
Abstract
N6-methyladenosine (m6A) messenger RNA methylation is the most widespread gene regulatory mechanism affecting liver functions and disorders. However, the relationship between m6A methylation and arsenic-induced hepatic insulin resistance (IR), which is a critical initiating event in arsenic-induced metabolic syndromes such as type 2 diabetes (T2D) and non-alcoholic fatty liver disease (NAFLD), remains unclear. Here, we showed that arsenic treatment facilitated methyltransferase-like 14 (METTL14)-mediated m6A methylation, and that METTL14 interference reversed arsenic-impaired hepatic insulin sensitivity. We previously showed that arsenic-induced NOD-like receptor protein 3 (NLRP3) inflammasome activation contributed to hepatic IR. However, the regulatory mechanisms underlying the role of arsenic toward the post-transcriptional modification of NLRP3 remain unclear. Here, we showed that NLRP3 mRNA stability was enhanced by METTL14-mediated m6A methylation during arsenic-induced hepatic IR. Furthermore, we demonstrated that arsenite methyltransferase (AS3MT), an essential enzyme in arsenic metabolic processes, interacted with NLRP3 to activate the inflammasome, thereby contributing to arsenic-induced hepatic IR. Also, AS3MT strengthened the m6A methylase association with NLRP3 to stabilize m6A-modified NLRP3. In summary, we showed that AS3MT-induced m6A modification critically regulated NLRP3 inflammasome activation during arsenic-induced hepatic IR, and we identified a novel post-transcriptional function of AS3MT in promoting arsenicosis.
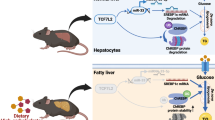
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exposure to inorganic arsenic (iAs) poses an environmental threat to public health (Argos et al. 2010, Zhang et al. 2020). Over 200 million people ingesting iAs from drinking water at level above the World Health Organization (WHO) standard of 10 μg/L (Naujokas et al. 2013). Therefore, it is of great public health importance to investigate the mechanism of inorganic arsenic-induced type 2 diabetes (T2D). Hepatic insulin resistance (hepatic IR) is an important pathology of T2D, and a key pathophysiological step in non-alcoholic steatohepatitis (NASH) and T2D pathogenesis (Allen and Neuschwander-Tetri 2021; Ferguson and Finck 2021; Loomba et al. 2021). We previously demonstrated an association between iAs and hepatic IR (Gao et al. 2019, Jia et al. 2020); however, the mechanism of early onset of arsenic-induced hepatic IR remains unclear. Therefore, it is important to explore the molecular mechanisms and therapeutic targets of early onset hepatic IR to prevent and treat arsenicosis.
The NOD-like receptor protein 3 (NLRP3) inflammasome is a protein complex composed of NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-1, and is a core molecule of pyroptosis (Schroder and Tschopp 2010; Shi et al. 2017). Inflammasome activation is a critical regulatory mechanism in several inflammatory diseases, including coronavirus disease 2019 (COVID-19), atherosclerosis, neurodegeneration, and T2D (Vandanmagsar et al. 2011; Grebe et al. 2018; Ising et al. 2019, Rodrigues et al. 2021). We previously demonstrated that iAs induced NLRP3 inflammasome activation via the autophagic-lysosomal pathway and mitochondrial reactive oxygen species (mtROS) in hepatocytes, and ER stress in hepatic stellate cells (HSCs) (Qiu et al. 2018, Jia et al. 2020, Tao et al. 2021). However, the molecular mechanism whereby iAs affects the post-transcriptional modification of NLRP3 remains elusive.
Among more than 100 distinct in vivo chemical modifications, N6-methyladenosine (m6A) is the most common, abundant, and conserved post-transcriptional modification in eukaryotic cells (Nombela et al. 2021). Methyltransferases (e.g., writers such as METTL14, METTL3, and WTAP), binding proteins such as YTH domain-containing family proteins (YTHDFs) and Insulin-like growth factor 2 mRNA-binding proteins (IFG2BPs), and demethylase such as FTO and ALKBH5 mediate m6A modification in messenger RNA (mRNA) (Huang et al. 2018a; Zong et al. 2021). m6A regulates the RNA decay, translation, transcription, and RNA–protein interactions (Zhao et al. 2017; Frye et al. 2018; Liu et al. 2020) and plays an essential roles in the development and progression of several disease processes, including cancer, Alzheimer’s disease, and non-alcoholic fatty liver disease (NALFD) (Wang et al. 2020; Yi et al. 2020; Zhao et al. 2020b; Wang et al. 2021). For example, METTL14 knockdown or deficiency increases mitochondrial antiviral signaling (MAVS) mRNA stability and enhances downstream TBK1/IRF3 phosphorylation and interferon-β production in response to RNA viruses (Qin et al. 2021). In some endometrial cancers, reduced m6A mRNA methylation is a putative oncogenic mechanism, with m6A methylation regulating the AKT pathway and cell growth (Liu et al. 2018). However, the role of m6A methylation in arsenicosis remains unknown.
