
Abstract
The phylum Cressdnaviricota includes viruses with circular single-stranded DNA (ssDNA) genomes and icosahedral capsids. These viruses display global environmental distribution and infect diverse eukaryotic hosts, including animals, plants, and fungi. Here, we report on the formal creation of two new orders, Rivendellvirales and Rohanvirales, and three new families, Naryaviridae, Nenyaviridae, and Vilyaviridae, of ssDNA viruses associated with protozoan parasites belonging to the genera Entamoeba and Giardia. We describe a sequence-based taxonomic framework, which was used to classify 60 ssDNA viruses into 12 genera (with 18 species) within the family Vilyaviridae; four genera (with five species) within the family Naryaviridae; and five genera (with six species) within the family Nenyaviridae. We also highlight the challenges associated with the classification of chimeric virus genomes, such as those in the families Naryaviridae and Nenyaviridae, where the replication initiation and capsid protein genes have undergone several independent non-orthologous replacements. The described taxonomic changes have been ratified by the International Committee on Taxonomy of Viruses (ICTV) and expand the phylum Cressdnaviricota to eight orders and 11 families.
Similar content being viewed by others

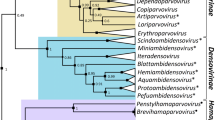

The phylum Cressdnaviricota, created in 2019, includes viruses with single-stranded DNA (ssDNA) genomes and icosahedral capsids that infect diverse eukaryotes, including algae, fungi, plants, insects, and vertebrates [1]. A characteristic feature of viruses in this phylum is the presence of homologous replication-associated proteins (Reps) with an N-terminal rolling-circle replication initiation endonuclease domain of the HUH superfamily [2] and a C-terminal superfamily 3 helicase domain [3]. By contrast, the capsid proteins encoded by viruses from different families can be non-orthologous, although all cressdnaviruses for which structural information is available appear to use single jelly-roll capsid proteins for virion formation [4, 5]. The phylum consists of two classes, the class Repensiviricetes, which currently includes all fungal and plant viruses of the families Genomoviridae and Geminiviridae, and the class Arfiviricetes, which includes six virus families (Bacilladnaviridae, Circoviridae, Smacoviridae, Nanoviridae, Metaxyviridae, and Redondoviridae). However, many groups of related ssDNA viruses, informally referred to as CRESSV1 to CRESSV6, remain unclassified [6, 7].
Recently, Kinsella et al. [8] identified three groups of ssDNA viruses associated with protozoan parasites of the genera Entamoeba and Giardia. The authors suggested that viruses associated with the Entamoeba hosts could constitute two families, “Naryaviridae” and “Nenyaviridae”, whereas those associated with Giardia hosts could form the family “Vilyaviridae” [8]. The families Naryaviridae, Nenyaviridae, and Vilyaviridae are named after three rings from the Middle-earth canon [9, 10] (also known as Tolkien’s canon) [8]. Here, we report on the formal establishment of the three families and a sequence-based taxonomic framework and demarcation criteria for classification of viruses within these families.
We assembled a dataset of unclassified ssDNA virus genome sequences from the GenBank database displaying similarity to those of representative members of the originally proposed Naryaviridae, Nenyaviridae, and Vilyaviridae [8]. The Rep sequences of these 60 viruses were analyzed together with those of Bacilladnaviridae, Circoviridae, Geminiviridae, Genomoviridae, Nanoviridae, Metaxyviridae, Redondoviridae, and Smacoviridae as well as those of CRESSV1 to CRESSV6 [6, 7]. The sequences were aligned using MAFFT v7.490 [11], and the resulting alignment was trimmed with TrimAL v1.2 [12] with a gap threshold of 0.2. A maximum-likelihood phylogenetic tree was constructed using IQtree2 [13] with automatic selection of the best-fit substitution model for a given alignment, which was rtREV+F+R6. The maximum-likelihood phylogenetic analysis of the corresponding Rep proteins in the framework of other members of the phylum Cressdnaviricota confirmed that the three groups form monophyletic clades distinct from the previously classified viruses (Fig. 1). Whereas the Vilyaviridae fell within the established order Cirlivirales, the Naryaviridae and Nenyaviridae formed distinct branches within the class Arfiviricetes (Fig. 1). Thus, to bridge the gap between the family and class taxa, we created the orders Rivendellvirales and Rohanvirales, which accommodate the families Naryaviridae and Nenyaviridae, respectively. The name of the order Rivendellvirales is derived from Rivendell, an Elven refuge in a steep and hidden valley to the west of the Misty Mountains, founded in the Second Age by Elrond; Rivendell was home to a number of great Elven lords [14]. The name of the order Rohanvirales, is derived from Rohan, a kingdom of the Rohirrim, bounded by the Anduin, the Misty Mountains, and Fangorn Forest, among others; once a province of Gondor, the land was given to the Men of Eotheod in return for their aid to Gondor in a battle [14].

