
Abstract
Since the 1980s, the MAO-B inhibitors have gained considerable status in the therapy of the Parkinson’s disease. In addition to the symptomatic effect in mono- and combination therapies, a neuroprotective effect has repeatedly been a matter of some discussion, which has unfortunately led to a good many misunderstandings. Due to potential interactions, selegiline has declined in significance in the field. For the MAO-B inhibitor safinamide, recently introduced to the market, an additional inhibition of pathological release of glutamate has been postulated. At present, rasagiline and selegiline are being administered in early therapy as well as in combination with levodopa. Safinamide has been approved only for combination therapy with levodopa when motor fluctuations have occurred. MAO-B inhibitors are a significant therapeutic option for Parkinson’s disease, an option which is too often not appreciated properly.
Similar content being viewed by others
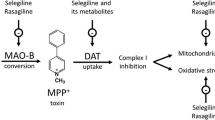
Introduction
MAO-A (Monoamine oxidase inhibitor-B) inhibitors are used in anti-depressive therapy and MAO-B inhibitors in Parkinson therapy. At present, the MAO-B inhibitors selegiline, rasagiline and safinamide have been approved for treatment of Parkinson’s disease. Clinical studies were initiated in the 1970s (Knoll 1978; Riederer et al. 1978), with the original idea of Riederer and Youdim (Tábi et al. 2020). Selegiline was then introduced in the 1980s and 20 years later rasagiline internationally. Finally, safinamide was approved in numerous countries and launched on the market as of 2015.
Selegiline
The first description of a levodopa-reinforcing or levodopa-reducing effect appeared in the 1970s, the decade when the first approval for levodopa preparations was granted in combination with a decarboxylase inhibitor (Birkmayer et al. 1985; Knoll 1978). Selegiline, also known since its introduction as l-Deprenyl, has been approved for mono- and combination therapies.
Selegiline is a relatively selective, irreversible inhibitor for central MAO-B. The half-life period of the substance ranges around 40 h, and this short period distinguishes it clearly from the extremely long half-life of 40 days in the case of cerebral MAO-B. This longer half-life in turn results from a neosynthesis of the enzyme because selegiline is an example of a so-called “suicide” inhibitor (Fowler et al. 1987, 1994). And precisely, this long time of recovery should be taken into account when administering selegiline with other drugs in PD patients.
Selegiline is rapidly reabsorbed and demonstrates a strong plasma–protein binding. Due to a high degree of lipophilicity, the blood–brain barrier can easily be overcome. Studies have shown that after intravenous administration, a maximum concentration can already be measured in the striatum after only five minutes (Fowler et al. 1987]. A maximum in plasma concentration is reached after 30 to 120 min. The substance demonstrates a high first-pass effect and is mainly metabolized in the liver (probably the cytochrome-P450-system) and eliminated renally.
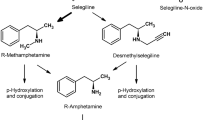
Metabolisation proceeds over methamphetamine to amphetamine whereby under therapeutic doses no relevant amphetamine effect is to be expected (Fig. 1).

Metabolism of selegiline and rasagiline (after Chen et al. 2007)
As a further preparation, a dissolving tablet was also available. This preparation was given at a lower dose, and the switch from traditional selegiline proved unproblematical (Ondo et al. 2011). A dissolving tablet (lyophilisate) contains 1.25 mg of selegiline HCL and corresponds to 10 mg in the usual selegiline tablet form (comparative plasma concentrations) (Clarke et al. 2003; Saeger 1998). The advantage of the sublingual application lies in by-passing first-pass metabolism and thus in avoiding amphetamine derivates. At present, the preparation is not available.
Mode of action of selegiline
The first step in increasing cerebral levodopa concentrations was to inhibit decarboxylase. Thus, the attempt was made, in a second step, to inhibit the cerebral metabolism of levodopa by means of extra- and intraneuronal MAO. This idea was largely driven by Peter Riederer (Riederer et al. 1978). Clinical research succeeded with selegiline (deprenyl). Under the use of this selective MAO-B inhibitor, the dopamine concentrations were increased in the synaptic gap. In addition, a weak inhibition of dopamine uptake occurs.
To achieve the pharmacodynamic effect, the MAO-B has to be inhibited by at least 80% (Green et al. 1977). In therapeutic doses (10 mg), MAO-B is completely inhibited in thrombocytes. This then is the rationale for deciding on the routine dosage. The recommendation of using 1 mg of selegiline per 10 kg of body weight is relatively arbitrary and has not been scientifically corroborated. In principle, the decision for five or 10 mg is not based on scientific data (LeWitt 2009).
Clinical effects of selegiline
Selegiline has been approved for both mono- and combination therapies. The symptomatic effect of monotherapy can be assessed as low to moderate, but the substance achieves a better rating in combination with levodopa. In that case, selegiline demonstrates, among others, a levodopa-smoothing effect and is beneficial for treating end-of-dose akinesia. The usual dose is 10 mg/die, given in either one or two portions. Higher doses are not recommended (see above).
In a recent study, the UPDRS score improved under selegiline monotherapy by 6.26 ± 7.86 (versus 3.14 ± 6.98 with a placebo) (Mizuno et al. 2017). In addition to the symptomatic effect, both a levodopa-saving effect and a positive effect on late motor complications have been described for selegiline (Dashtipour et al. 2015). There are also long-term studies which describe an increased rate of dyskinesia, which may best be explained by the dopaminergic effect (Shoulson et al. 2002). Freezing phenomena is said to occur less frequently (Shoulson et al. 2002; Zhang et al. 2016).
Data on the amount of levodopa saved differ strongly. Most studies claim an average savings effect of 20 und 30%. It is noteworthy that higher savings are seen especially in open studies. Here the overall dosage has to be taken into consideration. A saving of 20% under an overall dose of 300 mg of levodopa has to be seen as substantially different when compared to an overall dose of 1000 mg.
A double-blind study by Myllylä et al. (1997) of 5 years duration was able to demonstrate that without selegiline 725 mg of levodopa were necessary, while with selegiline only 405 mg of levodopa were required, that is, 45% less. In this study, the effect even increased over time (Myllylä et al. 1997).
Various publications from the DATATOP study (PSG 1989a, b, 1993, 1998) have frequently been cited. The most relevant finding was that with the administration of selegiline, giving levodopa can be delayed by several months (PSG 1989a, b, 1993, 1998, Tertrud and Langston 1989). But the conclusion that a neuroprotective effect is at work has to be viewed critically.
The effect of selegiline has been described as most noticeable for the first year (PSG 1989a) and as declining in long-term use (Rinne 1987; Yahr 1989). This has led to the frequent conclusion that selegiline should only be administered in the initial stage of the disease. But the results from Myllylä et al. (1997) as well as those from the SELEDO study (Przuntek et al. 1999) and one from Pålhagen et al. (2006) are in clear contradiction here. There is no decline over time. In addition, being convinced that the substance does have a neuroprotective effect automatically implies that its discontinuation in the course of treatment does not make sense. The long-term study by Mizuno et al. (2019) exhibited the best results after 20 weeks, whereby however the side effects were relatively strong (44.3%), and only 67.9% of the patients could be treated for more than 56 weeks (while in the control phase, tolerability was very good (Mizuno et al. 2017)).
In this context, a publication from Larsen et al. (1999) should be mentioned: In this double-blind study extending five years, the levodopa savings (of 424 vs. 506 mg/day) and in addition a milder course of the disease could be demonstrated. The mild course became even more apparent with time and persisted after discontinuation of selegiline. This effect cannot simply be explained by the slight symptomatic effect and may perhaps be an indication of a neuroprotective effect.
In therapeutic, but especially in over-therapeutic doses, an antidepressant effect has been observed, but is not covered by sufficient study data.
Adverse effects of selegiline
The most frequent adverse effect is insomnia which particularly occurs when the substance is given in the late afternoon or the evening. For this reason, drug administration is recommended for the morning or at noon post-prandial. In combination with levodopa, confusional states and hallucinations can occur, and in addition, levodopa-induced hyper- or dyskinesias can be aggravated.
As opposed to the d-form, the therapeutically used l-form has no relevant amphetaminergic effect. Potential for addiction does not exist for selegiline (Yasar et al. 1996).
Interactions of selegiline
One interaction holds for tyramine. The much-feared “cheese-effect” (which can, among others, induce hypertensive crises) is not to be expected under therapeutic doses. At most a minor amplification occurs in the sympathicomimetic effect of tyramine.
Unfortunately, selegiline exhibits further medicinal interactions (Csoti et al. 2012). The substance should not be given with sympathomimetic drugs, dampening neuropharmacological drugs and pethidin.
Caution is further recommended when jointly combining the substance with SSRI (serotonin reuptake inhibitors). The risk thereby is admittedly slight, but in combination with SSRI a reinforcement of the serotonergic effect can occur. Before giving selegiline, SSRI should be discontinued at the least 2 weeks beforehand, and in the case of fluoxetine by even as much as 5 weeks. In addition to SSRI, selegiline should not be combined with MAO-A inhibitors, triptans (in particular rizatriptan) and sibutramine.
