
Abstract
A series of compounds containing a 1,7,7-trimethylbicyclo[2.2.1]heptane fragment were evaluated for their antiviral activity against influenza A virus strain A/Puerto Rico/8/34 (H1N1) in vitro. The most potent antiviral compound proved to be a quaternary ammonium salt based on (-)-borneol, 10a. In in vitro experiments, compound 10a inhibited influenza A viruses (H1, H1pdm09, and H3 subtypes), with an IC50 value of 2.4-16.8 µM (depending on the virus), and demonstrated low toxicity (CC50 = 1311 µM). Mechanism-of-action studies for compound 10a revealed it to be most effective when added at the early stages of the viral life cycle. In direct haemolysis inhibition tests, compound 10a was shown to decrease the membrane-disrupting activity of influenza A virus strain A/Puerto Rico/8/34. According to molecular modelling results, the lead compound 10a can bind to different sites in the stem region of the viral hemagglutinin.
Similar content being viewed by others

Introduction
Influenza is a serious challenge to medical science and human health worldwide. Influenza A virus belongs to the family Orthomyxoviridae; it is an enveloped virus containing a segmented single-stranded RNA genome of negative polarity. Because of its segmented nature and high rate of polymerase errors, influenza virus demonstrates a high rate of mutation, leading to the emergence of novel variants, including those with pandemic potential. Four influenza pandemics have occurred in the past 100 years: H1N1 Spanish influenza in 1918, H2N2 Asian influenza in 1957, H3N2 Hong Kong influenza in 1968, and H1N1 swine influenza in 2009 [1]. Influenza virus infection results in high morbidity and about 250,000 to 500,000 fatal cases annually. It can cause multiple complications for the patient, resulting in multi-organ failure, thereby leading to high pathogenicity and mortality [2]. Usually, influenza A virus infects the upper respiratory tract and causes mild respiratory symptoms. However, in risk groups, the infection spreads to the lower respiratory tract, resulting in viral pneumonia. This occurs mainly in patients with a weakened immune system, including the elderly. The limited effectiveness of current treatments in the late phase of the disease highlights the need for additional therapies.
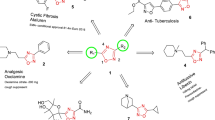
Four classes of antiviral drugs have been approved for treating influenza: M2 ion channel inhibitors (adamantane derivatives and the isoborneol derivative deitiforin), neuraminidase inhibitors, membrane fusion inhibitors, and RNA-dependent RNA polymerase inhibitors (Fig. 1). Of these, only the adamantane derivatives (amantadine and rimantadine) and neuraminidase inhibitors (oseltamivir, zanamivir, and peramivir) are approved by the Food and Drug Administration (FDA). In addition, laninamivir, a long-acting neuraminidase inhibitor, was launched in Japan to treat influenza A and B in 2010. Favipiravir (T-705) and ribavirin are nucleoside analogues with a broad range of antiviral activity. They inhibit the viral polymerase enzymatic complex and induce lethal mutagenesis in the viral genome, thus decreasing the infectivity of viral progeny [3,4,5,6]. Umifenovir (arbidol), an inhibitor of the fusion of the viral envelope with the endosome membrane, was developed in Russia and was recently approved for the treatment of influenza in China [7]. Recently, an inhibitor of viral endonuclease activity, baloxavir marboxil (Xofluza®), was approved for the treatment of influenza infection [8].

Anti-influenza drugs approved for clinical use to treat influenza virus infection
As mentioned above, owing to the error-prone activity of its viral polymerase and its segmented genome, influenza virus is able to mutate to generate drug-resistant variants. In particular, due to the widespread resistance of influenza viruses to adamantane derivatives, M2 ion channel inhibitors are currently not recommended for therapy [9]. Also, resistance to oseltamivir (Tamiflu) is observed to be spreading rapidly [10]. Since influenza virus is capable of developing resistance to the available antivirals, there is a real need to develop novel inhibitors of this virus.
In previous studies, our research group showed that (+)-camphor and (-)-borneol are potentially useful scaffolds for synthesizing antiviral drugs. In particular, the camphor imine derivatives, including compounds 1 and 2 were found to possess high antiviral activity (Fig. 2) [11,12,13]. The hit compound camphecene (Camphecin®) was shown to directly inhibit the acid-induced membrane-disrupting activity of the viral haemagglutinin (HA) of influenza A viruses [14]. Also, dimeric quaternary ammonium camphor derivatives were demonstrated to be highly effective as inhibitors of influenza A virus replication (strain California/07/09 (H1N1)pdm09), with compound 3 being the most active [15]. Moreover, for the past decade, our group has been involved in the synthesis of biologically significant novel (-)-borneol derivatives. We have discovered several borneol esters with activity against Ebola virus (EBOV), Marburg virus (MARV), and influenza virus – for example, compounds 4 and 5 [16,17,18]. Moreover, we have shown that the possible mechanism of inhibition of EBOV is binding to the active site of the EBOV glycoprotein [19]. Members of the family Filoviridae, including EBOV and MARV, and of the family Orthomyxoviridae, including influenza virus, have class I fusion proteins with similar pre- and post-fusion forms [20]. It can be assumed that bicyclic monoterpenoids, including the 1,7,7-trimethylbicyclo[2.2.1]heptane structural fragment, are promising scaffolds for the synthesis of inhibitors of viruses with class I fusion proteins, such as influenza virus.

(+)-Camphor and (-)-borneol derivatives with potent activity against influenza viruses A(H1N1)pdm09. SI is the selectivity index.