Maximum-likelihood phylogenetic tree of Rep proteins from members of the phylum Cressdnaviricota. Closely related sequence groups are collapsed into triangles, the side lengths of which are proportional to the distances between the closest and farthest leaf nodes. The Rep alignment used for tree reconstruction was taken from Krupovic et al. [1] and supplemented with sequences of members of the newly established families Naryaviridae, Nenyaviridae, and Vilyaviridae. The latter dataset included previously reported sequences [8] as well as related sequences downloaded from the GenBank database. Numbers at the nodes represent aLRT branch support. The scale bar represents the number of substitutions per site.
Viruses of the family Vilyaviridae encode capsid proteins distantly related to those characteristic of circovirids, consistent with their placement within the order Cirlivirales. Notably, due to high sequence divergence, the similarity was only detectable when the corresponding profile hidden Markov models (HMM) were compared using HHpred [15]. In the sequence-similarity-based network constructed using CLANS [16], members of the families Naryaviridae and Nenyaviridae formed several mixed clusters of non-orthologous proteins (Fig. 2), consistent with the phylogenetic analysis reported previously [8]. In particular, members of the family Naryaviridae formed four separate clusters, two of which also included nenyavirids. Profile-profile comparisons revealed a further distant relationship between these CP clusters and CPs of ssDNA viruses of the families Geminiviridae, Nanoviridae, and Redondoviridae (Fig. 2). The replacement of the capsid gene on several occasions emphasizes the importance of recombination in the evolution of the Naryaviridae and Nenyaviridae. Even viruses of the same species, as in the case of Nimphelosvirus isildur, can encode highly distinct CPs. Notably, members of both the Naryaviridae and the Nenyaviridae are associated with Entamoeba sp., and thus the interfamily gene exchange is likely to be enabled by the shared host range.

Diversity of capsid proteins encoded by members of the families Naryaviridae, Nenyaviridae, and Vilyaviridae. Protein sequences were clustered by their pairwise sequence similarity. Lines connect sequences with P-value ≤ 1e−05. Species of the Naryaviridae (red), which form several distinct clusters, are labeled. Affiliation of the clusters to CPs of other ssDNA viruses is indicated with bold text.
To determine meaningful demarcation criteria within the families Naryaviridae, Nenyaviridae, and Vilyaviridae, we analyzed the relationships between the corresponding viruses within each of the three families by performing all-against-all genome and Rep sequence comparisons as well as phylogenetic analysis (Figs. 3, 4, 5). For species demarcation, we used 78% pairwise nucleotide sequence identity, similar to what was used for other cressdnaviricots, including genomovirids [17, 18] and smacovirids [19, 20]. Thus, all viral genomes showing sequence identity higher than 78% should be considered variant members of the existing species. Nonetheless, there may be situations where it is difficult to assign species because a particular new sequence is
-
(1) >78% identical to sequences from a particular species but <78% identical to other variants belonging to that same species;
-
(2) >78% identical to sequences from two or more different species.

Genera and species of the family Vilyaviridae. (A) Pairwise all-against-all comparison of the vilyavirid genomes. Genome maps are shown on the left with amino acid identity values shown for the open reading frames generated using Clinker [44]. A heat map showing the corresponding identity values is shown on the right. (B) Maximum-likelihood tree of the family Vilyaviridae (left) and a heat map showing pairwise comparison of the Rep amino acid sequences (right). Species belonging to the same genus are indicated with the same color; the genera are listed on the right. The phylogeny was inferred using IQtree2 [13], with automatic selection of the best-fit substitution model for a given alignment, which was Q.pfam+F+I+G4. Numbers at the nodes represent aLRT branch support. The cyan line shows the established demarcation of genera. The pairwise identities were calculated using SDT [45].

Genera and species of the family Naryaviridae. (A) Pairwise all-against-all comparison of the naryavirid genomes. Genome maps are shown on the left with amino acid identity values shown for the open reading frames generated using Clinker [44]. A heat map showing the corresponding identity values is shown on the right. (B) Maximum-likelihood tree of the family Naryaviridae (left) and a heat map showing pairwise comparison of the Rep amino acid sequences (right). Species belonging to the same genus are indicated with the same color; the genera are listed on the right. The phylogeny was inferred using IQtree2 [13], with automatic selection of the best-fit substitution model for a given alignment, which was LG+G4. Numbers at the nodes represent aLRT branch support. The cyan line shows a proposed demarcation of genera. The pairwise identities were calculated using SDT [45].