Arsenite methyltransferase (AS3MT) catalyzes the transfer of a methyl group from S-adenosylmethionine (SAM) to trivalent arsenicals to produce methylated and dimethylated arsenicals (Zhang et al. 2015; Chen et al. 2017). Although there remain different opinions, the conversion of iAs to DMA is considered a detoxification mechanism because the toxicity of DMA is lower than that of iAs. However, arsenic methylation can also be a bioactivation process because the toxicity of intermediate metabolites such as monomethylarsonous acid (MMA) is much higher than that of iAs in terms of acute toxicity(Styblo et al. 2000). Epidemiological studies have found that AS3MT is closely associated with iAs-induced IR and diabetes, but the mechanism remains unclear (Grau-Perez et al. 2017, Huang et al. 2018b). Changes in the expression of m6A regulators induce noticeable pathological and physiological aberrations in liver function (Zhao et al. 2020b). However, whether AS3MT as a methyltransferase plays a crucial roles in the changes induced by iAs is unknown. Therefore, it is important to ascertain whether iAs affect liver disease progression by regulating m6A modification levels and whether AS3MT plays a role in this process.
Thus, we hypothesized that iAs induced NLRP3 inflammasome activation by regulating AS3MT-mediated m6A modification to facilitate early onset of hepatic IR.
Methods and materials
Animals
Six-week-old C57BL/6 J male mice were purchased from the Institute of Genome Engineered Animal Models for Human Disease of Dalian Medical University. Mice were exposed to drinking water containing As2O3 (Sigma-Aldrich, 1327–53-3) at 4 mg/l for 6 weeks to investigate the effects of As2O3 on hepatic insulin sensitivity. The control group was given distilled water (n = 6 for each group). After 6 weeks, animals were humanely euthanized and liver and whole blood collected for study. The Animal Ethics Committee of Dalian Medical University approved all animal care and study procedures (AEE18065).
Cell culture and pretreatment
Human fetal hepatocyte cells, L-02 (Advanced Research Center of Central South University) were cultured in RPMI-1640 medium (Eallbio, 03.4007C) supplemented with 10% fetal bovine serum (Biological Industries, 1,707,254) and 1% antibiotics (Hyclone, HYC-SV30010) in a 5% CO2 humidified atmosphere at 37 °C. Additionally, 3.96 mg As2O3 was dissolved in 1 ml phosphate-buffered saline (PBS) to prepare a 20 mM stock solution. L-02 cells were treated with 0 or 4 μM As2O3 24 h.
For studies, L-02 cells were pre-treated with 1 μM of the NLRP3 inhibitor, MCC950 (MCE, HY-12815A) before As2O3 treatment, or 5 μg/ml actinomycin D (ActD, AbMole, M4881) for 0 h, 3 h, and 6 h after As2O3 treatment.
Oral glucose tolerance test (OGTT)
After As2O3 exposure for 6 weeks, 6 mice/group were fasted overnight and received a glucose challenge via oral gavage (2 g/kg body weight). Blood samples were collected before and at 15, 30, 60, and 120 min after glucose loading. Glucose levels in tail blood were determined using an Accusure-580 glucose analyzer (Yuwell, China).
Insulin tolerance test (ITT)
After As2O3 exposure for 6 weeks, 6 mice/group were fasted for 6 h and received insulin (0.5 U/kg body weight) via intraperitoneal injection. Glucose levels in tail blood were determined at 0, 15, 30, and 60 min after insulin injection using the Accusure-580 glucose analyzer (Yuwell, China).
Histopathological examination
Liver tissues from mice were fixed in 10% neutral buffered formalin, dehydrated in graded ethanol concentrations, embedded in paraffin, sectioned at 4 µm, and stained with hematoxylin and eosin (H&E) for blinded assessment under light microscopy (40 × 10, Olympus BX63, Japan).