In the present work, we describe the synthesis and antiviral activity of quaternary ammonium salts with groups found in our previous research to be responsible for antiviral activity, such as the 1,7,7-trimethylbicyclo[2.2.1]heptane fragment, quaternary nitrogen atom, and imino and ester group. In addition, we have conducted mechanism-of-action studies with the most potent compound.
Materials and methods
Chemistry
General
Reagents and solvents were purchased from commercial suppliers and used as received. Dry solvents were obtained according to standard procedures. Column chromatography was performed on Macherey-Nagel 60–200 µm silica gel. All of the target compounds reported in this publication were of at least 98% purity. The synthesis of derivatives 1 and 2 [11] and 8a-c and 9a-c [19] were described previously. Compounds 6-7, 10a and c, and 11a and b, have not been described previously in the literature. 1H and 13C NMR spectra were recorded on Bruker AV-300 (300.13 and 75.47 MHz, respectively), AV400 (400.13 and 100.78 MHz, respectively), and DRX 500 (500.13 and 125.76 MHz, respectively) spectrometers in CDCl3; chemical shifts δ, in ppm relative to residual [δ(CHCl3) 7.24, δ(CDCl3) 76.90 ppm]. The atom numbering in the compounds is given for assigning the signals in the NMR spectra and does not match the standard nomenclature of compounds. Elemental analysis was carried out using a Euro EA 3000 C, H, N, S-analyser. Analysis of Br was carried out by the mercurimetric titration method. Analysis of I was carried out by the iodometric titration method. X-ray data were collected at room temperature using a Bruker Kappa Apex II CCD diffractometer with graphite monochromated MoKα radiation (λ = 0.71073 Å) by the φ, ω-scan method. The data were corrected for absorption using a multi-scan method with the SAINT program. The structure was solved by direct methods using SHELXS97, and refinement was carried out by a full-matrix least-squares technique, using SHELXL97. Anisotropic displacement parameters were included for all non-hydrogen atoms. All H atoms were positioned geometrically and treated as riding on their parent C atoms.
General procedure for the target camphor derivatives 6 and 7
A solution of compounds 1 or 2 (4 mmol) and anhydrous CH3CN (10 mL) was treated with an excess of iodomethane and heated in a bath at 70–75°C for 6 h. The solvent was removed at reduced pressure. The resulting precipitate was purified via silica gel column chromatography (CHCl3/MeOH eluent, (100:0→0:100)).
N,N,N-Trimethyl-2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethanaminium iodide (6).
Yield: 46%; mp: 195–197°C; 1H NMR (400 MHz, CDCl3, J Hz) δ ppm: 0.67 (3H, s, Me-9), 0.84 (3H, s, Me-8), 0.88 (3H, s, Me-10), 1.17-1.27 (2H, m, H-4endo, H-5endo), 1.59-1.68 (1H, m, H-5exo), 1.77-1.86 (2H, m, H-2endo, H-4exo), 1.94-1.99 (1H, m, H-3), 2.36-2.46 (1H, m, H-2exo), 3.52 (9H, s, Me-13, Me-14, Me-15), 3.64-3.75 (2H, m, H-12), 3.85-3.95 (2H, m, H-11). 13C NMR (75 MHz, CDCl3) δ: 186.36 s (C-1), 66.41 t (C-12), 54.61 q (Me-13, Me-14, Me-15), 53.90 s (C-6), 46.97 s (C-7), 46.47 t (C-11), 43.35 d (C-3), 35.61 t (C-2), 31.54 t (C-5), 26.71 t (C-4), 19.20 q (Me-9), 18.39 q (Me-10), 10.81 q (Me-8). Anal. Calcd for C15H29IN2 C, 49.45; H, 8.02; I, 34.83; N, 7.69 %. Found, %: C 48.54; H 8.10; N 7.64; I 34.57.
N,N,N-Trimethyl-3-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)propan-1-aminium iodide (7)
Yield, 76%; mp: 231°C; 1H NMR (400 MHz, CDCl3) δ: 0.71 (3H, s, Me-9), 0.88 (3H, s, Me-8), 1.00 (3H, s, Me-10), 1.23-1.39 (2H, m, H-4endo, H-5endo), 1.64-1.73 (1H, m, H-5exo), 1.76-1.85 (1H, m, H-4exo), 1.98-2.02 (1H, m, H-3), 2.11 (1H, d, 2J=19.6, H-2endo), 2.20-2.29 (2H, m, H-12), 2.54-2.62 (1H, m, H-2exo), 3.41 (9H, s, Me-14, Me-15, Me-16), 3.41-3.47 (2H, m, H-13), 3.71-3.77 (2H, m, H-11). 13C NMR (75 MHz, CDCl3) δ: 191.13 s (C-1), 64.86 t (C-13), 55.35 t (C-11), 53.93 q (Me-14, Me-15, Me-16), 48.13 s (C-6), 46.96 s (C-7), 43.46 d (C-3), 36.75 t (C-2), 31.86 t (C-5), 26.56 t (C-4), 23.94 t (C-12), 19.58 q (Me-9), 18.52 q (Me-10), 11.40 q (Me-8). Anal. Calcd for C16H31IN2 C, 50.79; H, 8.26; I, 33.54; N, 7.40%. Found, %: C 49.68; H 8.19; N 7.32; I 33.82.