Genera and species of the family Nenyaviridae. (A) Pairwise all-against-all comparison of the nenyavirid genomes. Genome maps are shown on the left with amino acid identity values shown for the open reading frames generated using Clinker [44]. A heat map showing the corresponding identity values is shown on the right. (B) Maximum-likelihood tree of the family Nenyaviridae (left) and a heat map showing pairwise comparison of the Rep amino acid sequences (right). Species belonging to the same genus are indicated with the same color; the genera are listed on the right. The phylogeny was inferred using IQtree2 [13], with automatic selection of the best-fit substitution model for a given alignment, which was LG+G4. Numbers at the nodes represent aLRT branch support. The cyan line shows a proposed demarcation of genera. The pairwise identities were calculated using SDT [45].
To resolve the above conflicts, we suggest adopting an approach similar to that proposed for alphasatellites [21], circoviruses [22], geminiviruses [23, 24], and genomoviruses [18]. To resolve conflict 1, we suggest that the new virus be classified within any species in which it shares >78% sequence identity with any one variant already classified as belonging to that species, even if it is <78% identical to other viruses within that species. To resolve conflict 2, we suggest that the new virus be considered a member of the species with whose members it shares the highest degree of sequence similarity.
Given the interfamilial recombination observed within the Naryaviridae and Nenyaviridae, which produces genomes with diverse combinations of CPs and Reps (Fig. 2), we chose to define genera based on the cohesive phylogenetic lineages of the Rep, because this protein is generally more conserved within other ssDNA virus families than the CP and is the only protein shared by all members of the phylum Cressdnaviricota [1]. Notably, pairwise comparison of the Rep amino acid sequences fully recapitulated the Rep-phylogeny-based classification (Figs. 3, 4, 5). Using the criteria outlined above, the family Vilyaviridae was divided into 12 genera with 18 species (Fig. 3; Table 1, 2); the family Naryaviridae was divided into four genera with five species (Fig. 4; Tables 1, 2), and the family Nenyaviridae was divided into five 5 genera with six species (Fig. 5; Tables 1, 2).
Since the families Naryaviridae, Nenyaviridae, and Vilyaviridae are named after three rings from the Middle-earth canon, we followed the Tolkien Middle-earth [25] theme for genus and species names (Table 1). Furthermore, for species names, we used a binomial format with the “genus name + free-form epithet” [26, 27], where epithets are derived from various characters from the Tolkien canon. All species, genera, and families and their members are listed in Table 2.
The taxonomic changes described above were ratified by the International Committee on Taxonomy of Viruses (ICTV) in 2022 [46]. With the creation of the orders Rivendellvirales and Rohanvirales and the families Naryaviridae, Nenyaviridae, and Vilyaviridae, the phylum Cressdnaviricota now includes eight orders and 11 families. Yet, many more groups of ssDNA viruses remain to be discovered and classified, including the previously recognized CRESSV1-6. Notably, CRESSV2 [1, 6, 7] forms a sister group to the Nenyaviridae in the Rep phylogeny (Fig. 1) and, once formally classified, is likely to represent another family within the order Rohanvirales, whereas CRESSV1 and CRESSV3 [1, 6, 7] fall within the order Cirlivirales, together with the Vilyaviridae and Circoviridae. The families Naryaviridae and Nenyaviridae highlight the chimerism of certain ssDNA virus genomes, which has been discovered previously among the ssDNA viruses known as ‘cruciviruses’ [28,29,30,31,32,33,34]. We chose here to classify naryavirids and nenyavirids based on their Rep phylogeny, whereas the cruciviruses, as a group, are united by homologous tombusvirus-like CPs and encode non-orthologous Reps, which fall into different clades of ssDNA viruses. It remains to be decided what is the best approach to classify cruciviruses and other viruses with highly chimeric genomes.