Immunohistochemistry (IHC) staining
Paraffin-embedded sections were deparaffinized and rehydrated. Sections were microwaved in 0.01 M citrate buffer, washed in PBS for 15 min, and then treated with a blocking buffer containing 5% bovine serum albumin for 20 min at room temperature. After this, sections were incubated with a rabbit polyclonal anti-AS3MT antibody (1:200) (Santa Cruz, sc-376537) at 4℃ overnight. After washing three times in PBS, a secondary antibody was added and immunostaining was performed using a DAB kit (ZSGB-BIO, ZLI-9108). AS3MT positive areas were measured using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Western blotting
Proteins were extracted from L-02 cells or mouse liver sections using RIPA lysis buffer (Merck Millipore, 92,590). Cytoplasm and nucleoprotein fractions were extracted and separated using a nuclear and cytoplasmic protein extraction kit (KeyGEN BioTECH, KGP1100). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and blotted onto polyvinylidene fluoride membranes (Merck Millipore, ISEQ00010) using a wet electrophoretic transfer method. Membranes were blocked for 1 h in 10% powdered skimmed milk at 37 °C and then incubated with primary antibodies (Table 1) at 4 °C overnight. After washing in PBS, membranes were incubated with a horse radish peroxidase-conjugated secondary antibody (Table 1) for 1.5 h at room temperature or 4 °C overnight. Protein expression was assessed using an enhanced chemiluminescence kit (Beyotime, P0018), imaged on a Bio-Rad ChemiDoc TMMP system, and analyzed by ImageJ software (National Institutes of Health).
Quantitative real-time PCR (RT-qPCR)
Total RNA from mouse liver was isolated using an RNA extraction kit (Accurate Biology, AG21017). Samples were prepared for RT-qPCR using the Evo M-MLV RT Premix for qPCR (Accurate Biology, AG11706) and SYBR Premix Ex TaqTM (Accurate Biology, AG11702). RT-qPCR was performed on a Rotor-Gene Q instrument (Qiagen, Germany). The relative expression of target genes was calculated using the 2-ΔΔCt method, and each sample was performed in triplicate. Primer sequences are shown (Tables 2 and 3).
RNA lifetime measurement
L-02 cells were seeded in 6-cm plates. After 24 h, cells were transfected with small interfering RNA (siRNA). After As2O3 treatment, ActD was added at 5 μg/ml 6 h, 3 h, and 0 h before collection. Total RNAs were extracted using an RNA extraction kit (Accurate Biology) and analyzed by RT-qPCR.
Enzyme-linked immunosorbent assay (ELISA)
Culture supernatant and serum insulin IL-1β and IL-18 levels were detected using ELISA kits (Longton, BPE20353, BPE10083, BPE10092, BPE20532, BPE11385) according to manufacturer’s instructions. The absorbance was measured on a microplate reader (Spectra MR, Dynex, USA).
Glucose uptake assay
Insulin sensitivity was evaluated by insulin‐stimulated glucose intake in L-02 cells seeded in 96‐well plates, incubated with 100 nM insulin for 30 min in a serum‐free medium at the end of treatment. Glucose levels were measured using a glucose assay kit (Nanjing Jiancheng Bioengineering Institute, F006).
Measurement of m 6 A content
Total RNA from mouse livers or L-02 cells was isolated using an RNA extraction kit (Accurate Biology) and mRNA purified with a Dynabeads™ mRNA DIRECT™ purification kit (Invitrogen, 61,012). Then, m6A levels in total RNA and mRNA from samples were detected using the RNA methylation assay kit (Abcam, ab185912) according to manufacturer’s instructions. RNA (200 ng), capture, and detection antibodies were added to wells, and m6A levels measured at 450 nm using a microplate reader (Spectra MR).
