
Abstract
This post-hoc analysis investigated the long-term effects of safinamide on the course of dyskinesia and efficacy outcomes using data from a phase III, open-label 52-week study of safinamide 50 or 100 mg/day in Japanese patients with Parkinson’s disease (PD) with wearing-off. Patients (N = 194) were grouped using the UPDRS Part IV item 32: with and without pre-existing dyskinesia (pre-D subgroup; item 32 > 0 at baseline [n = 81], without pre-D subgroup; item 32 = 0 at baseline [n = 113]). ON-time with troublesome dyskinesia (ON-TD) increased significantly from baseline to Week 4 in the pre-D subgroup (+ 0.25 ± 0.11 h [mean ± SE], p = 0.0355) but gradually decreased up to Week 52 (change from baseline: − 0.08 ± 0.17 h, p = 0.6224); ON-TD did not change significantly in the Without pre-D subgroup. UPDRS Part IV item 32 score increased significantly at Week 52 compared with baseline in the Without pre-D subgroup, but no UPDRS Part IV dyskinesia related-domains changed in the pre-D subgroup. Both subgroups improved in ON-time without TD, UPDRS Part III, and Part II [OFF-phase] scores. The cumulative incidence of new or worsening dyskinesia (adverse drug reaction) at Week 52 was 32.5 and 5.0% in the pre-D and Without pre-D subgroups, respectively. This study suggested that safinamide led to short-term increasing dyskinesia but may be not associated with marked dyskinesia at 1-year follow-up in patients with pre-existing dyskinesia, and that it improved motor symptoms regardless of the presence or absence of dyskinesia at baseline. Further studies are warranted to investigate this association in more details.
Trial registration: JapicCTI-153057 (Registered: 2015/11/02).
Similar content being viewed by others


Introduction
Levodopa has yet to be surpassed as the most effective oral treatment for motor symptom control in Parkinson’s disease (PD) (Armstrong and Okun 2020; LeWitt and Fahn 2016). Motor complications such as involuntary movements (i.e., dyskinesias) and wearing-off often occur in PD as the disease progresses and within an estimated 6.5 years of chronic treatment with levodopa (Tran et al. 2018). Such dyskinesias can lead to impaired activities of daily living (ADL) (Pahwa et al. 2019) and quality of life (QoL) (Montel et al. 2009; Péchevis et al. 2005).
In addition to a reduction in levodopa dose, a dose reduction or discontinuation of monoamine oxidase (MAO)-B inhibitors or other dopaminergic drugs may be considered for patients with advanced PD who develop troublesome dyskinesia. As such, it is often difficult to choose a treatment that improves wearing-off without the patient newly developing dyskinesia or aggravating pre-existing dyskinesias (Hinson 2010).
The development of levodopa-induced dyskinesia (LID) is attributed to a decrease in dopamine neurons or stimulation of D1 receptors upon intermittent stimulation of direct pathway striatal neurons by levodopa (Cenci and Lundblad 2006; Ding et al. 2015). Furthermore, both animal and clinical studies have revealed that LID is linked to increases in extracellular glutamate. However, the exact pathophysiological mechanisms driving LID are still elusive, and those mentioned here are among many possible speculations.
Safinamide is a selective and reversible MAO-B inhibitor approved as an add-on therapy for patients with PD who are experiencing motor fluctuations with levodopa (Kurihara et al. 2021). In addition to MAO-B inhibition, safinamide modulates glutamate release by inhibiting sodium channels in vivo (Morari et al. 2018). In a preclinical study using a primate model of macaque monkeys, safinamide suppressed LID but increased the period of antiparkinsonian response to levodopa (Grégoire et al. 2013).
Several clinical studies have examined the effects of safinamide on dyskinesias, but the findings have been controversial. Study 018 reported that the change in the Dyskinesia Rating Scale was not significantly different between the safinamide and placebo groups after 24 months of treatment. However, in a subpopulation with moderate or severe dyskinesia, there was an improvement in the Dyskinesia Rating Scale with safinamide 100 mg/day (Borgohain et al. 2014; Cattaneo et al. 2015). In a large European observational study (SYNAPSES) in PD patients with wearing-off, the proportion of patients with dyskinesia tended to decrease from baseline (from 39.2 to 27.8%) after 12 months’ administration of safinamide 50 mg/day or 100 mg/day; however, in 13.7% of patients, dyskinesia was reported as a common adverse event (AE) after safinamide administration (Abbruzzese et al. 2021).
