2009) Aminophospholipid translocase and phospholipid scramblase activities in sickle erythrocyte subpopulations. Br J Haematol 146:447–455
Boas FE, Forman L, Beutler JA (1998) Phosphatidylserine exposure and red cell viability in red cell ageing and in hemolytic anemia. Proc Natl Acad Sci U S A 95:3077–3081
Bunn HF, Forget BG (1986) Hemoglobin: molecular, genetic and clinical aspects. Saunders, Philadelphia
Cytlak UM, Hannemann A, Rees DC, Gibson JS (2013) Identification of the Ca2+ entry pathway involved in deoxygenation-induced phosphatidylserine exposure in red blood cells from patients with sickle cell disease. Pflugers Archiv - European Journal of Physiology
de Jong K, Larkin SK, Styles LA, Bookchin RM, Kuypers FA (2001) Characterization of the phosphatidylserine-exposing subpopulations of sickle cells. Blood 98:860–867
Dunham PB, Ellory JC (1981) Passive potassium transport in low potassium sheep red cells: dependence upon cell volume and chloride. J Physiol 318:511–530
Flatman PW (1980) The effect of buffer composition and deoxygenation on the concentration of ionized magnesium inside human red blood cells. J Physiol 300:19–30
Foller M, Kasinathan RS, Duranton C, Wieder T, Huber SM, Lang F (2006) PGE2-induced apoptotic cell death in K562 human leukaemia cells. Cell Physiol Biochem 17:201–210
Gardos G (1958) The function of calcium and potassium permeability of human erythrocytes. Biochimica Biophysica Acta 30:653–654
Haest CWM (2003) Distribution and movement of membrane lipids. In: Bernhardt I, Ellory JC (eds) Red cell membrane transport in health and disease. Springer Verlag, Berlin, pp 1–25
Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH (1980) Erythrocyte adherence to endothelium in sickle cell anaemia: a possible determinant of disease severity. New Engl J Med 302:992–995
Hoffman JF, Joiner W, Nehrke K, Potapova O, Foye K, Wickrema A (2003) The hSK4 (KCNN4) isoform is the Ca2+-activated K+ channel (Gardos channel) in human red blood cells. Proc Natl Acad Sci U S A 100:7366–7371
Joiner CH (1990) Deoxygenation-induced cation fluxes in sickle cells: II Inhibition by stilbene disulfonates. Blood 76:212–220
Joiner CH (1993) Cation transport and volume regulation in sickle red blood cells. Am J Physiol 264:C251–C270
Kaestner L (2011) Cation channels in erythrocytes — historical and future perspective. Open Biol J 4:27–34
Kaestner L, Bernhardt I (2002) Ion channels in the human red blood cell membrane: their further investigation and physiological relevance. Bioelectrochemistry 55:71–74
Kaestner L, Tabellion W, Lipp P, Bernhardt I (2004) Prostaglandin E2 activates channel-mediated calcium entry in human erythrocytes: an indication of a blood clot formation supporting process. Thromb Haemost 92:1269–1272
Kuypers FA (2008) Red cell membrane lipids in hemoglobinopathies. Curr Mol Med 8:633–638
Kuypers FA, Lewis RA, Hua M, Schott MA, Discher D, Ernst JD, Lubin BH (1996) Detection of altered membrane phospholipid asymmetry in subpopulations of human red blood cells using fluorescently labeled Annexin V. Blood 87:1179–1187
Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D (1998) Functional significance of cell volume regulatory mechanisms. Physiol Rev 78:247–306
Lang F, Gulbins E, Lerche H, Huber SM, Kempe DS, Foller M (2008) Eryptosis, a window to systemic disease. Cell Physiol Biochem 22:373–380
Lang PA, Kaiser S, Myssina S, Wieder T, Lang F, Huber SM (2003) Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am J Cell Physiol 285:C1553–C1560