m 6 A-seq
Three replicate samples from C57BL/6 J mice, treated with or without As2O3, were submitted to Gene Pioneer Company (Nanjing, China). Total RNA was isolated and purified using TRIzol (Invitrogen, 15,596,018) following manufacturer's instructions. Poly (A) RNA was purified from total RNA using Dynabeads Oligo (dT) 25 (Thermo Fisher, 61,005) over two purification rounds. Then, cleaved RNA fragments were incubated for 2 h at 4 °C with an m6A-specific antibody (Synaptic Systems, No. 202003) in IP buffer. Then, IP RNA was reverse-transcribed to generate cDNA using SuperScript™ II Reverse Transcriptase (Invitrogen, 1,896,649) and A bases added to the blunt ends of each strand for ligation to indexed adapters. After treatment of the heat-labile UDG enzyme (NEB, m0280) of U-labeled second-stranded DNAs, the ligated products were amplified by PCR. Finally, we performed 2 × 150 base pair (bp) paired-end sequencing (PE150) using the Illumina Novaseq™ 6000 (LC-Bio Technology Co., Ltd., Hangzhou, China) platform following recommended protocols.
m 6 A-seq data analysis
m6A-seq data were analyzed according to a previous protocol (Meng et al. 2014). We used HISAT2 to map reads to the reference Mus musculus genome (Version: v101). Mapped IP reads and input libraries were provided by R package exome Peak (Version 3.7), which identified m6A peaks with a bed or bigwig format that could be adapted for visualization on IGV software (http://www.igv.org). HOMER (Version 4.9) was used for de novo and known motif identification, followed by motif localization with respect to the peak summit. Next, Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) enrichment analyses were performed using the KEGG Orthology-Based Annotation System (KOBAS, http://kobas.cbi.pku.edu.cn/kobas3) or DAVID bioinformatics resources 6.8 (https://david.ncifcrf.gov/). Differentially expressed mRNAs were selected at log2 (fold change) > 1 or log2 (fold change) < -1 and a p value < 0.05 using R package edgeR (https://bioconductor.org/packages/edgeR).
Co-immunoprecipitation (Co-IP)
According to manufacturer's instructions, mouse liver or L-02 protein complexes were evaluated by Co-IP combined with immunoblotting (Proteintech, PK-10007). Protein lysates (1 mg) were incubated with a precipitating antibody (1 μg) and 150 μl incubation buffer overnight at 4 °C. The next day, complexes were immunoprecipitated with protein A + G-sepharose beads at room temperature for 2 h with shaking. Protein-bead complexes were washed five times in washing buffer and eluted twice in 40 μl elution buffer. Elution fractions were analyzed by western blotting.
Small interfering RNA transfections
We purchased siRNAs from General Biosystems Co., Ltd. Negative control siRNAs acted as siRNA controls. Valid sequences were selected from three sequences according to RT-qPCR data. Sequences for siAS3MT, siMETTL14, and siIGF2BP2 are shown (Table 4). Lipofectamine 3000 was used for transfections (Invitrogen, L3000015) following manufacturer’s instructions.
Plasmid transfections
The recombinant plasmids were purchased from General Biosystems Co., Ltd. An empty vector was used as a negative control. Transfection was achieved by using P3000 and Lipofectamine 3000 (Invitrogen, L3000015) following the manufacturer’s protocols.
Statistical analysis
Data were presented as the means ± standard error of the mean (SEM). Statistically significant differences between groups were identified by the unpaired Student’s t-test, one-way ANOVA followed by an LSD post hoc test, or two-way ANOVA in SPSS 26 (IBM, Inc., NY, USA). Statistical information is outlined in figure legends; differences were significant at P < 0.05.
Results
Arsenic exposure impaired hepatic glucose metabolism and insulin sensitivity in mice in the early onset of hepatic IR model
To investigate the molecular mechanisms underpinning arsenic-induced early onset IR, mice were treated with iAs for 6 weeks. Although the limit of arsenic content in groundwater set by the WHO is 10 μg/L, the arsenic content of groundwater in most arsenic-contaminated areas is much higher than this limit. Therefore, in the current study, we selected a concentration of 4 mg/L for animal experiments, based on the concentration of arsenic in arsenic-contaminated areas and the results of previous literature studies (Perry et al. 2013; Rodriguez et al. 2016; Castriota et al. 2020; Xie et al. 2020).
At study end, the fasting blood glucose levels of exposed animals were significantly higher than control animals (Fig. 1A), but no significant differences were observed in fasting blood insulin levels between groups (Fig. 1B). However, the HOMA-IR of the exposed group was significantly higher than controls (Fig. 1C). Additionally, Pck1 and G6pc mRNA levels were significantly higher in exposed mice than controls (Fig. 1D). Also, glucose tolerance, insulin sensitivity, and insulin signaling were all significantly impaired in exposed mice when compared with controls (Fig. 1E–K). Differences in body weight and organ coefficients for liver, kidney, and pancreas between groups were not observed (Fig. S1A-D). No significant differences were observed for liver pathological, fibrosis, and lipid accumulation indices between groups, as indicated by H&E-, Masson-, and Oil red O-staining of liver sections (Fig. S1E–G). Collectively, these data indicated our mouse model represented the early stages of arsenic-induced IR.