Although there have been long-term studies conducted to assess the effect of safinamide on dyskinesia after 24 months (Borgohain et al. 2014; Cattaneo et al. 2015), none of these reports analyzed the effects of safinamide on dyskinesia over time. Therefore, we considered it meaningful to explore the profile of safinamide according to the presence/absence of dyskinesia for treatment selection in a clinical setting. Thus, in this post-hoc study, we aimed to investigate the long-term effect of safinamide on the course of dyskinesia using the results of a Japanese phase III 52-week study of safinamide 50 mg/day or 100 mg/day in Japanese patients with PD who had wearing-off (Tsuboi et al. 2020). We also explored the safety and efficacy of safinamide for patients with dyskinesia prior to administration (pre-D group), as well as the incidence rate of new-onset dyskinesia.
Methods
Study design
The study was a phase III, multicenter, open-label study conducted at 29 centers in Japan between December 2015 and November 2017. Details of the study design are published elsewhere (Tsuboi et al. 2020). Briefly, the study included a 4-week observation (screening) period and a 52-week treatment period (4 weekly follow-ups from baseline [Visit 2] to the end of study drug administration or drug discontinuation [Visit 15]; Online Resource 1).
The study protocol was approved by an ethics committee at each study site, and all patients provided informed consent before study initiation. The study conduct adhered to the ethical principles of the Declaration of Helsinki, Good Clinical Practice Guidelines, and local laws and regulations. The study was registered in the Japan Pharmaceutical Information Center under the identifier JapicCTI-153057.
Patients
The study’s target population included Japanese patients aged ≥ 30 years diagnosed with PD according to the UK Parkinson’s Disease Society Brain Bank diagnostic criteria. All patients received a levodopa combination drug with a stable dose regimen (three doses/day or more and 300 mg/day or more) and must not have started treatment with an anti-PD drug other than levodopa combination drugs or must not have undergone a change in the dose regimen of such therapy during the observation period. Additionally, patients were required to have a mean daily OFF time of 2 or more hours. All patients had a modified Hoehn and Yahr stage of II–IV during the OFF-phase. Patients with evidence of dementia, major psychiatric illnesses, and/or severe and progressive medical illnesses, as well as those receiving antipsychotics, antidepressants, or drugs with antagonistic dopamine action, were excluded.
Treatment
Patients were administered safinamide at a dosage of 50 mg/day; the dose could be increased up to 100 mg once daily from Week 4 onward based on the following criteria: (1) there was no safety concern; (2) the therapeutic response to 50 mg/day was poor; and (3) the patient wanted to increase the dose. The criteria for dose reduction were as follows: (1) AEs attributable to excessive dopamine action made it difficult for the patient to continue the study; and (2) AEs did not improve even after reducing the dose of the levodopa combination drugs. A subsequent dose increase was prohibited.
Clinical features associated with dyskinesia
The clinical features associated with dyskinesia were evaluated based on the change from baseline to Week 52 in the mean daily ON-time with troublesome dyskinesia (ON-TD), which was assessed using a 24-h symptom diary, and changes in the Unified Parkinson’s Disease Rating Scale (UPDRS) Part IV items 32 (duration of dyskinesia, score 0–4), 33 (severity of dyskinesia, score 0–4), or 34 (dyskinesia with pain, score 0–4) as evaluated by a physician. Physicians explained to the subjects how to complete the 24-h symptom diary until sufficient understanding was achieved. The cumulative incidence rate of dyskinesia as an adverse drug reaction (ADR) from baseline was also evaluated.
Effects on wearing-off, motor symptoms, and ADL
The effects of wearing-off and motor symptoms were evaluated based on the change from baseline to Week 52 in the mean daily ON-time without troublesome dyskinesia (ON-WOTD), which was assessed using a 24-h symptom diary, and changes from baseline to Week 52 in UPDRS Part II (OFF-phase and ON-phase) and Part III (ON-phase).
Statistical analysis
As this was a post-hoc analysis, statistical methods were not prespecified. Imputation for drop-out and missing data was not conducted. The significance level of all testing was 5% (two-tailed) and no adjustments were made for multiplicity. SAS version 9.4 (SAS Institute, Cary, NC, USA) was used for the statistical analysis.
The full analysis set (FAS) included all subjects who received at least one dose of the study drug, had available evaluations of ON-time at baseline, and had at least one follow-up evaluation. The safety analysis set consisted of all patients who received at least one dose of the study drug.
Categorical variables were summarized as frequencies using number (n) and percentage (%), and continuous variables were presented using summary statistics. Changes from baseline to each time point were summarized and compared using a paired t test with data from patients who had both baseline and post-baseline time point evaluations.