In their material on technical information, the manufacturers make mention of numerous contraindications when combining selegiline with levodopa.
-
hypertonia
-
hyperthyreosis
-
phaeochromocytoma
-
narrow angle glaucoma
-
prostatic hyperplasia with residual urine
-
tachykardia
-
cardiac arrhythmias
-
angina pectoris
-
psychosis
-
dementia
Contraindications for selegiline
Contraindications are listed for: severe hypertonia, cardiac arrhythmias and severe angina pectoris. Patients who have had an exogenic psychosis or who have extensive cerebral damage or advanced dementia should not be treated with selegiline. Further contraindications include patients with gastric or duodenal ulcers.
Rasagiline
Rasagiline (N-Propargyl-1-[R]-aminoindan) is a reversible, selective MAO-B inhibitor (Finberg et al. 1996) and has five to ten times stronger MAO-B inhibition than selegiline (Finberg et al. 1996; Youdim et al. 2001). Its bioavailability reaches approximately 36% and is not compromised in any way by food ingestion. Metabolisation occurs mainly in the liver, and the metabolite is aminoindane, not amphetamine derivates (see Fig. 1). The precise significance of aminoindane has not yet been clarified (Müller and Reichmann 2012).
In the meantime, a number of studies have been published on rasagiline, some of which extended over several years (Hauser et al. 2016; Jankovic et al. 2014; Jiang et al. 2020; Lew et al. 2010; PSG 2002; Peretz et al. 2016; Rascol et al. 2005, 2016; Stern et al. 2004), notably: TEMPO, PRESTO, LARGO und ADAGIO:
The TEMPO study (Hauser et al. 2016; PSG 2002), which included 404 patients, aimed at identifying the effect and the safety of the medication in monotherapy. A distinct improvement in motor symptoms could be demonstrated for both the 1 mg and the 2 mg dose. The study extension proved interesting: Here all the patients, even the placebo group, received rasagiline (PSG 2004). After a year, the two groups differed profoundly, showing a decided advantage for the patients who had been given rasagiline from the very start. In the course of the trial period, just short of half, the patients received monotherapy after 2 years, 23% after four years and 13% after 6 years (Lew et al. 2010).
LARGO (Rascol et al. 2005) and PRESTO (PSG 2005) are studies which clearly demonstrated that rasagiline shows a good effect in combination therapy as well (for example with an increase in on-time). The LARGO study was able to show that the effects of rasagiline and entacapone are comparable (Rascol et al. 2005). According to these studies, the savings effect for levodopa in the case of rasagiline is higher than for selegiline.
The ADAGIO study (Hauser et al. 2016; Jankovic et al. 2014; Olanow et al. 2009; Rascol et al. 2011), a prospective study with delayed-start design, enrolled 1176 patients who had been diagnosed with Parkinson’s disease for an average of four-and-a-half months (SD ± 4.6) and had an UPDRS score of 20.4 (SD ± 8.5). The study extended over a period of 72 weeks and was able to demonstrate that with 1 mg of rasagiline the clinical course was significantly better, even after the placebo group had been given rasagiline after 36 weeks. The group with 2 mg failed to reach the study objective. There was considerable speculation on the reasons for this effect, and finally the so-called “floor effect” was favored, indicating that the diagnostic sensitivity of the UPDRS scale was insufficient in the lower range of the scale. 683 patients were kept under further observation. Unfortunately, there was no significant difference between the two groups (Rascol et al. 2016).
Rasagiline has a symptomatic mode of treatment in the early as well in the later stages (McCormack 2014). Clinical experience reveals that a major percentage of patients profits from administering rasagiline in vigilance, mood and quality of life. A rather large open study has substantiated these observations (Jost et al. 2008). In another study (Barone et al. 2015), the effect of rasagiline on depression in PD patients is less clear: At week 4, there was a significant difference in favor of rasagiline, while at week 12, rasagiline did not differ from placebo in improving depressive symptoms, as assessed by the Beck Depression inventory scale. These studies demonstrated improvements not only in motor behavior and non-motor disturbances, but also importantly in cognition and other non-motor aspects (Hanagasi et al. 2011; Olanow et al. 2009). One study that targeted changes in cognitive functioning under rasagiline however could not demonstrate any improvement (Weintraub et al. 2016). Rasagiline has been administered successfully in older patients (Tolosa and Stern 2012). In a study on preladenant, rasagiline was selected as the active comparison, and interestingly neither of the two substances, not even rasagiline, was superior to the placebo (Stocchi et al. 2017).
Some patients may experience a delay in the onset of action which means that a final evaluation of the therapy can only be done after up to four weeks of the intervention.
The tolerability of rasagiline is categorized as good (Jiang et al. 2020), and the adverse reactions correspond to those of selegiline (both substances are irreversible MAO-B inhibitors). The substance is well tolerated by older patients (Goetz et al. 2006). There is discussion on potential interactions with foodstuffs containing teramine, but this appears to be merely a theoretical risk (deMarcaida et al. 2006; LeWitt 2009). A “serotonine effect” of rasagiline is also possible, but basically very improbable (Montgomery and Panisset 2009; Panisset et al. 2014); the STACCATO study at any rate found no such symptoms in 471 patients (Panisset et al. 2014). Patients in the ADAGIO study profited from a combination of an SSRI with rasagiline (Smith et al. 2015). Nonetheless, combining with SSRI requires some amount of caution.
Health economic evaluations indicate that rasagiline can be classified as beneficial (Farkouh et al. 2012; Haycox et al. 2009; Hudry et al. 2006; McCormack 2014).
Safinamide
Safinamide was introduced to the market in 2015 and was at the time the first market launch for a specific Parkinson treatment for over 10 years. In principle, the substance can be categorized with drugs with a MAO-B inhibiting effect, but is treated as distinct because of the complex dual mechanism of action (Caccia et al. 2006; Olanow and Stocchi 2016), whose clinical importance we still cannot explain conclusively. As in the case of the two other MAO-B inhibitors, a neuroprotective capability is under discussion (Sadeghian et al. 2016).
Pharmacology
Safinamide is an α-aminoamide derivate and has a MAO-B inhibiting effect and both a dopaminergic and a non-dopaminergic mode of action, by among others blocking the voltage-dependent sodium channels.
Safinamide blocks the voltage-dependent sodium and, to a lesser degree, the calcium channels (Olanow et al. 2016). Due to the application-dependent modulation of voltage-dependent sodium channels, the excessive release of glutamate is reduced without influencing the basal glutamate levels. The clinical significance of this lies in the fact that only the pathological glutamate release is arrested.
Mode of action of safinamide
Safinamide is a highly selective, reversible MAO-B inhibitor which inhibits MAO-B in the human brain one thousand times more strongly than MAO-A. This MAO-B inhibition is exclusively pharmacodynamic, does not in any way induce structural modifications in the MAO-B enzyme and is thus completely reversible. There is a complete inhibition of platelet-derived MAO-B in the nanomolar range without any negative impact on MAO-A. Other dopaminergic mechanisms are either not influenced or at most only to a slight degree. Safinamide does not influence the enzymes that are involved in levodopa metabolism, such as aromatic l-amino acid decarboxylase (AADC) and catechol-O-methyltransferase (COMT).
The substance is water-soluble, demonstrates a high capacity for permeability and is quickly reabsorbed. The plasma–protein bond is approximately 90% and is not subject to a significant first-pass effect. There is dose linearity for Cmax and AUC. The bioavailability is constant over all different doses. After administering 50 mg safinamide under fasting conditions, the absolute bioavailability of safinamide is high (on the average 95%) and after administering an individual dose of 100 mg, the maximum plasma concentration (Tmax) is reached after 2 to 2.5 h (Cmax at approximately 650 ng/ml and AUC at 19.000 ng/ml × h) (Fariello 2007). The half-life period is about 24 h, a steady-state is dose-dependent with but minor interindividual variability (Marzo et al. 2004). The pharmacokinetics is not subject to influence from the variables of age, gender or race of the patient. The elimination half-life (T1/2) of safinamide amounts to approximately 20 to 24 h (Marzo et al. 2004).
Within a range of between 300 and 10 mg/kg, safinamide has linear pharmacokinetics. Under joint administration with levodopa and/or dopamine agonists, no effect could be seen on the clearance of safinamide, and the pharmacokinetic profile for the simultaneous application of levodopa was not altered (Borgohain et al. 2014b). The so-called “cocktail study” with CYP1A2 and CYP3A4 substrates (coffein and midazolam) showed no clinically significant effect on the pharmacokinetic profile of safinamide. Further studies demonstrated that CYP enzymes play only a subordinate role in the biotransformation of safinamide. Because safinamide can temporarily inhibit BCRP (Breast Cancer Resistance Protein), an interval of 5 h should be adhered to between administering safinamide and then giving medication that has a BCRP substrate with a Tmax of ≤ 2 h (such as pitavastatin, pravastatin, ciprofloxacin, methotrexate, topotecan, diclofenac or glyburide).