Synthesis of (-)-borneol derivatives 10a, 10c and 11a-b
N,N,N-Trimethyl-2-oxo-2-((1S,2R,4S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yloxy)ethanaminium iodide (10a)
A solution of compound 8a (4 mmol) and anhydrous CH3CN (10 mL) was treated with an excess of iodomethane and heated in a bath at 70–75°C for 6 h. The solvent was removed at reduced pressure. The crude product was purified by recrystallisation from CH3CN. Yield: 62%; mp:242°C; 1H NMR (400 MHz, DMSO-d6, J Hz) δ ppm: 0.83 (3H, s, Me-9), 0.87 (3H, s, Me-8), 0.89 (3H, s, Me-10), 1.04(1H, dd, 2J = 13.7, J2endo,1exo=3.5, H-2endo), 1.17-1.25 (1H, m, H-4endo), 1.27-1.37 (1H, m, H-5exo), 1.67-1.77 (2H, m, H-3, H-4exo), 1.79-1.87 (1H, m, H-5endo), 2.27-2.37 (1H, m, H-2exo), 3.24 (9H, s, Me-13, Me-14, Me-15), 4.50 (1H, AB-d, J1,2=16.7 Hz, H-12), 4.55 (1H, AB-d, J2,1=16.7 Hz, H-12), 4.95 (1H, m, H-1exo). 13C NMR (125 MHz, DMSO-d6) δ: 165.28 s (C-11), 81.75 d (C-1), 62.89 t (C-12), 53.46 q (Me-13, Me-14, Me-15), 48.86 s (C-6), 47.79 s (C-7), 44.32 d (C-3), 36.03 t (C-2), 27.69 t (C-4), 26.85 t (C-5), 19.69 q (Me-9), 18.77 q (Me-10), 13.61 q (Me-8). Anal. Calcd for C15H28INO2 C, 47.25; H, 7.40; I, 33.28; N, 3.67 %. Found, %: C 47.19; H 7.26; I 33.21; N 3.73.
Crystal data for compound 10a
C15H28INO2, M = 381.28, monoclinic, space group P21, a = 12.2791(5), b = 7.1686(3), c = 20.9901(9) Å, β = 101.030(2)°, V = 1813.50(13) Å3, Z = 4, Dcalc = 1.396 mg•m-3, μ = 1.766 mm-1. Data collection yielded 45735 reflections with θ < 30.2° resulting in 10595 unique, averaged reflections, 7365 with I > 2σ(I). Full-matrix least-squares refinement led to a final R = 0.0440, wR2 = 0.1375, GOF = 0.936 for I > 2σ(I). CCDC 1813408 contains supplementary crystallographic data for the structure. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html.
N,N,N-Trimethyl-4-oxo-4-((1S,2R,4S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yloxy)butan-1-aminium iodide (10c)
The synthesis of compound 10c was performed in analogy to the synthesis compound 10a. Yield: 43%; mp:171.5°C; 1H NMR (400 MHz, CDCl3, J Hz) δ ppm: 0.76 (3H, s, Me-9), 0.81 (3H, s, Me-8), 0.83 (3H, s, Me-10), 0.90 (1H, dd, 2J=13.7, J2endo,1exo=3.5, H-2endo), 1.12-1.28 (2H, m, H-4endo, H-5exo), 1.60-1.73 (2H, m, H-3, H-4exo), 1.76-1.86 (1H, m, H-5endo), 2.00-2.10 (2H, m, H-13), 2.22-2.32 (1H, m, H-2exo), 2.49 (2H, t, J=6.7 Hz, H-12), 3.42 (9H, s, Me-15, Me-16, Me-17), 3.66-3.75 (2H, m, H-14), 4.76-4.84 (1H, m, H-1exo). 13C NMR (75 MHz, CDCl3, J Hz) δ ppm: 171.9 s (C-11), 80.4 d (C-1), 65.4 t (C-14), 53.4 q (Me-15, Me-16, Me-17), 48.3 s (C-6), 47.3 s (C-7), 44.3 d (C-3), 36.2 t (C-2), 29.4 t (C-12), 27.4 t (C-4), 26.5 t (C-5), 19.2 q (Me-9), 18.3 q (Me-10), 18.0 t (C-13), 13.1 q (Me-8). Anal. Calcd for C17H32INO2 C, 49.88; H, 7.88; N, 3.42 %. Found, %: C 49.87; H 7.71; N 3.20.
N,N-Diethyl-N-methyl-2-oxo-2-((1S,2S,4S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yloxy)ethanaminium iodide (11a)
A solution of compound 9a (0.2 mmol) and anhydrous CH3CN (5 mL) was treated with an excess of iodomethane and refluxed for 6 h. The solvent was removed at reduced pressure. The crude product was purified by recrystallisation from CH3CN. Yield: 51%; mp:159.2-159.3°C; 1H NMR (400 MHz, CDCl3, J Hz) δ ppm: 0.80 (3H, s, Me-9), 0.83 (3H, s, Me-8), 0.85 (3H, s, Me-10), 1.01 (1H, dd, 2J=13.7, J2endo,1exo=3.5, H-2endo), 1.17-1.35 (2H, m, H-4endo, H-5exo), 1.42 (6H, t, J=7.2, Me-15, Me-16), 1.62-1.76 (2H, m, H-3, H-4exo), 1.78-1.90 (1H, m, H-5endo), 2.25-2.38 (1H, m, H-2exo), 3.48 (3H, s, Me-17), 3.75-3.95 (4H, m, H-13, H-14), 4.55 (2H,AB, H-12), 4.91-5.00 (1H, m, H-1exo).13C NMR (100 MHz, CDCl3, J Hz) δ ppm: 164.3 s (C-11), 83.2 d (C-1), 58.7 t (C-12), 57.8 t (C-13, C-14), 48.8 s (C-6), 48.6 q (Me-17), 47.8 s (C-7), 44.4 d (C-3), 36.2 t (C-2), 27.6 t (C-4), 26.8 t (C-5), 19.4 q (Me-9), 18.5 q (Me-10), 13.5 q (Me-8), 8.3 q (Me-15, Me-16). Anal. Calcd for C17H32INO2 C 49.87; H 7.71; N 3.20 %. Found, %: C 50.29; H 8.08; N 3.14.