For subgroup analyses, patients were categorized into two groups based on the UPDRS Part IV item 32: those with pre-existing dyskinesia (pre-D; UPDRS Part IV item 32 > 0 at baseline) and those without pre-existing dyskinesia (Without pre-D; UPDRS Part IV item 32 = 0 at baseline). To compare the differences in baseline values between the two subgroups, a Welch’s t test was used for continuous variables, a Fisher’s exact test was used for categorical variables, and a Wilcoxon rank-sum test was used to compare the number of concomitant non-levodopa antiparkinsonian drugs. Additional subgroup analyses were also conducted in patients who completed 52 weeks of treatment without any changes in the dose of levodopa combination drugs.
Results
Patient characteristics
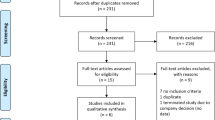
A total of 194 patients were included in the FAS, 132 patients completed the 52-week treatment without any changes in dose of levodopa combination drugs, and 203 were included in the safety analysis set (Fig. 1). Compared with the Without pre-D subgroup, the pre-D subgroup had a significantly higher proportion of women (75.3 vs 47.8%, p = 0.0001), significantly longer mean [standard deviation] duration of levodopa treatment (9.58 [4.41] years vs 5.44 [3.93] years, p < 0.0001), significantly higher levodopa doses (508.64 [183.34] mg vs 403.54 [129.04] mg, p < 0.0001), and significantly lower ADL according to the UPDRS Part II (OFF-phase) assessment (16.07 [6.86] vs 12.90 [7.71], p = 0.0029) at baseline (Table 1).

Flowchart showing patient disposition in this post-hoc analysis. Among AEs that led to discontinuation, dyskinesia was reported in 4 cases in the pre-D subgroup and 0 cases in the Without pre-D subgroup. All other AEs had different symptoms. AE adverse event, PD Parkinson’s disease, pre-D pre-existing dyskinesia W week
Clinical features associated with dyskinesia
In the pre-D subgroup, ON-TD increased significantly from baseline to Week 4 but gradually decreased up to Week 52 (Fig. 2). The mean (standard error) change from baseline to 52 weeks was − 0.08 (0.17) h (p = 0.6224). After 52 weeks, about half (FAS: 49%, Completed: 50%) of patients were taking the 100-mg/day dosage. In the Without pre-D subgroup, ON-TD did not change significantly compared with baseline throughout the observation period. A similar tendency was observed in the population who completed the study without any changes in the dose of levodopa combination drugs.

Average daily ON-time with troublesome dyskinesia (ON-TD) in the FAS. FAS full analysis set, pre-D pre-existing dyskinesia, SE standard error, W week. The p values indicate the difference at Week 4 vs baseline and were calculated using a paired t test based on patients who had both evaluations at baseline and each timepoint. *p = 0.0355; **p = 0.0246
Regarding changes in UPDRS Part IV (Table 2), no significant changes were observed from baseline to Week 52 for items 32, 33, 34, or 32–34 together in the pre-D subgroup. However, the UPDRS Part IV item 32 score at Week 52 had significantly increased when compared with baseline in the Without pre-D subgroup.
The cumulative incidence of new or worsening dyskinesia as an ADR in both subgroups is shown in Fig. 3. The cumulative incidence of new or worsening dyskinesia as an ADR at Week 52 was lower in the Without pre-D subgroup (5.0%, 6 of 120 patients) compared with the pre-D subgroup (32.5%, 27 of 83 patients). In the pre-D subgroup, 19 of the 27 patients experienced dyskinesia as an ADR within 12 weeks.

Progression of dyskinesia as an adverse drug reaction (safety analysis set). ADR adverse drug reaction, pre-D pre-existing dyskinesia, W week
Effects on wearing-off, motor symptoms, and ADL
ON-WOTD was significantly increased at each time points from Week 4 to Week 52 compared to baseline in both pre-D and Without pre-D patients (p < 0.05, each time point) (Fig. 4). A similar trend was observed in the population who completed the study without any changes in the dose of levodopa combination drugs.

Average daily ON-time without troublesome dyskinesia (ON-WOTD) in the FAS. FAS full analysis set, pre-D pre-existing dyskinesia, SE standard error, W week. The p values indicate the difference from baseline and were calculated using a paired t-test based on patients who had both evaluations at baseline and each timepoint. *p < 0.05; **p < 0.01
Online Resource 2 shows the changes in UPDRS Part III, Part II (ON-phase), and Part II (OFF-phase). Both UPDRS Part II (OFF-phase) and Part III showed significant improvement (p < 0.01, each time point for both parts) from Week 4 to Week 52 compared to baseline, regardless of the presence of dyskinesia at baseline. UPDRS Part II (ON-phase) did not change from baseline in the pre-D subgroup, but improved significantly from Week 4 to Week 52 compared to baseline in the Without pre-D subgroup.