Safinamide is eliminated exclusively by being metabolised and subsequently it is predominantly eliminated in the urine. An extensive biotransformation leads to a slight elimination of the unmodified substance (approximately 2% in the faeces and 7% in the urine) (Onofrj et al. 2008). All essential metabolites (NW-1153, NW-1199 and NW-1689 glucuronide) are considered inactive as far as effectiveness and safety are concerned.
In the case of patients presenting with mild or moderate renal dysfunction, dosage adjustments are not necessary. In moderate liver dysfunction (Child–Pugh B), safinamide can increase exposition by ~ 80%. And patients with moderate curtailments in liver functioning require a lower dose (50 mg/day).
Safinamide does not have any clinically relevant influence on the depletion of tyramine (Cattaneo et al. 2003; Di Stefano and Rusca 2011; Marquet et al. 2012). 100 mg/day of safinamide reinforces the circulatory effects of oral tyramine vs. placebo by 1.6 times. A supra-therapeutic dose of 350 mg/day led to a 1.8 potentiation (Marquet et al. 2012). A possible potentiation of the hypertensive effect of oral tyramine by safinamide was studied in 20 healthy subjects who received safinamide at a dose of 300 mg/day for six to seven days (Di Stefano and Rusca 2011). Safinamide failed to show any clinically relevant tyramine-induced rise in blood pressure.
It is important to note that safinamide does not have any relevant risk for a substance-induced Torsade-de-pointes syndrome and thus has a favourable cardiac safety profile. There is no negative influence on the QT interval. In therapeutic (100 mg/day) as well as supra-therapeutic (350 mg/day) doses safinamide can in fact induce a slight dose-dependent shortening of the QT interval (~ 5 ms). The SETTLE and the MOTION studies did not show any increase in the QTc interval.
Studies on safinamide with special clinical relevance
The first clinical studies suggest that safinamide might have an effect more than just MAO-B inhibition, because, under complete MAO-B inhibition through increase in dosage, further positive effects were observed on the fluctuations (Fariello 2007; Stocchi et al. 2006).
In further clinical studies, over 3000 patients were examined and over 500 patients were seen for a period of 2 years (Cattaneo et al. 2020; Olanow and Stocchi 2016). The most relevant studies here are: 016, 018, SETTLE, SYNAPSES and MOTION (Abbruzzese et al. 2021; Barone et al. 2013; Borgohain et al. 2014a, b, Schapira et al. 2013b, 2017).
In the phase III study 016 (Borgohain 2014a), effectivity and safety were examined in a double-blind, placebo-controlled parallel group study in Parkinson patients who were in intermediate to late stages with motor fluctuations and who before study begin had been receiving only levodopa or levodopa in combination with other Parkinson’s medications. In addition to their levodopa dosage, the patients were then given for 24 weeks either: 100 mg/day (n = 224), 50 mg/day safinamide (n = 223) or placebo (n = 222). The primary outcome was the change in on-time without disturbing dyskinesias (in the DRS: the Dyskinesia Rating Scale). At week 24, an improvement was found for the duration of on-time: by 1.36 ± 2.6 h for the 100 mg/day arm, by 1.37 ± 2.7 h for the 50 mg/day arm and by 0.97 ± 2.4 h for the placebo group. Significance was found for both the treatment arms. In addition, the secondary target parameters, off-time, UPDRS Part III and CGI-C, were also significantly improved in both arms of the study. There were no differences in the side effects in the three study groups. It is important to note that "placebo" here in this discussion did not mean that the patients were given a specific placebo substance but rather that they kept receiving their standard therapy without any additional safinamide.
The study was conducted as an extension study (018) (Borgohain et al. 2014b; Cattaneo et al. 2015). Dyskinesias made up the primary outcome as evaluated by the Dyskinesia Rating Scale. The placebo group consisted initially of 175 patients (142 at the end), 189 (148) were given 50 mg safinamide, 100 mg received 180 (150) patients. The participants remained in the same treatment group to which they had been randomised in Study 016. In both safinamide groups, a decrease in their DRS total score was seen compared to baseline:
-
31% reduction—Safinamide 50 mg/day
-
27% reduction—Safinamide 100 mg/day
-
3% reduction—placebo
The primary outcome was not in fact achieved, but at the time of the baseline measurements, the majority of the patients (64%) had either no or at most only mild dyskinesias (DRS ≤ 4), so that here no improvement was to be expected. Therefore, a post hoc analysis of the DRS data was performed for 242 patients who had already presented with moderate to severe dyskinesias at the start of the study 016 (DRS total score > 4). This time significance was clearly seen in the 100 mg arm (p = 0.0317) but not in the 50 mg arm (Cattaneo et al. 2015). This positive effect could be seen in all subgroups, even for all combinations of medications (Cattaneo et al. 2015). The improvement in on-time amounted to over one hour in both arms.
The SETTLE Study (Schapira et al. 2017) was initiated in 2009, and their data became decisive for the final approval of safinamide. The study design itself was in fact even included in the approval text and is to a considerable extent identical with that of Study 016. Between 2009 and 2012 Parkinson patients were assigned in equal numbers to either a safinamide or a placebo treatment group for a duration of 24 weeks. In the first two weeks, the patients received 50 mg safinamide and subsequently this dose was raised to 100 mg (in tablet form in all cases).
After reaching the optimized medication setting/dosage, the patients had to have at least 1.5 h of off-time daily (with the one exception of early morning off-time) and also had to have received levodopa (with a decarboxylase inhibitor) for at least 4 weeks in a fixed dose. 851 patients were examined as to their eligibility for being selected for the final study and thus 549 were found for randomization. 245 safinamide patients (89.4%) and 241 placebo-treated (87.6%) completed the course of the study. Due to adverse side effects, 12 patients in the safinamide group (4.4%) and 10 placebo-treated (3.6%) ones withdrew prematurely.
The primary outcome was the amount of improvement in on-time without any afflicting dyskinesias. In the safinamide group, on-time could be extended by 1.42 h (after having started with approximately nine hours, and 0.57 h in the placebo group).
The MOTION study (Barone et al. 2013) examined safinamide in combination with dopamine agonists. In the group with dopamine agonists (n = 66), 100 mg/day of safinamide significantly improved the UPDRS score (p = 0.0396). The 50 mg arm failed to show significant improvement (p = 0.2280).
The SYNAPSES study (Abbruzzese et al. 2021) aimed at checking the safety profile, particularly in older patients and those with psychiatric and other comorbidities. Altogether 1610 patients were enrolled in eight different European countries with 82.4% evaluable after 12 months. 25.1% of patients were > 75 years, 42.4% with psychiatric conditions, and 70.8% with relevant comorbidities. Clinically significant improvements were seen in the UPDRS motor score and in the UPDRS total score in ≥ 40% of patients. In the patient group > 75 years, no relevant differences were found for AEs frequency, severity or action taken. 13.6% experienced SAE vs 7.7% of younger patients. In the group with relevant comorbidities, 49.1% experienced AEs vs 37.8% of patients without comorbidities. 11.1% experienced SAE vs 4.6% of patients without comorbidities. In the patients with psychiatric conditions, no relevant differences were found for AEs and SAEs frequency, severity and action taken. 58% patients had a dose increase from 50 to 100 mg/day (decision of the physician), 6% patients had a dose decrease from 100 to 50 mg/day (by decision/wish of the patient). In summary, neither age, comorbidities, nor psychiatric conditions seem to have any relevant effect on its safety profile. The final conclusion of the EMA-Type II variation assessment report was that “The benefit-risk balance of safinamide remains positive.” Another recent trial confirmed the safety of safinamide when administered with SSRIs and SNRIs in PD depressed patients (Pérez-Torre et al. 2021). No cases of serotonin syndrome were recorded, even in the group of subjects who were taking opioids. The authors concluded that concomitant use of safinamide with antidepressant drugs seems to be safe and well tolerated, in the long-term as well.
The X-TRA study (Jost et al. 2018) observed 297 patients between 2015 and 2017 under everyday conditions ("real life") exclusively in Germany and thus constitutes the largest completed study of its kind on safinamide. The motor and non-motor symptoms as well as the quality of life improved while unknown adverse side effects have not yet been observed.
Recently, Guerra and co-workers demonstrated the positive effect of safinamide on the relative excess of glutamate in patients (Guerra et al. 2019). In another trial, Geroin and co-workers found that after 12 weeks of safinamide therapy, a significant improvement was noted in several scales for pain (King’s Pain Scale for Parkinson’s Disease, Brief Pain Inventory Intensity and Interference, and Numerical Rating Scale) and also in UPDRS III and IV, CGI, and PDQ39 (Geroin et al. 2020).
No significant changes in LEP complexes were observed. They concluded that safinamide may be effective for the management of pain in PD patients with motor fluctuations.