N,N-Diethyl-N-methyl-3-oxo-3-((1S,2S,4S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yloxy)propan-1-aminium iodide (11b)
The synthesis of compound 11b was performed in analogy to the synthesis compound 11a. Yield: 53%; mp:147.3-147.7°C; 1H NMR (400 MHz, CDCl3, J Hz) δ ppm: 0.81 (3H, s, Me-9), 0.84 (3H, s, Me-8), 0.86 (3H, s, Me-10), 1.00 (1H, dd, 2J=13.7, J2endo,1exo=3.5, H-2endo), 1.21-1.34 (2H, m, H-4endo, H-5exo), 1.42 (6H, t, J=7.2, Me-16, Me-17), 1.65-1.76 (2H, m, H-3, H-4exo), 1.82-1.91 (1H, m, H-5endo), 2.26-2.36 (1H, m, H-2exo), 2.96 (2H, t, J=7.1 Hz, H-12), 3.29 (3H, s, Me-18), 3.59-3.71 (2H, m, H-14, H-15), 3.75 (2H, t, J=7.1, H-13), 4.85-4.90 (1H, m, H-1exo). 13C NMR (100 MHz, CDCl3, J Hz) δ ppm: 169.5 s (C-11), 81.9 d (C-1), 57.3 t (C-14, C-15), 56.0 t (C-13), 48.6 s (C-6), 48.2 q (Me-18), 47.8 s (C-7), 44.5 d (C-3), 36.4 t (C-2), 28.1 t (C-12), 27.7 t (C-4), 26.9 t (C-5), 19.5 q (Me-9), 18.6 q (Me-10), 13.5 q (Me-8), 8.3 q (Me-16, Me-17). Anal. Calcd for C18H34INO2 C 51.06; H 8.09; N 3.42 %. Found, %: C 50.50; H 8.22; N 3.17.
Biological assays
Cells and viruses
Influenza A viruses A/Puerto Rico/8/34 (H1N1) (PR8 (H1N1)), A/California/07/09 (H1N1)pdm09 (Cal (H1N1)pdm09), and A/Aichi/2/68 (H3N2) (Aichi (H3N2)) were obtained from the collection of viruses of St Petersburg Pasteur Institute, Russia, and used in the study. Prior to the experiment, the virus was propagated in the allantoic cavity of 10 to 12-day-old chicken embryos for 48 h at 36°C. The titer of the virus was determined in MDCK cells (ATCC no. CCL-34) in 96-well plates in alpha-MEM medium (Biolot, St. Petersburg, Russia).
Cytotoxicity assay
The microtetrazolium test (MTT) was used to study the cytotoxicity of the compounds. Briefly, a series of threefold dilutions of each compound in MEM were prepared. MDCK cells were incubated for 48 h at 36°C in 5% CO2 in the presence of the dissolved substances. The cells were washed twice with phosphate-buffered saline (PBS), and a solution of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (ICN Biochemicals Inc. Aurora, Ohio) (0.5 μg/mL) in PBS was added to the wells. After incubation for 1 h, the wells were washed, and the formazan residue was dissolved in DMSO (0.1 mL per well). The optical density in the wells was then measured on a Victor2 1440 multifunctional reader (Perkin Elmer, Finland) at a wavelength of 535 nm and plotted against the concentration of compounds. Each concentration was tested in triplicate. The 50% cytotoxic concentration (CC50) of each compound was calculated from the data obtained.
In vitro antiviral activity
The compounds were dissolved in 0.1 mL of DMSO to prepare stock solutions, and working solutions (1000.0–4.0 μM) were prepared by adding MEM with 1 μg of trypsin per mL. The compounds were incubated with MDCK cells for 1 h at 36°C. Each concentration of the compounds was tested in triplicate. The cell culture was then infected with influenza virus A/PR/8/34 (H1N1) (MOI, 0.01) for 24 h at 36°C in the presence of 5% CO2. The virus titer in the supernatant was determined after cultivation of the virus in the MDCK cells for 72 h at 36°C in the presence of 5% CO2. The presence of virus in the wells was detected using a hemagglutination test. For this purpose, 100 µL of culture medium was transferred to round-bottom wells and mixed with an equal volume of 1% chicken erythrocytes, followed by incubation for 1 hour at 20°C. Rimantadine, amantadine, deitiforin, and ribavirin were used as reference drugs. The 50% inhibitory concentration (IC50) and the selectivity index (SI, the ratio of CC50 to IC50) were calculated from the obtained data.
Time-of-addition experiments
To determine which stage of the viral life cycle is affected by the compound, cells were seeded into 24-well plates and incubated with influenza virus A/PR/8/34 (H1N1) (at an MOI of 10) for 1 h at 4°C. After washing away unabsorbed virions for 5 min with MEM, the plates were incubated for 8 h at 36°C at 5% CO2. The starting point of this incubation was referred to as 0 hours. The cells were treated with compound 11 for the following time periods: -2 to -1 (before infection); -1 to 0 (simultaneously with absorption); 0 to 2; 2 to 4; 4 to 6; 6 to 8; and -2 to 8 hours. In each case, after the appropriate incubation time, the compound was removed and cells were washed for 5 min with MEM. After 8 hours of growth, the titer of the virus in the culture medium was determined as described above.