References
Krupovic M, Varsani A, Kazlauskas D, Breitbart M, Delwart E, Rosario K, Yutin N, Wolf YI, Harrach B, Zerbini FM, Dolja VV, Kuhn JH, Koonin EV (2020) Cressdnaviricota: a Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes. J Virol 94:e00582-20
Chandler M, de la Cruz F, Dyda F, Hickman AB, Moncalian G, Ton-Hoang B (2013) Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat Rev Microbiol 11:525–538
Gorbalenya AE, Koonin EV, Wolf YI (1990) A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett 262:145–148
Krupovic M, Koonin EV (2017) Multiple origins of viral capsid proteins from cellular ancestors. Proc Natl Acad Sci U S A 114:E2401–E2410
Krupovic M (2013) Networks of evolutionary interactions underlying the polyphyletic origin of ssDNA viruses. Curr Opin Virol 3:578–586
Kazlauskas D, Varsani A, Koonin EV, Krupovic M (2019) Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat Commun 10:3425
Kazlauskas D, Varsani A, Krupovic M (2018) Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses 10:187
Kinsella CM, Bart A, Deijs M, Broekhuizen P, Kaczorowska J, Jebbink MF, van Gool T, Cotten M, van der Hoek L (2020) Entamoeba and Giardia parasites implicated as hosts of CRESS viruses. Nat Commun 11:4620
Tolkien JRR (2002) The history of Middle-earth. HarperCollins Publishers, London
Tolkien JRR (1977) The Silmarillion, 1st American, ed. Houghton Mifflin, Boston
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R (2020) IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37:1530–1534
Tolkien JRR (1965) The lord of the rings. Ballantine Books, New York
Gabler F, Nam SZ, Till S, Mirdita M, Steinegger M, Soding J, Lupas AN, Alva V (2020) Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr Protoc Bioinformatics 72:e108
Frickey T, Lupas A (2004) CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20:3702–3704
Varsani A, Krupovic M (2021) Family Genomoviridae: 2021 taxonomy update. Arch Virol 166:2911–2926
Varsani A, Krupovic M (2017) Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol 3:vew037
Krupovic M, Varsani A (2021) A 2021 taxonomy update for the family Smacoviridae. Arch Virol 166:3245–3253
Varsani A, Krupovic M (2018) Smacoviridae: a new family of animal-associated single-stranded DNA viruses. Arch Virol 163:2005–2015
Briddon RW, Martin DP, Roumagnac P, Navas-Castillo J, Fiallo-Olive E, Moriones E, Lett JM, Zerbini FM, Varsani A (2018) Alphasatellitidae: a new family with two subfamilies for the classification of geminivirus- and nanovirus-associated alphasatellites. Arch Virol 163:2587–2600
Rosario K, Breitbart M, Harrach B, Segales J, Delwart E, Biagini P, Varsani A (2017) Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch Virol 162:1447–1463
Muhire B, Martin DP, Brown JK, Navas-Castillo J, Moriones E, Zerbini FM, Rivera-Bustamante R, Malathi VG, Briddon RW, Varsani A (2013) A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch Virol 158:1411–1424
Varsani A, Martin DP, Navas-Castillo J, Moriones E, Hernandez-Zepeda C, Idris A, Murilo Zerbini F, Brown JK (2014) Revisiting the classification of curtoviruses based on genome-wide pairwise identity. Arch Virol 159:1873–1882
Tolkien C (1978) Middle Earth. Houghton Mifflin Company, Boston
Siddell SG, Walker PJ, Lefkowitz EJ, Mushegian AR, Dutilh BE, Harrach B, Harrison RL, Junglen S, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Nibert ML, Rubino L, Sabanadzovic S, Simmonds P, Varsani A, Zerbini FM, Davison AJ (2020) Binomial nomenclature for virus species: a consultation. Arch Virol 165:519–525
Zerbini FM, Siddell SG, Mushegian AR, Walker PJ, Lefkowitz EJ, Adriaenssens EM, Alfenas-Zerbini P, Dutilh BE, Garcia ML, Junglen S, Krupovic M, Kuhn JH, Lambert AJ, Lobocka M, Oksanen HM, Robertson DL, Rubino L, Sabanadzovic S, Simmonds P, Suzuki N, Van Doorslaer K, Vandamme AM, Varsani A (2022) Differentiating between viruses and virus species by writing their names correctly. Arch Virol 167:1231–1234
de la Higuera I, Kasun GW, Torrance EL, Pratt AA, Maluenda A, Colombet J, Bisseux M, Ravet V, Dayaram A, Stainton D, Kraberger S, Zawar-Reza P, Goldstien S, Briskie JV, White R, Taylor H, Gomez C, Ainley DG, Harding JS, Fontenele RS, Schreck J, Ribeiro SG, Oswald SA, Arnold JM, Enault F, Varsani A, Stedman KM (2020) Unveiling Crucivirus Diversity by Mining Metagenomic Data. mBio 11:e01410-20