Discussion
In this post-hoc analysis, safinamide temporarily worsened ON-TD, but this effect gradually eased during the 52-week treatment in the pre-D subgroup. An analysis of the subpopulation that completed 52 weeks of administration and did not undergo any changes in the dose of levodopa combination drugs showed that dyskinesia did not worsen in the long term, indicating that the long-term ON-TD reduction tendency did not result from patient drop-out or levodopa dose reduction. In the pre-D subgroup, none of the UPDRS Part IV dyskinesia related-domains worsened at Week 52 compared with baseline, which is consistent with the ON-TD result, and the incidence of new-onset dyskinesia reported as an ADR tended to be low. There were few cases of withdrawal due to AEs at the initial stage of safinamide administration. The number of dyskinesia cases that led to discontinuation of administration was small in both subgroups (n = 4) (Tsuboi et al. 2020). In addition, patients showed improvements in wearing-off, motor symptoms, and ADL (UPDRS Part II [OFF-phase]) regardless of the presence or absence of dyskinesia at baseline.
The definition of the pre-D subgroup was based on the clinician-reported UPDRS item 32 (duration of dyskinesia), with a score of 1 used as the cut-off. Of the 81 patients in the pre-D subgroup, 38 patients had troublesome dyskinesia at baseline (ON-TD > 0). In the pre-D subgroup, the proportion of women was higher, the duration of levodopa treatment was longer, and the dose of levodopa was higher than those in the Without pre-D subgroup, all of which are consistent with the reported risk factors for dyskinesia (Eusebi et al. 2018).
From the results of UPDRS Part IV, the duration of dyskinesia (item 32) was extended in the Without pre-D subgroup. In contrast, ON-TD was relatively stable, and UPDRS part II score (ON-phase) was significantly improved from baseline in this subgroup, which suggests that even though the time of dyskinesia increased, ADL were not impaired by this effect. However, the results of this study should be interpreted with caution because UPDRS Part IV and ADR were clinician-reported, while ON-TD and ON-WOTD were patient-reported using 24-h symptom diaries. In fact, a gap between physician assessment and patient self-awareness concerning the presence of dyskinesia has been recently reported (Ogura et al. 2021).
As safinamide inhibits the degradation of dopamine by inhibiting MAO-B (Kurihara et al. 2021) and an excess of dopamine causes dyskinesia, it is considered that a slight increase in ON-TD during the initial treatment phase is caused by the dopaminergic action of safinamide. Nonetheless, dyskinesia was not reported to worsen in the long term. Involvement of glutamate signaling has also been suggested in dyskinesias, given that glutamatergic striatal neurons are hypertrophied, and signal transduction is enhanced in the direct pathway (Cenci and Konradi 2010; Cenci and Lundblad 2006; Cerasa et al. 2014; Holtmaat and Svoboda 2009). Constitutive changes in the subunits of NMDA and AMPA receptors suggest an association with glutamate. In particular, it has been observed in animal and human studies that changes in the subunit of the NMDA receptor in the striatum are associated with the onset of dyskinesia (Gardoni et al. 2006, 2012; Mellone et al. 2015).
Safinamide has non-dopaminergic actions, such as its action as a sodium channel blocker, and it has been reported to suppress changes in the GluN2A/GluN2B ratio caused by chronic levodopa administration (Gardoni et al. 2018). Patients with LID have abnormal cortical facilitation, suggesting overactive glutamatergic neurotransmission in the cortex, and this dysfunction was restored via modulation of synaptic plasticity mechanisms by the long-term safinamide effect (Guerra et al. 2019, 2021). It is not clear whether this action of safinamide is mediated by blocking sodium channels; however, safinamide may affect dyskinesia. Studies have reported that amantadine, which has an NMDA receptor antagonistic effect, is effective against LID in the long term (Ory-Magne et al. 2014).
Because ON-TD may temporarily worsen after the administration of safinamide in the pre-D subgroup, it is essential to evaluate the location and time of dyskinesia carefully. During this study period, levodopa and other anti-PD drugs were prescribed at a set dosage, but it is necessary to consider adjusting the doses of these drugs during clinical practice.