Hemagglutination inhibition assay and haemolysis assay
In order to assess the ability of compounds to interfere directly with HA receptor binding, we performed a hemagglutination inhibition test. Twofold dilutions of influenza-A/PR/8/34-virus-containing culture medium (1:8 to 1:256) were mixed with the lead compound at a range of concentrations and incubated for 1 hour at 36°C at 5% CO2, followed by addition of an equal volume of 1% chicken erythrocytes. After incubation for 1 hour at 20°C, the results were checked visually. Anti-hemagglutinin activity was evaluated by the ability of specimens to prevent virus-driven hemagglutination.
The membrane-disrupting activity of the viral hemagglutinin was measured according to Maeda and Ohnishi [21] with slight modifications. Briefly, chicken erythrocytes were washed twice with PBS and resuspended to make a 0.75% (vol./vol.) suspension in PBS, which was stored at 4°C until use. One hundred microliters of compound diluted in PBS to appropriate concentrations was mixed with an equal volume of influenza virus (128 hemagglutinating units per 0.1 mL) or PBS as a negative control. After incubating the virus-compound mixture at room temperature for 30 min, the mixture was mixed with 300 µL of 0.75% chicken erythrocytes. The mixture was incubated for 1 hour at +4°C for absorption of virions to the erythrocytes. Five hundred µL of MES buffer (0.1 M MES, 0.15 M NaCl, 0.9 mM CaCl2, 0.5 mM MgCl2, pH 5.0) was added, mixed, and incubated for 1 hour at 37°C for HA acidification and hemolysis. To separate unlysed erythrocytes at the end of the incubation period, tubes were centrifuged at 1,200 rpm for 6 min. After sedimentation of erythrocytes, 100 µL of supernatant was transferred to the wells of a flat-bottom plate, and the optical density in the wells was measured on Multiscan FC plate reader (Thermo Scientific) at a wavelength of 405 nm. The activity of the compounds was evaluated by their ability to suppress the destruction of membranes and thus decrease the concentration of free hemoglobin and the optical density in the wells compared to the control wells without additives. The percent activity of HA was calculated as (ODc-ODb)/(ODp-ODb) × 100, where ODp and ODs are mean optical density in the wells with PBS and with the compound under investigation, respectively, and ODb (background) is the mean optical density in the wells with erythrocytes but without virus and compounds. The activity of HA in control wells without any virus was calculated by comparison to the HA activity of influenza A virus.
Molecular docking study
The crystal structures of haemagglutinins of different strains were used for the docking procedure. Their PDB codes were 1RU7 (A/PR/8/34 (H1N1)) [22], 3EYM (A/Aichi/2/68 (H3N2)) [23], and 3LZG (A/California/07/09 (H1N1)pdm09) [21]. The geometric parameters of the HA protein were prepared for calculation and optimized using the OPLS3 force field algorithm.
To find the active site in the HA corresponding to PDB code 3LZG, we analyzed the binding site of the well-known HA inhibitor TBHQ in PDB code 3EYM [23]. This necessary procedure is one of the stages of molecular modelling. The TBHQ binding site is located at the interface between two monomers of the HA trimer. Based on an analysis of the active site, the area around the native ligand, with a radius of 5Å, was selected. The functional amino acid sequence was compared in two HA structures and the area of TBHQ binding was identified in protein 3LZG and was used for further docking procedures
The CPH-binding site was located near the site of proteolysis, next to the amino acid valine at position 615 (the numbering corresponds to structure 1RU7 [22]). Details have been described previously [24].
Ligand preparation (optimization, which took into account all possible conformations of the ligand) and all of the docking procedures were carried out using the Schrodinger Small-Molecule Drug Discovery Suite 2018-4, Schrödinger, LLC, New York, NY, 2018 program packages. The docking was performed under the following conditions: ligand and protein were flexible and induced-fit docking protocols were used for standard prediction accuracy.
Results and discussion
Chemistry
The synthesis of camphor derivatives 6 and 7 was performed as outlined in Scheme 1. The key intermediate N,N-disubstituted camphor imines 1 and 2 were synthesised according to our recently reported approach [11]. Subsequently, the methylation of the tertiary amino group of intermediates 1 and 2 was performed using excess methyl iodide, leading to the desired compounds 6 and 7 in moderate yields.

Reagents and conditions: (i) N,N-dimethylethylenediamine or N,N-dimethylpropane-1,3-diamine, ZnCl2, reflux under solvent-free conditions; (ii) CH3I, CH3CN, reflux
Target borneol derivatives were synthesised via a sequence of reactions starting with commercially available (-)-borneol. Borneol derivatives 8a-c and 9a-c with a tertiary amino group were prepared in two steps as described previously [19]. Subsequent methylation, using iodomethane under reflux, gave the corresponding quaternary ammonium salts 10a, 10c and 11a-b (Scheme 2).

Synthesis of the (-)-borneol derivatives 8a-c, 9a-c, 10a and c and 11a and b. Reagents and conditions: (i) corresponding chloroacetyl chloride, Et3N, CH2Cl2, r.t.; (ii) the corresponding amine, Et3N, CH2Cl2, r.t.; CH3I, CH3CN, reflux
The structures of the synthesised compounds were confirmed by NMR spectra (1H, 13C NMR). The structure of compound 10a was confirmed by X-ray diffraction. The bond lengths of 10a are close to the lengths of the analogous bonds in betaine hydrobromide and (cyclopentadienyl)-tricarbonyl-(2-((-)-borneolato)-2-oxoethyl)-molybdenum. In the crystal structure, the iodine anion is located close to the trimethylammonium group. The shortest I---H contact of 3.04 Å is shown in Figure 3. Between the cations, there is a weak hydrogen bond, C10-H---O=C (H---O distance 2.59 Å and C-H---O angle 167°).