Lang PA, Kempe DS, Myssina S, Tanneur V, Birka C, Laufer S, Lang F, Wieder T, Huber SM (2005) PGE2 in the regulation of programmed erythrocyte death. Cell Death Differ 12:415–428
Lang PA, Kempe DS, Tanneur V, Eisele K, Klarl BA, Myssina S, Jendrossek V, Ishii S, Shimizu T, Waidmann M, Hessler G, Huber SM, Lang F, Wieder T (2005) Stimulation of erythrocyte ceramide formation by platelet-activating factor. J Cell Sci 118:1233–1243
Lang KS, Myssina S, Brand V, Sandu C, Lang PA, Berchtold S, Huber SM, Lang F, Wieder T (2004) Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ 11:231–243
Lang KS, Roll B, Myssina S, Schittenhelm M, Scheel-Walter H-G, Kanz L, Fritz J, Lang F, Huber SM, Wieder T (2002) Enhanced apoptosis in sickle cell anemia, thalassemia and glucose-6-phosphate dehydrogenase deficiency. Cell Physiol Biochem 12:365–372
Lew VL, Bookchin RM (2005) Ion transport pathology in the mechanism of sickle cell dehydration. Physiol Rev 85:179–200
Ma Y-L, Rees DC, Gibson JS, Ellory JC (2012) The conductance of red blood cells from sickle cell patients: ion selectivity and inhibitors. J Physiol 590:2095–2105
Mandal D, Mazumber A, Das P, Kundu M, Basu J (2005) Fas-, caspase-8, and caspase-3-dependent signalling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J Biol Chem 280:39460–39467
Muzyamba MC, Campbell EH, Gibson JS (2006) Effect of intracellular magnesium and oxygen tension on K+–Cl− cotransport in normal and sickle human red cells. Cell Physiol Biochem 17:121–128
Nguyen DB, Wagner-Britz L, Maia S, Steffen P, Wagner C, Kaestner L, Bernhardt I (2011) Regulation of phosphatidylserine exposure in red blood cells. Cell Physiol Biochem 28:847–856
Rhoda MD, Apovo M, Beuzard Y, Giraud F (1990) Ca2+ permeability in deoxygenated sickle cells. Blood 75:2453–2458
Schneider J, Nicolay JP, Foller M, Wieder T, Lang F (2007) Suicidal erythrocyte death following cellular K+ loss. Cell Physiol Biochem 20:35–44
Speake PF, Roberts CA, Gibson JS (1997) Effect of changes in respiratory blood parameters on equine red blood cell K–Cl cotransporter. Am J Physiol 273:C1811–C1818
Suzuki J, Fuji T, Imao T, Ishihara K, Kuba H, Nagata S (2013) Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem 288:13305–13316
Suzuki J, Umeda M, Sims PJ, Nagata S (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468:834–838
Tosteson DC, Shea E, Darling RC (1952) Potassium and sodium of red blood cells in sickle cell anaemia. J Clin Investig 48:406–411
Wagner-Britz L, Wang J, Kaestner L, Bernhardt I (2013) Protein kinase Ca and P-type Ca2+ channel Cav2.1 in red blood cell calcium signalling. Cell Physiol Biochem 31:883–891
Weiss E, Cytlak UM, Rees DC, Osei A, Gibson JS (2012) Deoxygenation-induced and Ca2+-dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell Calcium 51:51–56
Wolfs JLN, Comfurius P, Bekers O, Zwaal RFA, Balasubramianian K, Schroit AJ, Lindhout T, Bevers EM (2009) Direct inhibition of phospholipid scrambling activity in erythrocytes by potassium ions. Cell Mol Life Sci 66:314–323
Acknowledgements
We thank MRC, Action Medical Research and British Heart Foundation for financial support. OTG is supported through the generosity of a Yousef Jameel Scholarship. UMC is a recipient of a BBSRC studentship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Gbotosho, O.T., Cytlak, U.M., Hannemann, A. et al. Inhibitors of second messenger pathways and Ca2+-induced exposure of phosphatidylserine in red blood cells of patients with sickle cell disease. Pflugers Arch - Eur J Physiol 466, 1477–1485 (2014). https://doi.org/10.1007/s00424-013-1343-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-013-1343-8