LID has been reported to develop in approximately 36% of patients 4–6 years after levodopa initiation (Ahlskog and Muenter 2001). It affects more than 50% of PD patients who have been treated with levodopa for 5 years or more (Grandas et al. 1999). The baseline PD morbidity of the Without pre-D population in this study was 8.01 years, and the mean levodopa treatment duration was 5.44 years; therefore, the patient background suggested that this population was likely to develop LID. However, the cumulative incidence of dyskinesia at Week 52 was 5.0%, which is approximately 6 times lower than that in the pre-D subgroup (32.5%). The incidence of new AEs within 12 weeks of istradefylline treatment was reported to be 4.8% at 20 mg/day and 7.2% at 40 mg/day (Elmer et al. 2020); hence, the risk of new-onset ADRs with safinamide is not high. Nevertheless, the cumulative incidence of new or worsening dyskinesia as an ADR, which was relatively high in the Pre-D subgroup, warrants caution.
It does not seem likely that the long-term effects on dyskinesias were caused by reduced dopaminergic stimulation of safinamide resulting from the prolongation of ON-WOTD and the improvement of UPDRS Part II (OFF-phase) or Part III continued for 52 weeks in this study. The improvement of wearing-off with safinamide reportedly continued for up to 2 years (Borgohain et al. 2014; Tsuboi et al. 2020). The presence of dyskinesia at baseline indicates dopaminergic nerve degeneration and loss of dopamine buffering capacity; nonetheless, we observed that motor symptoms and wearing-off were also improved in the pre-D subgroup. Abnormal glutamate receptor activity in the basal ganglia has previously been reported in advanced PD (Cenci and Lindgren 2007; Espay et al. 2018; Gardoni et al. 2006; Sgambato-Faure and Cenci 2012), and as such, it is possible that the non-dopaminergic action of safinamide (Cattaneo et al. 2018) contributed to its efficacy in the pre-D subgroup.
Limitations
The main limitations of this study were that it was a post-hoc analysis of an open-label, single-arm study, and statistical methods were not prespecified. Imputation for drop-out and missing data was not conducted, and no adjustments were made for multiplicity. It should be noted that few patients had troublesome dyskinesia at baseline in the pre-D group. Additionally, the treatment restrictions in this study are different from those in clinical practice. In clinical practice, measures such as increasing the number of doses of levodopa or reducing the daily dose should be considered. This interpretation of the FAS results is also limited because the dosage of safinamide and concomitant antiparkinsonian drugs could be adjusted at the onset of AEs, such as dyskinesia. Furthermore, the levodopa equivalent doses were not analyzed at each time point because data on concomitant antiparkinsonian drugs were often missing. Therefore, verification of our findings in clinical practice is also necessary. Finally, it should be noted that the definition of dyskinesia as an ADR includes onset and exacerbation of dyskinesia, but this distinction was not considered in this study.
Conclusions
Regardless of the presence or absence of dyskinesia at baseline, long-term 52-week safinamide adjunctive treatment (50 and 100 mg/day) did not cause marked dyskinesia and improved ON-WOTD, motor symptoms, and ADL based on the UPDRS Part II (OFF-phase). Conversely, a short-term, mild increase in ON-TD was observed. This tendency was also observed in the limited population without any changes in the dose of levodopa combination drugs. In addition, the incidence of new-onset dyskinesia tended to be low. Regarding the precautions and benefits of safinamide in the pre-D subgroup at baseline, as with other anti-PD drugs, the addition of safinamide could be a treatment option for patients with wearing-off. However, further prospective clinical trials are warranted to investigate these potential benefits and the clinical relevance of safinamide on dyskinesia, for example, the difference in effects between safinamide 50 and 100 mg/day.
Data availability
All data and materials support the reported claims and comply with standards of data transparency. Data will be made available on reasonable request.