ORTEP representations of compound 10a. The displacement ellipsoids are drawn at a probability of 30%. Only one of the two independent molecules is shown.
Biological activity
Anti-influenza activity of synthesised derivatives in vitro
The compounds that were obtained were studied as potential antiviral agents against influenza A virus strain A/Puerto Rico/8/34 (H1N1) (PR8 (H1N1)) (Table 1). An important characteristic of the biological activity of the compounds studied is the selectivity index (SI). The SI is the ratio between the concentration of a substance that causes 50% cellular damage (CC50) and the concentration required to reduce the viral activity by 50% (IC50). This ratio reflects the selectivity and safety of the compounds. As shown in Table 1, the transformation of camphor imine derivatives 1 and 2 into the corresponding quaternary ammonium salts 6 and 7 resulted in a loss of antiviral activity while maintaining low toxicity. Among the (-)-borneol esters 8a-c and 9a-c with a tertiary amino group, compounds 8a and c and 9a and b showed moderate antiviral activity with an SI value higher than 2. These derivatives were further converted to the quaternary ammonium salts 10a and c and 11a and b. Evaluation the antiviral activity and toxicity of the salts 10a and c and 11a and b revealed that compound 10a has a more potent effect against influenza A virus PR8 (H1N1).
In vitro screening allowed us to identify compound 10a as a lead compound, since this agent was found to have the lowest IC50 value and the highest SI value. For this compound, we performed specific assays to study its spectrum of activity and mechanism of action. We first examined the effect of 10a on the replication of influenza A viruses of distinct subtypes, namely, California/07/09 (H1N1)pdm09 (Cal (H1N1)pdm09) and Aichi/2/68 (H3N2) (Aichi (H3N2)), in MDCK cells (Table 2). Compound 10a potently inhibited the Aichi (H3N2) virus with an IC50 value of 9.2 μM and the pandemic influenza A virus Cal (H1N1)pdm09 with an IC50 value of 16.8 μM. These observations were seen in three different experiments.
Time-of-addition experiments
Time-of-addition experiments were performed to determine the target for the virus-inhibiting activity of compound 10a in the virus life cycle. The results are summarized in Figure 4. According to the data obtained, the compound turned out to be the most effective when added within 0–2 hours after infection. Over time, the efficacy of the drug decreased, and starting at 4 hours after infection, the infectious activity of the virus was not statistically different from the control values. Based on these results, compound 10a can be assumed to act at the initial stage of the influenza virus life cycle, which involves the attachment of virions to the surface of the target cell and penetration of the virion into the cell. At this stage, two viral proteins are essential. First, the viral HA mediates attachment of the virion to the cell surface and fusion of the viral envelope with the endosomal membrane. Second, the virus-specific proton channel M2 conducts protons into the virion interior, thus providing acidification of the core and allowing the dissociation of ribonucleoproteins (RNPs) from the envelope structures.

Time-of-addition activity of compound 10a against influenza A virus PR8 (H1N1). MDCK cells were infected with influenza virus, and the salt 10a was added and removed at the indicated time points, where 0 corresponds to the moment when the cells were infected. The infectious activity of viral progeny was tested by further titration on MDCK cells.
Study of the HA-mediated fusion process
Hemagglutination and haemolysis assays were performed to investigate the effect of compound 10a on HA-mediated fusion. The ability of compound 10a to interfere directly with the receptor-binding site of viral HA was evaluated by hemagglutination inhibition tests. Briefly, twofold dilutions of influenza A virus PR8 (H1N1) were mixed in round-bottom wells with compound 10a at a range of concentrations, followed by the addition of chicken red blood cells (cRBCs). Wells without compound 10a served as controls. The hemagglutination titer of the virus was found to be 64 hemagglutinating units (HAU) per 0.2 mL. The addition of compound 10a did not change this value even at the highest concentration used (1200 µM). Therefore, the results suggest that compound 10a does not inhibit the cell receptor-binding activity of the viral HA.
To determine how compound 10a affected viral fusion activity, a haemolysis assay was performed using influenza A virus PR8 (H1N1) (Fig. 5). Compound 10a can be seen to significantly decrease the ability of the viral HA to cause membrane damage and therefore may be considered an effective anti-influenza membrane fusion inhibitor.

Haemolysis-inhibiting activity of compound 10a against influenza A virus HA. The compound was mixed with 128 hemagglutinating units of influenza virus, incubated at room temperature for 30 min, mixed with 0.75% chicken erythrocytes, and incubated at +4°C. After incubation with MES buffer (0.1 M MES, 0.15 M NaCl, 0.9 mM CaCl2, 0.5 mM MgCl2, pH 5.0) and sedimentation of erythrocytes, the optical density in the wells was measured at 405 nm. The haemolysis-inhibiting activity of compound 10a was calculated by comparison to the haemolysis activity of influenza A virus without additives.
Molecular docking study
To investigate the details of the direct interaction of compound 10a with HA, docking of the lead compound 10a into the binding sites of HA was performed, and the calculated docking scores were compared with the values for the well-known HA inhibitors tert-butylhydroquinone (TBHQ) and the new anti-influenza drug camphecene (CPH) (Table 3). Two binding sites of HA were considered, the first of which is located in the binding region for TBHQ (the TBHQ site) [23] and the second of which is found near the proteolysis site, where camphecene binding occurs (the CPH site) [24, 25] (Fig. 6). The HA proteins of different strains and subtypes (PR8 (H1N1) (H1-PR), Cal (H1N1)pdm09 (H1-Cal) and Aichi (H3N2)) of influenza A virus were examined.