The so-called SAFINONMOTOR study was able to demonstrate positive effects on pain, sleep and mood (Grigoriou et al. 2021; Laban Deira et al. 2021, Santos Garcia et al. 2021a, b). The improvement seen on sleep which was confirmed by another study focused on Rapid Eye Movement (REM) sleep behaviour disorders (Plastino et al. 2021), exploring the clinical and video-polysomnographic changes occurred during safinamide treatment in 30 PD patients. Twenty-two of 30 patients reported clear improvement in symptoms during safinamide treatment, and 16 were absolutely free from clinical RBD-symptoms at the end of the treatment.
Goméz-Lopéz and co-workers investigate the effects of safinamide in patients with urinary problems, using the SCOPA-AUT-U scale, and found that the total score, as well as urgency, incontinence, frequency and nocturia subscales improved significantly after safinamide treatment. They concluded that, although the mechanism of this effect remains unknown, a nondopaminergic multimodal effect of the drug could be speculated on (Goméz-Lopéz et al. 2021).
At present, the so-called SUCCESS study (EUPASS Registry number 41428) is being conducted in five European countries as an observational study comparing safinamide, rasagiline and dopamine agonists/COMT inhibitors under real life conditions and measuring the effectiveness of the drugs on quality of life and healthcare resources consumption.
Further publications recently published show that for the category of mood a positive effect was also observed (Cattaneo et al. 2017b) and to be exact in the scores in both the PDQ-39 and in the GRID-HAMID (Hamilton Rating Scale for Depression).
Joint interpretation of the 016 and SETTLE studies
One interesting aspect stems from a post hoc analysis of the 016 and SETTLE studies (Cattaneo et al. 2017a, 2018). Because they have the same study design, 016 and SETTLE can readily be juxtaposed for comparison. In this analysis, the symptom of pain in the Parkinson’s Disease Quality of Life Questionnaire (PDQ-39) was specifically selected for review after six months into the study. In the safinamide, 100 mg arm fewer patients received an analgesic than in the comparison group. In week 24, a mere 23.9% of patients in the safinamide group and 30% of the placebo group were taking the pain reliever. Specifically reviewing individual parameters evaluated in the PDF-39, a distinct improvement can be seen in, for example, muscle cramps (item 37), joint pains (item 38) and unpleasant hot or cold sensations (item 39), and in the case of items 37 and 39 this difference is significant. Further clinical studies on pain symptoms have been initiated, some have already been concluded and confirm the positive effect (Grigoriou et al. 2021; Santos Garcia et al. 2021a, b).
Comparison of the MAO-B inhibitors
The question as to whether differences exist between selegiline and rasagiline might be the case is best responded to by referring to the fact that they are pharmacologically different medications (LeWitt 2009; Müller and Reichman 2012). The power of the inhibition for both MAO-A and MAO-B is clearly different and, to be exact, by either two powers of ten resp. five-fold (Müller and Reichmann 2012). The bioavailability is different as well (selegiline < 10%) and only the pharmacokinetics of rasagiline is linear in the therapeutic range (Müller and Reichmann 2012). The profile of side effects reveals distinct advantages for rasagiline (Csoti et al. 2012; Müller and Reichmann 2012). On this point the degradation of selegiline to amphetamine and metamphetamine should be recalled (Müller and Reichmann 2012).
The present assessment of the Movement Disorder Society (Fox et al. 2018) again reveals distinct differences, for example in the combination therapy with levodopa with motor complications (rasagiline is effective, but there is insufficient evidence on selegiline). A recent evaluation found a slight superiority for selegiline compared to the other MAO-B inhibitors available on the market (Binde et al. 2018).
Unfortunately, there are no head-to-head studies here. In one smaller study (n = 28) selegiline was switched to rasagiline. All examined parameters revealed improvement (Jost et al. 2008). A further switch study (Müller et al. 2013) found superiority for rasagiline. In a so-called retrospective head-to-head comparison, no relevant differences were found between selegiline and rasagiline but rather lower levodopa doses and fewer dyskinesias (Cereal et al. 2017).
To compare the symptomatic effects, we did a meta-analysis in which the 6 RCT (randomized, controlled studies) with rasagiline and the RCT 15 with selegiline were included (Jost et al. 2012). In monotherapy, there were significant differences favoring rasagiline in UPDRS total scores and in the motor score. The meta-analysis of all studies (mono- and combination therapy) confirmed significant differences in favor of rasagiline in the UPDRS total score.
In studies on rasagiline, the termination rate due to adverse reactions was on the level after placebo treatment while the rate for selegiline was significantly higher than for placebos (Jost et al. 2012).
Peretz et al. (2016) analysed the necessity for dopaminergic medication under the two substances in 349 (selegiline) vs. 485 (rasagiline) patients. Both groups received levodopa after approximately three years. In the selegiline group, dopamine agonists were introduced at a later time. The data supported the view that selegiline had a slightly better symptomatic effect (Peretz et al. 2016).
So far there are no comparative studies of rasagiline or selegiline with safinamide. The so-called SUCCESS study could close this gap somewhat.
Disease modification
No other substance has been examined as to the question of neuroprotection as early and as controversially as selegiline. At the present, there is no proof for either a relevant neuroprotective effect or a progression-delimiting effect (Fox et al. 2018; Schapira 2011). Nonetheless there are distinct theoretical as well as clinical indications for a disease-modifying effect (Marras et al. 2005; Naoi et al. 2020; Olanow et al. 1995; Riederer et al. 2018; Tábi et al. 2020). For example, the NET-PD LS1 study was able to demonstrate a retarded progression in the MAO-B inhibitor group.
The first speculations on a neuroprotective effect go as far back as Birkmayer, who found a lower rate of mortality among the patients in his selegiline group (Birkmayer et al. 1985). The fundamental explanation for this theoretical concept is the inhibition of oxidative stress in the dopaminergic neurones still intact, meaning that the concept is a model (the radikal hypothesis, H2O2). It is also a matter of discussion whether there is a neurotrophic and an anti-apoptotic effect [Naoi 78]. In the final analysis, however, the neuroprotective effect has only been demonstrated in the animal model, whereby in such models un-physiological conditions prevailed as for example in the MPTP model (neurotoxine hypothesis).
One other indication of a neuroprotective action can be seen in the quick recovery of patients after a cerebrovascular accident if they are treated with selegiline (Sivenius et al. 2001).
The frequently cited DATATOP study [PSG 1989a; PSG 1989b; PSG 1993; PSG 1998] has only limited use for supporting this neuroprotective effect because (1) on the one hand delaying the need for levodopa could be a purely symptomatic effect and (2) on the other hand even a renewed evaluation of this study’s results showed that there were no long-term relevant differences. We have to assume that the delayed use of levodopa by as much as several months is a result of the symptomatic effect of selegiline and not an inherently potentially neuroprotective capacity. And so no differences were found in the long-term course of the disease with or without selegiline.
Furthermore, a remark is in place here about selegiline being tested against vitamin E which has been reputed to have a neuroprotective effect and has in fact been supported in studies (de Rijk et al. 1997). Furthermore, in the DATATOP study, the above-average life expectancy of the patients was striking (PSG 1998), which stands in contradiction to other studies (Hely et al. 1999).
A contrary view on the effect of selegiline was published by a British workgroup (Lees 1995). In this particular study, no relevant symptomatic effect could be found and the mortality rate was in fact greater than in the control group. However, the study is debatable primarily due to its having a suboptimal study protocol (and the far-reaching conclusions of the publication should also be evaluated critically [31]). But their results nonetheless strongly unsettled any belief in a neuroprotective effect of selegiline permanently. Reviewing this study (Lees 1995), it is often overlooked that no routine levodopa increase had been required, and that could argue for a neuroprotective capacity. A publication by Margaret Thorogood relativized the assertions of the Lees team, but still came to the conclusion that mortality was discretely higher in the patients treated with selegiline than in levodopa monotherapy and in particular in patients with a younger age at onset (Thorogood et al. 1998). In the meantime, the data from the Lees study (Lees 1995) can be seen as sufficiently refuted (Marras et al. 2005).
A number of studies, for example by Olanow et al. (1995), demonstrated a slower rate of progression, as for example in the renewed analysis of the DATATOP study (PSG 1998). The question of a neuroprotective impact was again addressed in the study by Pålhagen et al. (2006) in a placebo-controlled study of 140 patients covering altogether 7 years. After 5 years, the UPDRS total score showed a difference of 28.7 ± 14.7 (selegiline) versus 38.6 ± 15.5 (placebo) and a difference in the levodopa dose 529 ± 145.6 mg (selegiline) versus 631 ± 186.3 mg (rasagiline), thus signifying an improvement in the UPDRS score of ten points and at the same time savings in levodopa of approximately 100 mg (Pålhagen et al. 2006).