Inorganic As (iAs) treatment induces early onset hepatic-IR. (A–C) Weekly fasting blood glucose levels (A), fasting insulin levels (B), and HOMA-IR comparisons between control and iAs-treated groups (n = 8) at study end. (D) Relative Pck1 and G6pc mRNA levels in the livers of indicated mouse groups (n = 3). (E–H) OGTT (2 g/kg) and ITT (0.5 U/kg) levels were measured in control and iAs groups at week 6. The corresponding area under the curve (AUC) of blood glucose levels was calculated (n = 8). (I–K) Total and phosphorylated AKT and GSK3β levels in control and iAs-treated mouse livers; some mice received intraperitoneal insulin injections 10 min before liver collection. GAPDH served as a loading control (n = 3). *P < 0.05 and **P < 0.01 versus the control group. The data represent the mean ± standard error of the mean (SEM). The two-way ANOVA (A) and Student's two-tailed t test (B–K) were used to determine statistical significance
The NLRP3 inflammasome promotes the early onset of iAs-induced hepatic IR
The NLRP3 inflammasome is a NOD-like receptor signaling process, with previous studies showing the NLRP3 inflammasome was involved in iAs-induced NASH-related IR and HSC activation. Here, the NLRP3 inflammasome was also activated at early onset iAs-induced hepatic IR stages (Fig. 2A–E), with increased serum IL-1β and IL-18 levels (Fig. 2F and G). The uniqueness of NLRP3 inflammasome in iAs-induced hepatic IR was proved by comparing two non-classical inflammasomes that have been extensively studied, NLRC4 and AIM2 inflammasome (Fig. S2). The role of the NLRP3 inflammasome in iAs-induced hepatic IR was further verified by inhibiting its activation with the NLRP3 inhibitor, MCC950 (Fig. 2H–K).

The NLRP3 inflammasome is involved in early onset iAs-induced hepatic IR. (A–E) NLRP3 inflammasome-associated protein levels in control and iAs-treated livers. GAPDH served as a loading control (n = 3). (E–F) Serum IL-1β and IL-18 levels in control and iAs-treated mice (n = 6). (H–K) Total and phosphorylated AKT and GSK3β expression levels (H–J) and glucose uptake (K) in iAs-treated and MCC950 pretreated L-02 cells (n = 3). **P < 0.01 versus the control group, ##P < 0.01 versus the iAs-treated group. The data represent the mean ± standard error of the mean (SEM), n = 3. Statistical significance was determined by Student's two-tailed t test (A–G) and one-way ANOVA (H–K)
AS3MT is involved in iAs-induced hepatic IR
We investigated AS3MT, which controls arsenic methyl metabolism, in early onset iAs-induced hepatic IR. From western blotting and IHC studies, AS3MT expression was upregulated in the liver of iAs-treated mice (Fig. 3A and B). IHC results also showed that AS3MT was increased in liver tissue and mainly expressed in hepatocytes (Fig. 3C). Consistently, iAs treatment triggered a marked upregulation of AS3MT expression in L-02 cells (Fig. 3D and E). Moreover, AS3MT mRNA levels were also increased in iAs-treated mouse liver when compared with controls (Fig. 3F). To further confirm the role of AS3MT in iAs-induced hepatic IR, AS3MT expression was silenced with siRNA; silencing efficiency was verified in L-02 cells (Fig. S3A). As expected, silencing improved iAs-blocked insulin signaling (Fig. 3G–I) and glucose uptake (Fig. 3J). Thus, AS3MT had a vital role in early onset iAs-induced hepatic IR.