Change history
08 September 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00702-022-02543-z
References
Abbruzzese G, Kulisevsky J, Bergmans B, Gomez-Esteban JC, Kägi G, Raw J, Stefani A, Warnecke T, Jost WH (2021) A european observational study to evaluate the safety and the effectiveness of safinamide in routine clinical practice: the SYNAPSES trial. J Parkinsons Dis 11:187–198. https://doi.org/10.3233/jpd-202224
Ahlskog JE, Muenter MD (2001) Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 16:448–458. https://doi.org/10.1002/mds.1090
Armstrong MJ, Okun MS (2020) Diagnosis and treatment of parkinson disease: a review. JAMA 323:548–560. https://doi.org/10.1001/jama.2019.22360
Borgohain R, Szasz J, Stanzione P, Meshram C, Bhatt MH, Chirilineau D, Stocchi F, Lucini V, Giuliani R, Forrest E, Rice P, Anand R (2014) Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov Disord 29:1273–1280. https://doi.org/10.1002/mds.25961
Cattaneo C, Kulisevsky J, Tubazio V, Castellani P (2018) Long-term efficacy of safinamide on parkinson’s disease chronic pain. Adv Ther 35(4):515–522. https://doi.org/10.1007/s12325-018-0687-z
Cattaneo C, Ferla RL, Bonizzoni E, Sardina M (2015) Long-term effects of safinamide on dyskinesia in mid- to late-stage Parkinson’s disease: a post-hoc analysis. J Parkinsons Dis 5:475–481. https://doi.org/10.3233/JPD-150569
Cenci MA, Konradi C (2010) Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res 183:209–233. https://doi.org/10.1016/s0079-6123(10)83011-0
Cenci MA, Lindgren HS (2007) Advances in understanding L-DOPA-induced dyskinesia. Curr Opin Neurobiol 17:665–671. https://doi.org/10.1016/j.conb.2008.01.004
Cenci MA, Lundblad M (2006) Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J Neurochem 99:381–392. https://doi.org/10.1111/j.1471-4159.2006.04124.x
Cerasa A, Fasano A, Morgante F, Koch G, Quattrone A (2014) Maladaptive plasticity in levodopa-induced dyskinesias and tardive dyskinesias: old and new insights on the effects of dopamine receptor pharmacology. Front Neurol 5:49. https://doi.org/10.3389/fneur.2014.00049
Ding S, Li L, Zhou FM (2015) Nigral dopamine loss induces a global upregulation of presynaptic dopamine D1 receptor facilitation of the striatonigral GABAergic output. J Neurophysiol 113:1697–1711. https://doi.org/10.1152/jn.00752.2014
Elmer L, Toyama K, Parno J, Braccia D, Ristuccia R, Mori A (2020) Safety and efficacy of istradefylline, an adenosine A2A receptor antagonist, as a function of baseline dyskinesia (BL-dyskinesia) in Parkinson’s disease (PD): A pooled analysis of 4 studies [abstract]. Mov Disord 35(suppl):1
Espay AJ, Morgante F, Merola A, Fasano A, Marsili L, Fox SH, Bezard E, Picconi B, Calabresi P, Lang AE (2018) Levodopa-induced dyskinesia in Parkinson disease: current and evolving concepts. Ann Neurol 84:797–811. https://doi.org/10.1002/ana.25364
Eusebi P, Romoli M, Paoletti FP, Tambasco N, Calabresi P, Parnetti L (2018) Risk factors of levodopa-induced dyskinesia in Parkinson’s disease: results from the PPMI cohort. NPJ Parkinsons Dis 4:33. https://doi.org/10.1038/s41531-018-0069-x
Gardoni F, Picconi B, Ghiglieri V, Polli F, Bagetta V, Bernardi G, Cattabeni F, Di Luca M, Calabresi P (2006) A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J Neurosci 26:2914–2922. https://doi.org/10.1523/jneurosci.5326-05.2006
Gardoni F, Sgobio C, Pendolino V, Calabresi P, Di Luca M, Picconi B (2012) Targeting NR2A-containing NMDA receptors reduces L-DOPA-induced dyskinesias. Neurobiol Aging 33:2138–2144. https://doi.org/10.1016/j.neurobiolaging.2011.06.019
Gardoni F, Morari M, Kulisevsky J, Brugnoli A, Novello S, Pisanò CA, Caccia C, Mellone M, Melloni E, Padoani G, Sosti V, Vailati S, Keywood C (2018) Safinamide modulates striatal glutamatergic signaling in a rat model of levodopa-induced dyskinesia. J Pharm Exp Ther 367:442–451. https://doi.org/10.1124/jpet.118.251645
Grandas F, Galiano ML, Tabernero C (1999) Risk factors for levodopa-induced dyskinesias in Parkinson’s disease. J Neurol 246:1127–1133. https://doi.org/10.1007/s004150050530
Grégoire L, Jourdain VA, Townsend M, Roach A, Di Paolo T (2013) Safinamide reduces dyskinesias and prolongs L-DOPA antiparkinsonian effect in parkinsonian monkeys. Parkinsonism Relat Disord 19:508–514. https://doi.org/10.1016/j.parkreldis.2013.01.009
Guerra A, Suppa A, D’Onofrio V, Di Stasio F, Asci F, Fabbrini G, Berardelli A (2019) Abnormal cortical facilitation and L-dopa-induced dyskinesia in Parkinson’s disease. Brain Stimul 12:1517–1525. https://doi.org/10.1016/j.brs.2019.06.012