A neuroprotective effect of rasagiline has also been discussed and was demonstrated in several studies (Finberg et al. 1996; Jenner 2012; Marras et al. 2005; Schulz 2012; Youdim et al. 2005). In particular, aminoindane may be playing a role here because, as opposed to amphetamine, it has potentially neuroprotective characteristics (Bar-Am and Amit 2004; Müller and Reichmann 2012). This possible neuroprotective potency was confirmed in a clinical study (PSG 2004) in patients with early onset who had a better course than patients whose therapy was delayed by 6 months. The ADAGIO study (Schapira et al. 2013a) furthermore demonstrated a disease-modifying effect at least for the 1 mg group of patients. This has not been found for any other substance. Unfortunately in the follow-up time, the two groups conformed more and more to each other (Rascol et al. 2016). The PROUD study, which had the same design using pramipexole, was negative, thus making the claims of the ADAGIO study all the more positive (Schapira et al. 2013a). But fundamentally it should be emphasized that all studies are initiated well into the course of the disease, that is, much too late as to ever show a clinically observable effect in neuroprotection (Naoi et al. 2020).
Further MAO-B inhibitors and the future of MAO-B inhibitors
In addition to selegiline, various other MAO-B inhibitors have found clinical use. Mention should be made of, for example: lazabemide, milacemide and mofegiline. The clinical studies were discontinued in the majority of the substances being tested (e.g. mofegilin). Further MAO-B inhibitors are in development (Binda et al. 2006; Yelekçi et al. 2013).
Even after four decades of clinical use of MAO-B inhibitors, there are still a lot of unsolved questions. Because rasagiline and selegiline are generic medications, however, larger clinical studies are no longer now being undertaken and the evidence from earlier studies on selegiline do not meet present-day standards. The potential in MAO-B inhibitors has definitely not yet been fully exhausted (Tábi et al. 2020). The topic of neuroprotection might very well once again gain momentum, because MAO-B inhibition modulates α-syn secretion and aggregation (Nakamura et al. 2021). There is the good possibility that a new patch application for rasagiline will be developed, which then poses the question as to just which clinical use this would have for Parkinson therapy (Bali and Salve 2020). Considerable hope can be placed especially in safinamide inasmuch as clinical studies are underway and because new approaches for non-motor dysfunctions can develop out of the dual approach (Pagonabarraga et al. 2021).
References
Abbruzzese G, Kulisevsky J, Bergmans B, Gomez-Esteban JC, Kägi G, Raw J, Stefani A, Warnecke T, Jost WH, SYNAPSES Study Investigators Group (2021) A European observational study to evaluate the safety and the effectiveness of safinamide in routine clinical practice: The SYNAPSES Trial. J Parkinsons Dis 11(1):187–198. https://doi.org/10.3233/JPD-219007
Bali NR, Salve PS (2020) Impact of rasagiline nanoparticles on brain targeting efficiency via gellan gum based transdermal patch: a nanotheranostic perspective for Parkinsonism. Int J Biol Macromol 164:1006–1024. https://doi.org/10.1016/j.ijbiomac.2020.06.261
Bar Am O, Amit T, Youdim MBH (2004) Contrasting neuroprotective and neurotoxic actions of respective metabolites of anti-Parkinson drugs rasagiline and selegiline. Neurosci Lett 355:169–172. https://doi.org/10.1016/j.neulet.2003.10.067
Barone P, Santangelo G, Morgante L, Onofrj M, Meco G, Abbruzzese G, Bonuccelli U, Cossu G, Pezzoli G, Stanzione P, Lopiano L, Antonini A, Tinazzi M (2015) A randomized clinical trial to evaluate the effects of rasagiline on depressive symptoms in non-demented Parkinson’s disease patients. Eur J Neurol 22(8):1184–1191. https://doi.org/10.1111/ene.12724
Barone P, Fernandez HH, Ferreira J et al (2013) Safinamide as an add-on therapy to a stable dose of a single dopamine agonist: results from a randomized, placebo-controlled, 24-week multicenter trial in early idiopathic Parkinson disease (PD) patients (MOTION Study). Neurology 80 (Meeting Abstracts 1): P01.061
Binda C, Hubálek F, Li M, Castagnoli N, Edmondson DE, Mattevi (2006) Structure of the human mitochondrial monoamine oxidase B: new chemical implications for neuroprotectant drug design. Neurology 67(Suppl. 2):S5–S7. https://doi.org/10.1212/wnl.67.7_suppl_2.s5
Binde CD, Tvete IF, Gåsemyr J, Natvig B, Klemp M (2018) A multiple treatment comparison meta-analysis of monoamine oxidase type-B inhibitors for Parkinson’s disease. Br J Clin Pharmacol 84:1917–1927. https://doi.org/10.1111/bcp.13651
Birkmayer W, Knoll J, Riederer P, Youdim MB, Hars V, Marton J (1985) Increased life expectancy resulting from addition of l-deprenyl to Madopar treatment in Parkinson’s disease: a longterm study. J Neural Transm 64:113–127. https://doi.org/10.1007/BF01245973
Borgohain R, Szasz J, Stanzione P, Meshram C, Bhatt MH, Chirilineau D, Stocchi F, Lucini V, Giuliani R, Forrest E, Rice P, Anand R, Study 018 Investigators (2014) Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov Disord 29:1273–1280. https://doi.org/10.1002/mds.25961
Borgohain R, Szasz J, Stanzione P, Meshram C, Bhatt M, Chirilineau D, Stocchi F, Lucini V, Giuliani R, Forrest E, Rice P, Anand R, Study 016 Investigators (2014) Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Mov Disord 29:229–237. https://doi.org/10.1002/mds.25751
Caccia C, Maj R, Calabresi M, Maestroni S, Faravelli L, Curatolo L, Salvati P, Fariello RG (2006) Safinamide: from molecular targets to a new anti-Parkinson drug. Neurology 67(7 Suppl. 2):S18–S23. https://doi.org/10.1212/wnl.67.7_suppl_2.s18
Cattaneo C, Caccia C, Marzo A, Maj R, Fariello RG (2003) Pressor response to intravenous tyramine in healthy subjects after safinamide, a novel neuroprotectant with selective, reversible monoamine oxidase B inhibition. Clin Neuropharmacol 26:213–217. https://doi.org/10.1097/00002826-200307000-00012
Cattaneo C, La Ferla R, Bonizzoni E, Sardina M (2015) Long-term effects of safinamide on dyskinesia in mid- to late stage Parkinson’s disease: a post-hoc analysis. J Parkinsons Dis 5:475–481. https://doi.org/10.3233/JPD-150569
Cattaneo C, Barone P, Bonizzoni E, Sardina M (2017a) Effects of safinamide on pain in fluctuating Parkinson’s disease patients: a post-hoc analysis. J Parkinsons Dis 7:95–101. https://doi.org/10.3233/JPD-160911
Cattaneo C, Müller T, Bonizzoni E, Lazzeri G, Kottakis I, Keywood C (2017b) Long-term effects of safinamide on mood fluctuations in Parkinson’s disease. J Parkinsons Dis 7:629–634. https://doi.org/10.3233/JPD-171143
Cattaneo C, Kulisevsky J, Tubazio V, Castellani P (2018) Long-term efficacy of safinamide on Parkinson’s Disease chronic pain. Adv Ther 35:515–522. https://doi.org/10.1007/s12325-018-0687-z
Cattaneo C, Jost WH, Bonizzoni E (2020) Long-term efficacy of safinamide on symptoms severity and quality of life in fluctuating Parkinson’s disease patients. J Parkinsons Dis 10:89–97. https://doi.org/10.3233/JPD-191765
Cereda E, Cilia R, Canesi M, Tesei S, Mariani CB, Zecchinelli AL, Pezzoli G (2017) Efficacy of rasagiline and selegiline in Parkinson’s disease: a head-to-head 3-year retrospective case-control study. J Neurol 264:1254–1263. https://doi.org/10.1007/s00415-017-8523-y
Chen JJ, Swope DM, Dashtipour K (2007) Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin Ther 9:1825–1849. https://doi.org/10.1016/j.clinthera.2007.09.021
Clarke A, Brewer F, Johnson ES, Mallard N, Hartig F, Taylor S, Corn TH (2003) A new formulation of selegiline: improved biovailability and selectivity for MAO-B inhibition. J Neural Transm (vienna) 110:1241–1255. https://doi.org/10.1007/s00702-003-0036-4