Quaiser A, Krupovic M, Dufresne A, Francez AJ, Roux S (2016) Diversity and comparative genomics of chimeric viruses in Sphagnum-dominated peatlands. Virus Evol 2:vew025
Krupovic M, Zhi N, Li J, Hu G, Koonin EV, Wong S, Shevchenko S, Zhao K, Young NS (2015) Multiple layers of chimerism in a single-stranded DNA virus discovered by deep sequencing. Genome Biol Evol 7:993–1001
Roux S, Enault F, Bronner G, Vaulot D, Forterre P, Krupovic M (2013) Chimeric viruses blur the borders between the major groups of eukaryotic single-stranded DNA viruses. Nat Commun 4:2700
Dayaram A, Galatowitsch ML, Arguello-Astorga GR, van Bysterveldt K, Kraberger S, Stainton D, Harding JS, Roumagnac P, Martin DP, Lefeuvre P, Varsani A (2016) Diverse circular replication-associated protein encoding viruses circulating in invertebrates within a lake ecosystem. Infect Genet Evol 39:304–316
Steel O, Kraberger S, Sikorski A, Young LM, Catchpole RJ, Stevens AJ, Ladley JJ, Coray DS, Stainton D, Dayaram A, Julian L, van Bysterveldt K, Varsani A (2016) Circular replication-associated protein encoding DNA viruses identified in the faecal matter of various animals in New Zealand. Infect Genet Evol 43:151–164
Diemer GS, Stedman KM (2012) A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol Direct 7:13
Khalifeh A, Blumstein DT, Fontenele RS, Schmidlin K, Richet C, Kraberger S, Varsani A (2021) Diverse cressdnaviruses and an anellovirus identified in the fecal samples of yellow-bellied marmots. Virology 554:89–96
Chrzastek K, Kraberger S, Schmidlin K, Fontenele RS, Kulkarni A, Chappell L, Dufour-Zavala L, Kapczynski DR, Varsani A (2021) Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs. Microorganisms 9:2602
Tisza MJ, Pastrana DV, Welch NL, Stewart B, Peretti A, Starrett GJ, Pang YS, Krishnamurthy SR, Pesavento PA, McDermott DH, Murphy PM, Whited JL, Miller B, Brenchley J, Rosshart SP, Rehermann B, Doorbar J, Ta’ala BA, Pletnikova O, Troncoso JC, Resnick SM, Bolduc B, Sullivan MB, Varsani A, Segall AM, Buck CB (2020) Discovery of several thousand highly diverse circular DNA viruses. Elife 9:e51971
Dayaram A, Potter KA, Pailes R, Marinov M, Rosenstein DD, Varsani A (2015) Identification of diverse circular single-stranded DNA viruses in adult dragonflies and damselflies (Insecta: Odonata) of Arizona and Oklahoma, USA. Infect Genet Evol 30:278–287
Pearson VM, Caudle SB, Rokyta DR (2016) Viral recombination blurs taxonomic lines: examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 4:e2585
Takano T, Yanai Y, Hiramatsu K, Doki T, Hohdatsu T (2018) Novel single-stranded, circular DNA virus identified in cats in Japan. Arch Virol 163:3389–3393
Siqueira JD, Dominguez-Bello MG, Contreras M, Lander O, Caballero-Arias H, Xutao D, Noya-Alarcon O, Delwart E (2018) Complex virome in feces from Amerindian children in isolated Amazonian villages. Nat Commun 9:4270
Phan TG, Kapusinszky B, Wang C, Rose RK, Lipton HL, Delwart EL (2011) The fecal viral flora of wild rodents. PLoS Pathog 7:e1002218
Rosario K, Mettel KA, Benner BE, Johnson R, Scott C, Yusseff-Vanegas SZ, Baker CCM, Cassill DL, Storer C, Varsani A, Breitbart M (2018) Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 6:e5761
Gilchrist CLM, Chooi YH (2021) Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 37:2473–2475
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9:e108277
Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Adriaenssens EM, Alfenas-Zerbini P, Dempsey DM, Dutilh BE, García ML, Curtis Hendrickson R, Junglen S, Krupovic M, Kuhn JH, Lambert AJ, Łobocka M, Oksanen HM, Orton RJ, Robertson DL, Rubino L, Sabanadzovic S, Simmonds P, Smith DB, Suzuki N, Van Doorslaer K, Vandamme AM, Varsani A, Zerbini FM (2022) Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses. Arch Virol. https://doi.org/10.1007/s00705-022-05516-5
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Handling Editor: Sead Sabanadzovic.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Krupovic, M., Varsani, A. Naryaviridae, Nenyaviridae, and Vilyaviridae: three new families of single-stranded DNA viruses in the phylum Cressdnaviricota. Arch Virol 167, 2907–2921 (2022). https://doi.org/10.1007/s00705-022-05557-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05557-w