Guerra A, Asci F, Zampogna A, D’Onofrio V, Suppa A, Fabbrini G, Berardelli A (2021) Long-term changes in short-interval intracortical facilitation modulate motor cortex plasticity and L-dopa-induced dyskinesia in Parkinson’s disease. Brain Stimul 15:99–108. https://doi.org/10.1016/j.brs.2021.11.016
Hinson VK (2010) Parkinson’s disease and motor fluctuations. Curr Treat Options Neurol 12:186–199. https://doi.org/10.1007/s11940-010-0067-8
Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10:647–658. https://doi.org/10.1038/nrn2699
Kurihara K, Mishima T, Fujioka S, Tsuboi Y (2022) Efficacy and safety evaluation of safinamide as an add-on treatment to levodopa for parkinson’s disease. Expert Opin Drug Saf 10(1080/14740338):1988926
LeWitt PA, Fahn S (2016) Levodopa therapy for Parkinson disease: a look backward and forward. Neurology 86:S3-12. https://doi.org/10.1212/wnl.0000000000002509
Mellone M, Stanic J, Hernandez LF, Iglesias E, Zianni E, Longhi A, Prigent A, Picconi B, Calabresi P, Hirsch EC, Obeso JA, Di Luca M, Gardoni F (2015) NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: from experimental models to patients. Front Cell Neurosci 9:245. https://doi.org/10.3389/fncel.2015.00245
Montel S, Bonnet AM, Bungener C (2009) Quality of life in relation to mood, coping strategies, and dyskinesia in Parkinson’s disease. J Geriatr Psychiatry Neurol 2:95–102. https://doi.org/10.1177/0891988708328219
Morari M, Brugnoli A, Pisanò CA, Novello S, Caccia C, Melloni E, Padoani G, Vailati S, Sardina M (2018) Safinamide differentially modulates in vivo glutamate and GABA release in the rat hippocampus and basal ganglia. J Pharm Exp Ther 364:198–206. https://doi.org/10.1124/jpet.117.245100
Ogura H, Nakagawa R, Ishido M, Yoshinaga Y, Watanabe J, Kurihara K, Hayashi Y, Nagaki K, Mishima T, Fujioka S, Tsuboi Y (2021) Evaluation of motor complications in Parkinson’s disease: understanding the perception gap between patients and physicians. Parkinsons Dis 2021:1599477. https://doi.org/10.1155/2021/1599477
Ory-Magne F, Corvol JC, Azulay JP, Bonnet AM, Brefel-Courbon C, Damier P, Dellapina E, Destée A, Durif F, Galitzky M, Lebouvier T, Meissner W, Thalamas C, Tison F, Salis A, Sommet A, Viallet F, Vidailhet M, Rascol O (2014) Withdrawing amantadine in dyskinetic patients with Parkinson disease: the AMANDYSK trial. Neurology 82:300–307. https://doi.org/10.1212/wnl.0000000000000050
Pahwa R, Isaacson S, Jimenez-Shaheed J, Malaty IA, Deik A, Johnson R, Patni R (2019) Impact of dyskinesia on activities of daily living in Parkinson’s disease: Results from pooled phase 3 ADS-5102 clinical trials. Parkinsonism Relat Disord 60:118–125. https://doi.org/10.1016/j.parkreldis.2018.09.005
Péchevis M, Clarke CE, Vieregge P, Khoshnood B, Deschaseaux-Voinet C, Berdeaux G, Ziegler M (2005) Effects of dyskinesias in Parkinson’s disease on quality of life and health-related costs: a prospective European study. Eur J Neurol 12:956–963. https://doi.org/10.1111/j.1468-1331.2005.01096.x
Sgambato-Faure V, Cenci MA (2012) Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol 96:69–86. https://doi.org/10.1016/j.pneurobio.2011.10.005
Tran TN, Vo TNN, Frei K, Truong DD (2018) Levodopa-induced dyskinesia: clinical features, incidence, and risk factors. J Neural Transm (vienna) 125:1109–1117. https://doi.org/10.1007/s00702-018-1900-6
Tsuboi Y, Hattori N, Yamamoto A, Sasagawa Y, Nomoto M (2020) Long-term safety and efficacy of safinamide as add-on therapy in levodopa-treated Japanese patients with Parkinson’s disease with wearing-off: results of an open-label study. J Neurol Sci 416:117012. https://doi.org/10.1016/j.jns.2020.117012
Acknowledgements
The authors would like to thank the patients who participated in this trial and their families, as well as the staff at all investigational sites. We also thank Michinori Koebis, PhD and Yuki Kogo of Eisai Co., Ltd. for contributing to the design of the study, and Keyra Martinez Dunn, MD, of Edanz (www.edanz.com) for providing medical writing support, which was funded by Eisai Co., Ltd through EP Croit. Employees of Eisai Co., Ltd. were involved in study design; in the collection, analysis, and interpretation of data; and in the decision to submit the article for publication. Employees of Meiji Seika Pharma Co., Ltd. were not involved in these aspects of the work.