Csoti I, Storch A, Müller W, Jost WH (2012) Drug interactions with selegiline versus rasagiline. Basal Ganglia 2(4):S27–S31. https://doi.org/10.1016/j.baga.2012.06.003
Dashtipour K, Chen JJ, Kani C, Bahjri K, Ghamsary M (2015) Clinical outcomes in patients with Parkinson’s disease treated with a monoamine oxidase type-B inhibitor: a cross-sectional, cohort study. Pharmacotherapy 35:681–686. https://doi.org/10.1002/phar.1611
de Rijk MC, Breteler MM, den Breeijen JH, Launer LJ, Grobbee DE, van der Meché FG, Hofman A (1997) Dietary antioxidants and Parkinson disease. The Rotterdam Study. Arch Neurol 54:762–765. https://doi.org/10.1001/archneur.1997.00550180070015
deMarcaida JA, Schwid SR, White WB, Blindauer K, Fahn S, Kieburtz K, Stern M, Shoulson I et al (2006) Effects of tyramine administration in Parkinson’s disease patients treated with selective MAO-B inhibitor rasagiline. Mov Disord 21:1716–1721. https://doi.org/10.1002/mds.21048
Di Stefano AF, Rusca A (2011) Pressor response to oral tyramine during co-administration with safinamide in healthy volunteers. Naunyn Schmiedebergs Arch Pharmacol 384:505–515. https://doi.org/10.1007/s00210-011-0674-2
Fariello RG (2007) Safinamide. Neurotherapeutics 4:110–116. https://doi.org/10.1016/j.nurt.2006.11.011
Farkouh RA, Wilson MR, Tarrants ML, Castelli-Haley J, Armand C (2012) Cost-effectiveness of rasagiline compared with first-line early Parkinson disease therapies. Am J Pharm Benefits 4(3):99–107
Finberg JP, Lamensdorf I, Commissiong JW, Youdim MB (1996) Pharmacology and neuroprotective properties of rasagiline. J Neural Transm Suppl 48:95–101. https://doi.org/10.1007/978-3-7091-7494-4_9
Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, Christman D, Logan J, Smith M, Sachs H et al (1987) Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science 235:481–485. https://doi.org/10.1126/science.3099392
Fowler JS, Volkow ND, Logan J, Wang GJ, MacGregor RR, Schyler D, Wolf AP, Pappas N, Alexoff D, Shea C et al (1994) Slow recovery of human brain MAO after l-deprenyl (selegeline) withdrawal. Synapse 18:86–93. https://doi.org/10.1002/syn.890180203
Fox SH, Katzenschlager R, Lim S-Y, Barton B, de Bie RM, Seppi K, Coelho M, Sampaio C (2018) International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord 33:1248–1266. https://doi.org/10.1002/mds.27372 (Epub 2018 Mar 23)
Geroin C, Di Vico IA, Squintani G, Segatti A, Bovi T, Tinazzi M (2020) Effects of safinamide on pain in Parkinson’s disease with motor fluctuations: an exploratory study. J Neural Transm (vienna) 127(8):1143–1152. https://doi.org/10.1007/s00702-020-02218-7
Goetz CG, Schwid SR, Eberly SW, Oakes D, Shoulson I, Parkinson Study Group TEMPO and PRESTO Investigators (2006) Safety of rasagiline in elderly patients with Parkinson disease. Neurology 66:1427–1429. https://doi.org/10.1212/01.wnl.0000210692.95595.1c
Gómez-López A, Sánchez-Sánchez A, Natera-Villalba A, Ros-Castelló V, Beltrán-Corbellini A, Fanjul-Arbós S, Pareés Moreno I, López-Sendon Moreno JL, Martínez Castrillo JC, Alonso-Canovas A (2021) SURINPARK: safinamide for urinary symptoms in Parkinson’s disease. Brain Sci 11(1):57. https://doi.org/10.3390/brainsci11010057
Green AR, Mitchell BD, Tordorff AF, Youdim MB (1977) Evidence that dopamine deamination by both type A and type B monoamine oxidase in rat brain in vivo and for the degree of enzyme inhibition necessary to increase functional activity of dopamine and 5-hydroxytryptamine. Br J Pharmacol 60:343–349. https://doi.org/10.1111/j.1476-5381.1977.tb07506.x
Grigoriou S, Martínez-Martín P, Ray Chaudhuri K, Rukavina K, Leta V, Hausbrand D, Falkenburger B, Odin P, Reichmann H (2021) Effects of safinamide on pain in patients with fluctuating Parkinson’s disease. Brain Behav 11:e2336. https://doi.org/10.1002/brb3.2336
Guerra A, Suppa A, D’Onofrio V, Di Stasio F, Asci F, Fabbrini G, Berardelli A (2019) Abnormal cortical facilitation and L-dopa-induced dyskinesia in Parkinson’s disease. Brain Stimul 12:1517–1525. https://doi.org/10.1016/j.brs.2019.06.012 (Epub 2019 Jun 11)
Hanagasi HA, Gurvit H, Unsalan P, Horozoglu H, Tuncer N, Feyzioglu A, Gunal DI, Yener GG, Cakmur R, Sahin HA, Emre M (2011) The effects of rasagiline on cognitive deficits in Parkinson’s disease patients without dementia: a randomized, double-blind, placebo-controlled, multicenter study. Mov Disord 26:1851–1858. https://doi.org/10.1002/mds.23738
Hauser RA, Abler V, Eyal E, Eliaz RE (2016) Efficacy of rasagiline in early Parkinson’s disease: a meta-analysis of data from the TEMPO and ADAGIO studies. Int J Neurosci 126:942–946. https://doi.org/10.3109/00207454.2016.1154552
Haycox A, Armand C, Murteira S, Cochran J, François C (2009) Cost effectiveness of rasagiline and pramipexole as treatment strategies in early Parkinson’s disease in the UK setting: an economic Markov model evaluation. Drugs Aging 26:791–801. https://doi.org/10.2165/11316770-000000000-00000
Hely MA, Morris JG, Traficante R, Reid WG, O’Sullivan DJ, Williamson PM (1999) The Sydney multicentre study of Parkinson’s disease: progression and mortality at 10 years. J Neurol Neurosurg Psychiatry 67:300–307. https://doi.org/10.1136/jnnp.67.3.300
Hudry J, Rinne JO, Keranen T et al (2006) Cost-utility model of rasagiline in the treatment of advanced Parkinson’s disease in Finland. Ann Pharmacother 40:651–657
Jankovic J, Berkovich E, Eyal E, Tolosa E (2014) Symptomatic efficacy of rasagiline monotherapy in early Parkinson’s disease: post-hoc analyses from the ADAGIO trial. Parkinsonism Relat Disord 20:640–643. https://doi.org/10.1016/j.parkreldis.2014.02.024
Jenner P (2012) Mitochondria, monoamine oxidase B and Parkinson’s disease. Basal Ganglia 2:S3–S7. https://doi.org/10.1016/j.baga.2012.06.006
Jiang DQ, Wang HK, Wang Y, Li MX, Jiang LL, Wang Y (2020) Rasagiline combined with levodopa therapy versus levodopa monotherapy for patients with Parkinson’s disease: a systematic review. Neurol Sci 41:101–109. https://doi.org/10.1007/s10072-019-04050-8
Jost WH, Klasser M, Reichmann H (2008) Rasagilin im klinischen Alltag. Fortschr Neurol Psychiatr 76:594–599. https://doi.org/10.1055/s-2008-1038249
Jost WH, Friede M, Schnitker J (2012) Indirect meta-analysis of randomised placebo-controlled clinical trials on rasagiline and selegiline in the symptomatic treatment of Parkinson’s disease. Basal Ganglia 2:S17–S26. https://doi.org/10.1016/j.baga.2012.05.006
Jost W, Kupsch A, Mengs J, Delf M, Bosse D (2018) Wirksamkeit und Sicherheit von Safinamid als Zusatztherapie zu Levodopa bei Parkinson-Patienten: eine nicht-interventionelle Beobachtungsstudie. Fortschr Neurol Psychiatr 86:624–634. https://doi.org/10.1055/a-0665-4667 (Epub 2018 Aug 24)
Knoll J (1978) The possible mechanisms of action of (-)deprenyl in Parkinson’s disease. J Neural Transm 43(3–4):177–198. https://doi.org/10.1007/BF01246955
Labandeira CM, Alonso Losada MG, Yáñez Baña R, Cimas Hernando MI, Cabo López I, Paz González JM, Gonzalez Palmás MJ, Martínez Miró C, Santos García D (2021) Effectiveness of safinamide over Mood in Parkinson’s disease patients: secondary analysis of the open-label study SAFINONMOTOR. Adv Ther 38:5398–5411. https://doi.org/10.1007/s12325-021-01873-w
Larsen JP, Boas J, Erdal JE (1999) Does selegiline modify the progression of early Parkinson’s disease? Results from a five-year study. Eur J Neurol 6:539–547. https://doi.org/10.1046/j.1468-1331.1999.650539.x
Lees AJ, Parkinson’s Disease Research Group of the United Kingdom (1995) Comparison of therapeutic effects and mortality data of levodopa and levodopa combined with selegiline in patients with early, mild Parkinson’s disease. Br Med J 311:1602–1607. https://doi.org/10.1136/bmj.311.7020.1602