Location of lead compound 10a in the binding sites of different HAs. The secondary structure of an HA subunit corresponding to PDB code 1RU7 (PR8 (H1N1)) is shown in blue, that of PDB code 3EYM (Aichi (H3N2)) is shown in orange, and that of PDB code 3LZG (Cal (H1N1)pdm09) is shown in green. H-bonds and salt-bridges are presented as yellow and pink dotted lines, respectively, and π-cation interactions are shown as green dotted lines.
The results presented in Table 3 and Fig. 6 show that the lead compound 10a can bind to the TBQH site in all three types of HA under study (H1-PR, H1-Cal, and H3N2). It can fit into the space between two monomers (the TBQH site) to form a number of intermolecular interactions: hydrogen bond bridges between the oxygen atom of compound 10a and amino acids (K558 – H1-PR, R54 – H3) and salt bridges between the protonated nitrogen atom and D21 (H1-PR), E57 (H1-Cal), E103 (H3). The calculated binding affinity of compound 10a to the TBQH sites of the studied types of HA showed similar values of Ebind, ranging from -6.28 to -6.65 kcal/mol, which is similar to the values of Ebind of the known HA inhibitors CPH and TBQH. The binding of compound 10a to the CPH site of different HAs revealed one striking feature: compound 10a can bind to the CPH site of only subtype H1 HAs and does not bind to the CPH-site of subtype H3, since there is no cavity that is favourable for such molecules to be located. The HA of subtypes H1 and H3 belong to different groups, and their structures are conformationally different in the regions required for membrane fusion. We compared the amino acid sequences of HAs of subtype H1 (strains PR8 (H1N1) and Cal (H1N1)pdm09) and subtype H3 (strain Aichi (H3N2)) in the region of the TBHQ site and CPH site (Fig. 7).

The result of protein alignment procedure. The secondary structure of an HA subunit corresponding to PDB code 1RU7 (PR8 (H1N1)) is shown in blue, that of PDB code 3EYM (Aichi (H3N2)) is shown in orange, and that of PDB code 3LZG (Cal (H1N1)pdm09) is shown in green. Amino acid sequence differences are highlighted in red.
The results of a comparison of the amino acid sequences of the PR8 (H1N1) and Aichi (H3N2) strains showed their structure in the CPH-site region to be significantly different, whereas in the TBHQ-site region, the amino acid sequences have only a few variations. The calculated binding affinities of compound 10a to the CPH site of HA of PR8 (H1N1) and Cal (H1N1)pdm09 strains are similar (-7.12 and -6.62 kcal/mol, respectively) and close to values of Ebind of CPH (-7.07 and -7.18 kcal/mol). In the case of strain PR8 (H1N1), compound 10a in the CPH site forms a series of π-cation stacking interactions between the protonated nitrogen atom and the aromatic amino acids Y619 and F509. In the case of the pandemic strain Cal (H1N1)pdm09, only hydrophobic contacts are observed. The in silico docking results are consistent with in vitro data; the IC50 value of compound 10a against PR8 (H1N1) is lower than that against Cal (H1N1)pdm09 (2.4 and 16.9 µM, respectively). Also, according to docking results, compound 10a in the CPH site of HA strain PR8 (H1N1) gives a more favourable docking score than in the CPH site of HA strain Cal (H1N1)pdm09.
From the above analysis, it can be concluded that compound 10a can bind to both sites of HA subtype H1 with equal probability, but with different affinity. Since the HA of subtype H3 lacks the cavity located near the site of proteolysis, the TBHQ site can be considered a possible binding site for similar structures.
Conclusion
We have synthesized quaternary ammonium salts based on (+)-camphor and (-)-borneol. It was found that the conversion of tertiary amines based on (+)-camphor to quaternary ammonium salts led to decreased antiviral activity, while the transformation of (-)-borneol derivatives to the corresponding salts increased their inhibitory activity. The derivative 10a showed the most significant antiviral activity against different subtypes of influenza A virus, with an IC50 value of 2.4 μM against PR8 (H1N1), 9.2 μM against the pandemic influenza A virus Cal (H1N1)pdm09, and 16.8 μM against Aichi (H3N2), and low toxicity with a CC50 value of 1311 μM. Based on the data obtained in the mechanism-of-action studies, we suggest that the virus-inhibiting activity of these compounds, in particular compound 10a, is due to the inhibition of viral HA. According to molecular docking study results, the compound 10a can bind to different stem part of HA depending on the HAs subtype, which leads to the blocking of the viral and cell membrane fusion process.