Funding
This research was funded by Eisai Co., Ltd. The phase III clinical trial was sponsored by Meiji Seika Pharma Co., Ltd.
Author information
Authors and Affiliations
Contributions
NH, TK, TI, IS, MN, and YT conceived of and designed the study, and interpreted the data. IS conducted the statistical analysis. TK prepared the first draft of the manuscript. NH, TI, IS, MN, and YT reviewed the manuscript drafts and provided important intellectual input. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
NH received personal fees from Meiji Seika Pharma Co., Ltd. during the conduct of the submitted work; grants from Ono Pharmaceutical Co., Ltd., FP Corp., Eisai Co., Ltd., and Nihon Mediphysics Co., Ltd.; personal fees from Sumitomo Dainippon Pharma Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Kyowa Hakko-Kirin Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., FP Corp., Eisai Co., Ltd., Kissei Pharmaceutical Co., Ltd., Nihon Medi-physics Co., Ltd., Novartis Pharma K.K., Biogen Idec Japan Ltd., AbbVie GK, Boston Scientific Japan K.K., Sanofi K.K., Alexion Pharmaceuticals, Inc., Mylan N.V., Daiichi Sankyo Co., Ltd., Hisamitsu Pharmaceuticals Co., Inc., and Kao Corp.; and reports donations to the department, endowed research departments and joint collaborative research departments from Sumitomo Dainippon Pharma Co., Ltd., Otsuka Pharmaceutical, Co., Ltd., Takeda Pharmaceutical Co., Ltd., Kyowa Hakko-Kirin Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., Eisai Co., Ltd., GSK K.K., Kissei Pharmaceutical Co. Ltd., Novartis Pharma K.K., Nihon Medi-physics Co., Ltd., Biogen Idec Japan Ltd., AbbVie GK, Medtronic, Inc., Boston Scientific Japan K.K., Ono Pharmaceutical Co., Ltd., Hydrogen Health Medical Labo Co., ABIST Co., Ltd., Daiwa Co., Ltd., Bayer Yakuhin Ltd., Nihon Pharmaceutical Co., Ltd., Asahi Kasei Medical Co., Ltd., MiZ Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Daiichi Sankyo Co. Ltd., and OHARA Pharmaceutical Co., Ltd. outside the submitted work. TK, TI, and IS are employees of Eisai Co., Ltd. MN received lecture fees from Eisai Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Takeda Pharmaceutical Co., Ltd., and consulting fees from Kissei Pharmaceutical Co. Ltd., and Hisamitsu Pharmaceutical Co., Ltd. YT received Honoraria from Eisai Co., Ltd., during the conduct of the submitted work, and Sumitomo Dainippon Pharma Co., Ltd., Takeda Pharmaceutical Co., Ltd., Novartis Pharma K.K., AbbVie GK., Otsuka Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., Kyowa Kirin Co., Ltd., Sunwels Co., Ltd., Eisai Co., Ltd. and Nipro Co., Ltd., outside the submitted work.
Ethical approval
The study protocol was approved by an ethics committee at each study site. The study conduct adhered to the ethical principles of the Declaration of Helsinki, Good Clinical Practice Guidelines, and local laws and regulations.
Informed consent
Informed consent was obtained from all the participants in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
702_2022_2532_MOESM2_ESM.pdf
Supplementary file2 Change from baseline in UPDRS Part III, Part II (ON), and Part II (OFF) The p values indicate the difference from baseline. *p < 0.05; **p < 0.01 Pre-D pre-existing dyskinesia, SE standard error, UPDRS unified Parkinson’s disease rating scale, W week (PDF 89 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hattori, N., Kamei, T., Ishida, T. et al. Long-term effects of safinamide adjunct therapy on levodopa-induced dyskinesia in Parkinson’s disease: post-hoc analysis of a Japanese phase III study. J Neural Transm 129, 1277–1287 (2022). https://doi.org/10.1007/s00702-022-02532-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02532-2