Lew MF, Hauser RA, Hurtig HI, Ondo WG, Wojcieszek J, Goren T, Fitzer-Attas CJ (2010) Long-term efficacy of rasagiline in early Parkinson‘s disease. Int J Neurosci 120:404–408. https://doi.org/10.3109/00207451003778744
LeWitt PA (2009) MAO-B inhibitor know-how: back to the pharm. Neurology 72:1352–1357. https://doi.org/10.1212/WNL.0b013e3181a0feba
Marquet A, Kupas K, Johne A, Astruc B, Patat A, Krösser S, Kovar A (2012) The effect of safinamide, a novel drug for Parkinson’s disease, on pressor response to oral tyramine: a randomized, double-blind, clinical trial. Clin Pharmacol Ther 92:450–457. https://doi.org/10.1038/clpt.2012.128 (Epub 2012 Sep 5)
Marras C, McDermott MP, Rochin PA, Tanner CM, Naglie G, Rudolph A, Lang AE, Parkinson Study Group (2005) Survival in Parkinson disease: thirteen-year follow-up of the DATATOP cohort. Neurology 64:87–93. https://doi.org/10.1212/01.WNL.0000148603.44618.19
Marzo A, Dal Bo L, Monte NC, Crivelli F, Ismaili S, Caccia C, Cattaneo C, Ruggero G Fariello RG (2004) Pharmacokinetics and pharmacodynamics of safinamide, a neuroprotectant with antiparkinsonian and anticonvulsant activity. Pharmacol Res 50:77–85
McCormack PL (2014) Rasagiline: a review of its use in the treatment of idiopathic Parkinson’s disease. CNS Drugs 28:1083–1097. https://doi.org/10.1007/s40263-014-0206-y
Mizuno Y, Hattori N, Kondo T, Nomoto M, Origasa H, Takahashi R, Yamamoto M, Yanagisawa N (2017) A randomized double-blind placebo-controlled phase III trial of selegiline monotherapy for early Parkinson disease. Clin Neuropharmacol 40:201–207. https://doi.org/10.1097/WNF.0000000000000239
Mizuno Y, Hattori N, Kondo T, Nomoto M, Origasa H, Takahashi R, Yamamoto M, Yanagisawa N (2019) Long-term selegiline monotherapy for the treatment of early Parkinson disease. Clin Neuropharmacol 42:123–130. https://doi.org/10.1097/WNF.0000000000000343
Montgomery EB, Panisset M (2009) Retrospective statistical analysis of the incidence of serotonin toxicity in patients taking rasagiline and anti-depressants in clinical trials. Mov Disord 24(S1):359
Müller WE, Reichmann H (2012) Pharmakokinetik und Pharmakodynamik von Selegilin und Rasagilin. Psychopharmakotherapie 19:191–201
Müller T, Hoffmann JA, Dimpfel W, Oehlwein C (2013) Switch from selegiline to rasagiline is beneficial in patients with Parkinson’s disease. J Neural Transm (vienna) 120:761–765. https://doi.org/10.1007/s00702-012-0927-3
Myllylä VV, Sotaniemi KA, Hakulinen P, Mäki-Ikola O, Heinonen EH (1997) Selegiline as the primary treatment of Parkinson’s disease—a long-term double-blind study. Acta Neurol Scand 95:211–218. https://doi.org/10.1111/j.1600-0404.1997.tb00101.x
Nakamura Y, Arawaka S, Sato H, Sasaki A, Shigekiyo T, Takahata K, Tsunekawa H, Kato T (2021) Monoamine oxidase-B inhibition facilitates α-synuclein secretion in vitro and delays its aggregation in rAAV-based rat models of Parkinson’s disease. J Neurosci 41:7479–7491. https://doi.org/10.1523/JNEUROSCI.0476-21.2021
Naoi M, Maruyama W, Shamoto-Nagai M (2020) Rasagiline and selegiline modulate mitochondrial homeostasis, intervene apoptosis system and mitigate α-synuclein cytotoxicity in disease-modifying therapy for Parkinson’s disease. J Neural Transm (vienna) 127:131–147. https://doi.org/10.1007/s00702-020-02150-w
Olanow CW, Stocchi F (2016) Safinamide—a new therapeutic option to address motor symptoms and motor complications in mid- to late-stage Parkinson’s disease. Eur Neurol Rev 11(Suppl. 2):2–15
Olanow CW, Hauser RA, Gauger L, Malapira T, Koller W, Hubble J, Bushenbark K, Lilienfeld D, Esterlitz J (1995) The effect of deprenyl and levodopa on the progression of Parkinson’s disease. Ann Neurol 38:771–777. https://doi.org/10.1002/ana.410380512
Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, Langston W, Melamed E, Poewe W, Stocchi F, Tolosa E, ADAGIO Study Investigators (2009) A double-blind, delayed start trial of rasagiline in Parkinson’s disease. N Engl J Med 361:1268–1278. https://doi.org/10.1056/NEJMoa0809335
Ondo WG, Hunter C, Isaacson SH, Silver DE, Stewart RM, Tetrud JW, Davidson A (2011) Tolerability and efficacy of switching from oral selegiline to Zydis selegiline in patients with Parkinson’s disease. Parkinsonism Relat Disord 17:117–118. https://doi.org/10.1016/j.parkreldis.2010.10.001
Onofrj M, Bonanni L, Thomas A (2008) An expert opinion on safinamide in Parkinson’s disease. Expert Opin Investig Drugs 17:1115–1125. https://doi.org/10.1517/13543784.17.7.1115
Pagonabarraga J, Tinazzi M, Caccia C, Jost WH (2021) The role of glutamatergic neurotransmission in the motor and non-motor symptoms in Parkinson’s disease: clinical cases and a review of the literature. J Clin Neurosci 90:178–183. https://doi.org/10.1016/j.jocn.2021.05.056
Pålhagen S, Heinonen E, Hägglund J, Kaugesaar T, Mäki-Ikola O, Palm R, Swedish Parkinson Study Group (2006) Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 66:1200–1206. https://doi.org/10.1212/01.wnl.0000204007.46190.54
Panisset M, Chen JJ, Rhyee SH, Jill Conner J, Mathena J, STACCATO study investigators (2014) Serotonin toxicity association with concomitant antidpressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy 34:1250–1258. https://doi.org/10.1002/phar.1500 (Epub 2014 Oct 14)
Parkinson Study Group (1989a) DATATOP: a multicenter controlled trial in early Parkinson’s disease. Arch Neurol 46:1052–1060. https://doi.org/10.1001/archneur.1989.00520460028009
Parkinson Study Group (1989b) Effect of deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med 321:1364–1371. https://doi.org/10.1056/NEJM198911163212004
Parkinson Study Group (1993) Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med 328:176–183. https://doi.org/10.1056/NEJM199301213280305
Parkinson Study Group (1998) Mortality in DATATOP: a multicenter trial in early Parkinson’s disease. Ann Neurol 43:318–325. https://doi.org/10.1002/ana.410430309
Parkinson Study Group (2002) A controlled trial of rasagiline in early Parkinson disease. The TEMPO study. Arch Neurol 59:1937–1943. https://doi.org/10.1001/archneur.59.12.1937
Parkinson Study Group (2004) A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol 61:561–566. https://doi.org/10.1001/archneur.61.4.561
Parkinson Study Group (2005) A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations. The PRESTO Study. Arch Neurol 62:241–248. https://doi.org/10.1001/archneur.62.2.241
Peretz C, Segev H, Rozani V, Gurevich T, El-Ad B, Tsamir J, Giladi N (2016) Comparison of selegiline and rasagiline therapies in Parkinson disease: a real-life study. Clin Neuropharmacol 39:227–231. https://doi.org/10.1097/WNF.0000000000000167
Pérez-Torre P, López-Sendón JL, Mañanes Barral V, Parees I, Fanjul-Arbós S, Monreal E, Alonso-Canovas A, Martínez Castrillo JC (2021) Concomitant treatment with safinamide and antidepressant drugs: safety data from real clinical practice. Neurologia (engl Ed). https://doi.org/10.1016/j.nrl.2021.08.004
Plastino M, Gorgone G, Fava A, Ettore M, Iannacchero R, Scarfone R, Vaccaro A, De Bartolo M, Bosco D (2021) Effects of safinamide on REM sleep behavior disorder in Parkinson disease: a randomized, longitudinal, cross-over pilot study. J Clin Neurosci 91:306–312. https://doi.org/10.1016/j.jocn.2021.07.011
Przuntek H, Conrad B, Dichgans J, Kraus PH, Krauseneck P, Pergande G, Rinne U, Schimrigk K, Schnitker J, Vogel HP (1999) SELEDO: a 5-year long-term trial on the effect of selegiline in early Parkinsonian patients treated with levodopa. Eur J Neurol 6:141–150. https://doi.org/10.1111/j.1468-1331.1999.tb00007.x
Rascol O, Brooks DJ, Melamed E, Oertel W, Poewe W, Stocchi F, Tolosa E, LARGO Study Group (2005) Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 365:947–954. https://doi.org/10.1016/S0140-6736(05)71083-7