AS3MT is associated with iAs-induced hepatic IR. (A–B) AS3MT expression in control and iAs-treated livers. GAPDH served as a loading control. (C) AS3MT expression by immunohistochemistry in control and iAs-treated livers. (D–E) AS3MT expression in iAs-treated L-02 cells. GAPDH served as a loading control. (F) Relative Pck1 and G6pc mRNA levels in livers from indicated groups. (G–J) Total and phosphorylated AKT and GSK3β expression levels (G-I) and glucose uptake (J) in iAs-treated L-02 cells, with or without siAS3MT transfection. *P < 0.05 and **P < 0.01 versus the control group, ##P < 0.01 versus the iAs-treated group. The data represent the mean ± standard error of the mean (SEM), n = 3. Scale bar = 100 μm. Statistical significance was determined by Student two-tailed t tests (A–F) and one-way ANOVA (G–J)
AS3MT activates the NLRP3 inflammasome by interacting with NLRP3 during iAs-induced hepatic IR
We next investigated the role of AS3MT in NLRP3 inflammasome activation and investigated how it functioned during iAs exposure. AS3MT interference inhibited iAs-induced NLRP3 inflammasome activation, but NLRP3 blockage with MCC950 did not affect AS3MT expression (Fig. 4A and B). Based on interaction prediction results from GeneMANIA and SWISS-MODEL, AS3MT (amino acid range 70–181) was predicted to interact with NLRP3 (amino acid range 217–241) (Fig. S4). Consequently, this AS3MT and NLRP3 interaction in iAs-treated mouse liver and L-02 cells was confirmed by Co-IP (Fig. 4C and D). Thus, AS3MT could be an upstream signal of the NLRP3 inflammasome to possibly promote NLRP3 inflammasome activation by interacting with NLRP3. To verify direct AS3MT and NLRP3 interactions, we performed IP studies in the HEK293T cell line. We generated serially truncated AS3MT and NLRP3 versions and performed Co-IP assays to identify which protein regions interacted (Fig. 4E and F); the AS3MT domain encompassing amino acids 201–375 bound to the NLRP3 domain encompassing amino acids 211–550 (Fig. 4G and H).

AS3MT interacts with NLRP3 and promotes inflammasome activation. (A) NLRP3 inflammasome-related proteins in iAs-treated L-02 cells, with or without siAS3MT transfection. (B) NLRP3 and AS3MT expression in iAs-treated L-02 cells, with or without MCC950 pretreatment. (C–D) AS3MT and NLRP3 interactions in iAs-treated liver or L-02 cells. (E–F) Schematic diagrams of AS3MT and NLRP3 recombinant plasmids. (G–H) AS3MT and NLRP3 binding domains were explored using full length and truncated AS3MT and NLRP3 expression plasmids and Co-IP assays, followed by western blotting
AS3MT regulates NLRP3 expression via m 6 A methylation
Although AS3MT interference inhibited NLRP3 protein expression, it did not affect NLRP3 transcription levels in iAs-treated L-02 cells (Fig. S3B). Therefore, we hypothesized that AS3MT affected NLRP3 inflammasome activation by regulating NLRP3 mRNA m6A methylation. To confirm this, we first explored the effects of AS3MT on iAs-regulated m6A methylation. Consistent with results in liver, iAs promoted METL14 and IGF2BP2 translocation to the nucleus in L-02 cells (Fig. 5A and B) and silenced AS3MT reduced METTL14 and IGF2BP2 expression in nucleus (Fig. 5A and B). Also, AS3MT interference significantly reduced m6A methylation content in total RNA and mRNA (Fig. 5C and D). Furthermore, AS3MT silencing significantly blocked interactions between NLRP3 and m6A methylase (Fig. 5E) and reduced NLRP3 mRNA stability (Fig. 5F).