References
Paules C, Subbarao K (2017) Influenza. Lancet 390:697–708. https://doi.org/10.1016/S0140-6736(17)30129-0
Yuan S (2013) Drugs to cure avian influenza infection multiple ways to prevent cell death. Cell Death Dis 4:e835. https://doi.org/10.1038/cddis.2013.367
Baranovich T, Wong S-S, Armstrong J et al (2013) T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J Virol 87:3741–3751. https://doi.org/10.1128/jvi.02346-12
Jin Z, Smith LK, Rajwanshi VK et al (2013) The ambiguous base-pairing and high substrate efficiency of T-705 (favipiravir) ribofuranosyl 5′-triphosphate towards influenza A virus polymerase. PLoS ONE 8:e68347
Pauly MD, Lauring AS (2015) Effective lethal mutagenesis of influenza virus by three nucleoside analogs. J Virol 89:3584–3597. https://doi.org/10.1128/jvi.03483-14
Binh NT, Wakai C, Kawaguchi A, Nagata K (2014) Involvement of the N-terminal portion of influenza virus RNA polymerase subunit PB1 in nucleotide recognition. Biochem Biophys Res Commun 443:975–979. https://doi.org/10.1016/j.bbrc.2013.12.071
Sun Y, He X, Qiu F, Zhu X, Zhao M, Li-Ling J, Su XZL (2013) Pharmacokinetics of single and multiple oral doses of arbidol in healthy Chinese volunteers. Int J Clin Pharmacol Ther 51:423–432. https://doi.org/10.5414/CP201843
Hayden FG, Sugaya N, Hirotsu N et al (2018) Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N Engl J Med 379:913–923. https://doi.org/10.1056/nejmoa1716197
Bright RA, Medina MJ, Xu X et al (2005) Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 366:1175–1181. https://doi.org/10.1016/S0140-6736(05)67338-2
Król E, Rychłowska M, Szewczyk B (2014) Antivirals—current trends in fighting influenza. Acta Biochim Pol 61:495–504. https://doi.org/10.18388/abp.2014_1870
Sokolova AS, Yarovaya OI, Shernyukov AV et al (2015) Discovery of a new class of antiviral compounds: camphor imine derivatives. Eur J Med Chem 105:263–273. https://doi.org/10.1016/j.ejmech.2015.10.010
Sokolova AS, Yarovaya OI, Baev DS et al (2017) Aliphatic and alicyclic camphor imines as effective inhibitors of influenza virus H1N1. Eur J Med Chem 127:661–670. https://doi.org/10.1016/j.ejmech.2016.10.035
Sokolova AS, Yarovaya OI, Korchagina DV et al (2014) Camphor-based symmetric diimines as inhibitors of influenza virus reproduction. Bioorganic Med Chem 22:2141–2148. https://doi.org/10.1016/j.bmc.2014.02.038
Zarubaev VV, Garshinina AV, Tretiak TS et al (2015) Broad range of inhibiting action of novel camphor-based compound with anti-hemagglutinin activity against influenza viruses in vitro and in vivo. Antiviral Res 120:126–133. https://doi.org/10.1016/j.antiviral.2015.06.004
Sokolova AS, Yarovaya OI, Shernyukov AV et al (2013) New quaternary ammonium camphor derivatives and their antiviral activity, genotoxic effects and cytotoxicity. Bioorganic Med Chem 21:6690–6698. https://doi.org/10.1016/j.bmc.2013.08.014
Sokolova AS, Yarovaya OI, Semenova MD et al (2017) Synthesis and in vitro study of novel borneol derivatives as potent inhibitors of the influenza A virus. Med Chem Comm 8:960–963. https://doi.org/10.1039/c6md00657d
Sokolova AS, Yarovaya OI, Shtro AA et al (2017) Synthesis and biological activity of heterocyclic borneol derivatives. Chem Heterocycl Compd 53:371–377. https://doi.org/10.1007/s10593-017-2063-3
Kononova AA, Sokolova AS, Cheresiz SV et al (2017) N-Heterocyclic borneol derivatives as inhibitors of Marburg virus glycoprotein-mediated VSIV pseudotype entry. Medchemcomm 8:2233–2237. https://doi.org/10.1039/c7md00424a
Sokolova AS, Yarovaya OI, Zybkina AV et al (2020) Monoterpenoid-based inhibitors of filoviruses targeting the glycoprotein-mediated entry process. Eur J Med Chem 207:112726. https://doi.org/10.1016/j.ejmech.2020.112726
Rey FA, Lok SM (2018) Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell 172:1319–1334. https://doi.org/10.1016/j.cell.2018.02.054
Maeda TOS (1980) Activation of influenza virus by acidic media causes hemolysis and fusion of erythrocytes. FEBS Lett 122:283–287. https://doi.org/10.1016/0014-5793(80)80457-1
Gamblin SJ, Haire LF, Russell RJ et al (2004) The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303:1838–1842. https://doi.org/10.1126/science.1093155
Russell RJ, Kerry PS, Stevens DJ et al (2008) Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci USA 105:17736–17741. https://doi.org/10.1073/pnas.0807142105
Zarubaev VV, Pushkina EA, Borisevich SS et al (2018) Selection of influenza virus resistant to the novel camphor-based antiviral camphecene results in loss of pathogenicity. Virology 524:69–77. https://doi.org/10.1016/j.virol.2018.08.011
Borisevich SS, Gureev MA, Yarovaya OI et al (2021) Can molecular dynamics explain decreased pathogenicity in mutant camphecene-resistant influenza virus? J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1871414
Acknowledgements
The authors would like to acknowledge the Multi-Access Chemical Service Centre SB RAS for spectral and analytical measurements. This work was supported by Russian Scientific Foundation grant 19-73-00125. The synthesis of compounds 6 and 7 was supported financially by the Ministry of Education and Science of the Russian Federation (award no. 0238-2021-0003). All of the calculations were carried out on a cluster in the regional centre for shared computer equipment at the Ufa Institute of Chemistry UFRC RAS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Ayato Takada.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sokolova, A.S., Yarovaya, O.I., Baranova, D.V. et al. Quaternary ammonium salts based on (-)-borneol as effective inhibitors of influenza virus. Arch Virol 166, 1965–1976 (2021). https://doi.org/10.1007/s00705-021-05102-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05102-1