Rascol O, Fitzer-Attas CJ, Hauser R, Jankovic J, Lang A, Langston JW, Melamed E, Poewe W, Stocchi F, Tolosa E, Eyal E, Weiss YM, Olanow CW (2011) A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol 10(5):415–423. https://doi.org/10.1016/S1474-4422(11)70073-4
Rascol O, Hauser RA, Stocchi F, Fitzer-Attas CJ, Sidi Y, Abler V, Olanow CW, Investigators AFU (2016) Long-term effects of rasagiline and the natural history of treated Parkinson’s disease. Mov Disord 31:1489–1496. https://doi.org/10.1002/mds.26724
Riederer P, Müller T (2018) Monoamine oxidase-B inhibitors in the treatment of Parkinson’s disease: clinical-pharmacological aspects. J Neural Transm (vienna) 125:1751–1757. https://doi.org/10.1007/s00702-018-1876-2
Riederer P, Youdim MB, Rausch WD, Birkmayer W, Jellinger K, Seemann D (1978) On the mode of action of l-deprenyl in the human central nervous system. J Neural Transm 43:217–226. https://doi.org/10.1007/BF01246958
Rinne UK (1987) R-(−)-deprenyl as an adjuvant to levodopa in the treatment of Parkinson’s disease. J Neural Transm Suppl 25:149–155
Sadeghian M, Mullali G, Pocock JM, Piers T, Roach A, Smith KJ (2016) Neuroprotection by safinamide in the 6-hydroxydopamine model of Parkinson’s disease. Neuropathol Appl Neurobiol 42:423–435
Saeger H (1998) Drug-delivery products and the Zydis fast-dissolving dosage form. J Pharm Pharmacol 50:375–382. https://doi.org/10.1111/j.2042-7158.1998.tb06876.x
Santos García D, Yáñez Baña R, Labandeira Guerra C, Cimas Hernando MI, Cabo López I, Paz González JM, Alonso Losada MG, Gonzalez Palmás MJ, Cores Bartolomé C, Martínez Miró C (2021a) Pain Improvement in Parkinson’s disease patients treated with safinamide: results from the SAFINONMOTOR study. J Pers Med 11:798. https://doi.org/10.3390/jpm11080798
Santos García D, Cabo López I, Labandeira Guerra C, Yáñez Baña R, Cimas Hernando MI, Paz González JM, Alonso Losada MG, Gonzalez Palmás MJ, Cores Bartolomé C, Martínez Miró C (2021b) Safinamide improves sleep and daytime sleepiness in Parkinson’s disease: results from the SAFINONMOTOR study. Neurol Sci 23:1–8. https://doi.org/10.1007/s10072-021-05607-2
Schapira AH (2011) Monoamine oxidase B inhibitors for the treatment of Parkinson’s disease: a review of symptomatic and potential disease-modifying effects. CNS Drugs 25:1061–1071. https://doi.org/10.2165/11596310-000000000-00000
Schapira AHV, McDermott MP, Barone P, Comella CL, Albrecht S, Hsu HH, Massey DH, Mizuno Y, Poewe W, Rascol O, Marek K (2013a) Pramipexole in patients with early Parkinson’s disease (PROUD): a randomised delayed-start trial. Lancet Neurol 12:747–755. https://doi.org/10.1016/S1474-4422(13)70117-0
Schapira AH, Stocchi F, Borgohain R, Onofrj M, Bhatt M, Lorenzana P, Lucini V, Giuliani R, Anand R, Study 017 Investigators (2013b) Long-term efficacy and safety of safinamide as add-on therapy in early Parkinson’s disease. Eur J Neurol 20:271–280. https://doi.org/10.1111/j.1468-1331.2012.03840.x (Epub 2012 Sep 12)
Schapira AHV, Fox S, Hauser R, Jankovic J, Jost WH, Kenney C, Kulisevsky J, Pahwa R, Poewe W, Anand R (2017) Assessment of safety and efficacy of safinamide as a levodopa adjunct in patients with Parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol 74:216–224. https://doi.org/10.1001/jamaneurol.2016.4467
Schulz JB (2012) Effects of selegiline and rasagiline on disease progression in Parkinson’s disease. Basal Ganglia 2:S41–S45. https://doi.org/10.1016/j.baga.2012.08.003
Shoulson I, Oakes D, Fahn S, Lang A, Langston JW, LeWitt P, Olanow CW, Penney JB, Tanner C, Kieburtz K, Rudolph A, Parkinson Study Group (2002) Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: a randomised placebo controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann Neurol 51:604–612. https://doi.org/10.1002/ana.10191
Sivenius J, Sarasoja T, Aaltonen H, Heinonen E, Kilkku O, Reinikainen K (2001) Selegiline treatment facilitates recovery after stroke. Neurorehabil Neural Repair 15:183–190. https://doi.org/10.1177/154596830101500305
Smith KM, Eyal E, Weintraub D, Investigators ADAGIO (2015) Combined rasagiline and antidepressant use in Parkinson disease in the ADAGIO study: effects on nonmotor symptoms and tolerability. JAMA Neurol 72:88–95. https://doi.org/10.1001/jamaneurol.2014.2472
Stern MB, Marek KL, Friedman J, Hauser RA, LeWitt PA, Tarsy D, Olanow CW (2004) Double-blind, randomized, controlled trial of rasagiline as monotherapy in early Parkinson’s disease patients. Mov Disord 19:916–923. https://doi.org/10.1002/mds.20145
Stocchi F, Vacca L, Grassini P, De Pandis MF, Battaglia G, Cattaneo C, Fariello RG (2006) Symptom relief in Parkinson disease by safinamid: Biochemical and clinical evidence of efficacy beyond MAO-B inhibition. Neurology 67(7 Suppl. 2):S24–S29. https://doi.org/10.1212/wnl.67.7_suppl_2.s24
Stocchi F, Rascol O, Hauser RA, Huyck S, Tzontcheva A, Capece R, Ho TW, Sklar P, Lines C, Michelson D, Hewitt DJ, Preladenant Early Parkinson Disease Study Group (2017) Randomized trial of preladenant, given as monotherapy, in patients with early Parkinson disease. Neurology 88:2198–2206. https://doi.org/10.1212/WNL.0000000000004003
Tábi T, Vécsei L, Youdim MB, Riederer R, Szökő E (2020) Selegiline: a molecule with innovative potential. J Neural Transm (vienna) 127:831–842. https://doi.org/10.1007/s00702-019-02082-0
Tetrud JW, Langston JW (1989) The effect of deprenyl (selegiline) on the natural history of Parkinson’s disease. Science 245:519–522. https://doi.org/10.1126/science.2502843
Thorogood M, Armstrong B, Nichols T, Hollowell J (1998) Mortality in people taking selegiline: observational study. Br Med J 317:252–254. https://doi.org/10.1136/bmj.317.7153.252
Tolosa E, Stern MB (2012) Efficacy, safety and tolerability of rasagiline as adjunctive therapy in elderly patients with Parkinson’s disease. Eur J Neurol 19:258–264. https://doi.org/10.1111/j.1468-1331.2011.03484.x
Weintraub D, Hauser RA, Elm JJ, Pagan F, Davis MD, Choudhry A, MODERATO Investigators (2016) Rasagiline for mild cognitive impairment in Parkinson’s disease: a placebo-controlled trial. Mov Disord 31:709–714. https://doi.org/10.1002/mds.26617 (Epub 2016 Mar 31)
Yahr MD, Elizan TS, Moros D (1989) Selegiline in the treatment of Parkinson’s disease long-term experience. Acta Neurol Scand Suppl. 126:157–161. https://doi.org/10.1111/j.1600-0404.1989.tb01796.x
Yasar S, Goldberg JP, Goldberg SR (1996) Are metabolites of L-deprenyl (selegiline) useful or harmful? Indications from preclinical research. J Neural Transm Suppl 48:61–73. https://doi.org/10.1007/978-3-7091-7494-4_6
Yelekçi K, Büyüktürk B, Kayrak N (2013) In silico identification of novel and selective monoamine oxidase B inhhibitors. J Neural Transm (vienna) 120:853–858. https://doi.org/10.1007/s00702-012-0954-0
Youdim MBH, Gross A, Finberg JP (2001) Rasagiline [N-propargyl-1R (+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol 132:500–506. https://doi.org/10.1038/sj.bjp.0703826
Youdim MBH, Bar Am O, Yogev-Falach M, Weinreb O, Maruyama W, Naoi M, Amit T (2005) Rasagiline: neurodegeneration, neuroprotection, and mitochondrial permeability transition. J Neurosci Res 79:172–179. https://doi.org/10.1002/jnr.20350
Zhang H, Yin X, Ouyang Z, Chen J, Zhou S, Zhang C, Pan X, Wang S, Yang J, Feng Y, Zhang Q (2016) A prospective study of freezing of gait with early Parkinson disease in Chinese patients. Medicine (baltimore) 95:e4056. https://doi.org/10.1097/MD.0000000000004056
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
W. Jost is or was a consultant and/or speaker for the following companies: Abbvie, Bial, Desitin, Kyowa Kirin, Stada, UCB, Zambon.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jost, W.H. A critical appraisal of MAO-B inhibitors in the treatment of Parkinson’s disease. J Neural Transm 129, 723–736 (2022). https://doi.org/10.1007/s00702-022-02465-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02465-w