AS3MT participates in the iAs-induced m6A methylation of NLRP3. (A–B) Expression of m6A-associated proteins in control and iAs-treated L-02 cells, cytoplasm (A) and nucleus (B). (C–D) m6A levels in total RNA (C) and mRNA (D) were compared between control and iAs-treated L-02 cells. (E) Endogenous protein interactions were examined in control and iAs-treated L-02 IP cell lysates using a rabbit anti-NLRP3 antibody and analyzed by western blotting to detect m6A methylation-associated proteins. (F) NLRP3 mRNA stability in siAS3MT transfected iAs-treated L-02 cells. **P < 0.01 versus the control group, ##P < 0.01 versus the iAs-treated group. The data represent the mean ± standard error of the mean (SEM), n = 3. One-way ANOVA was used to determine statistical significance
m 6 A methylation mediates the iAs-induced NLRP3 inflammasome activation
The autophagic-lysosomal pathway, mtROS, and ER stress are involved in iAs-induced NLRP3 inflammasome activation. However, the regulatory effects of iAs on NLRP3 transcription levels remain unclear. To explore the role of m6A methylation in the early onset of iAs-induced hepatic IR, we performed GO and KEGG enrichment analyses on m6A-seq results. Differential peaks were enriched for GO terms related to protein binding and transcription and were enriched in multiple pathways, including the NOD-like receptor signaling pathway (Fig. S5A–B), we speculated that m6A methylation could be an essential post-transcriptional modification mechanism in iAs-induced NLRP3 inflammasome activation. Therefore, we tested this by evaluating interactions between NLRP3 and m6A methylases by Co-IP in mouse liver. As shown (Fig. 6A), iAs treatment facilitated m6A methylase and NLRP3 binding. METTL14 interference also enhanced NLRP3 mRNA decay in iAs-treated L-02 cells (Fig. S6A and Fig. 6B). Moreover, METTL14 silencing blocked iAs-induced NLRP3 inflammasome activation (Fig. 6D–F) and IL-1β and IL-18 release (Fig. 6G and H) in L-02 cells. Additionally, NLRP3 mRNA attenuation and NLRP3 inflammasome activation upon IGF2BP2 silencing were identified in iAs-treated L-02 cells (Fig. S6B and Fig. 6C and I−M). These results revealed that iAs promoted NLRP3 expression and inflammasome activation via METTL14-dependent m6A methylation.

Inorganic As (iAs) induces NLRP3 inflammasome activation via m6A methylation. (A) Endogenous protein interactions were examined in control and iAs-treated liver lysates immunoprecipitated with rabbit anti-NLRP3 antibody and analyzed by western blotting to detect m6A methylation-associated proteins. (B) NLRP3 mRNA stability in iAs-treated L-02 cells, with or without siMETTL14 transfection. (C) NLRP3 mRNA stability in iAs-treated L-02 cells, with or without siIGF2BP2 transfection. (D–F) NLRP3 inflammasome-related protein expression in siMETTL14 transfected iAs-treated L-02 cells. GAPDH served as a loading control. (G–H) IL-1β (G) and IL-18 (H) secretion from indicated L-02 cells. (I–K) NLRP3 inflammasome-related protein expression in siIGF2BP2 transfected iAs-treated L-02 cells. GAPDH served as a loading control. (L–M) IL-1β (L) and IL-18 (M) secretion from indicated L-02 cells. *P < 0.05 and **P < 0.01 versus the control group, #P < 0.05 and ##P < 0.01 versus the iAs-treated group. The data represent the mean ± standard error of the mean (SEM), n = 3. One-way ANOVA was used to determine statistical significance
iAs upregulates the m 6 A methylation in mouse liver
To evaluate the effects of iAs on m6A methylation during hepatic IR, we assessed m6A methylation levels in iAs exposed and control groups. From m6A-seq results, we identified approximately 4,000 significant differences in m6A peaks between groups, with an overall increase in m6A mRNA methylation in iAs-treated group (Fig. 7A). To further confirm these results, m6A levels in total RNA and mRNA were higher in exposed animals than controls (Fig. 7B and C). Furthermore, when compared with controls, the expression of IGF2BP2 was consistently downregulated, and YTHDF2 and ALKBH5 were upregulated in cytoplasmic fractions iAs-treated group (Fig. 7D–F). However, in nuclear components, expression levels of METTL14 and IGF2BP2 were significantly upregulated and YTHDF2 was downregulated iAs-treated group (Fig. 7G–I).

Inorganic As (iAs) treatment upregulates m6A enrichment in mouse liver. (A) Histogram showing changes in m6A enrichment levels between control and iAs-treated samples of all peaks showing enrichment in the control group. The change in enrichment is represented by the median of n = 3 control-iAs pairs. (B–C) m6A levels in total RNA (B) and mRNA (C) were compared between control and iAs-treated groups (n = 6). (D–E) The expression of m6A-associated proteins in control and iAs-treated mouse livers; cytoplasm (D) and nuclei (E), respectively (n = 3). **P < 0.01 versus the control group. The data represent the mean ± standard error of the mean (SEM). The Student's two-tailed t test was used to determine statistical significance
m 6 A methylation regulates iAs-induced hepatic IR
To determine the role of METTL14 and IGF2BP2 in iAs-induced hepatic IR, we knocked down METTL14 and IGF2BP2 expression using siRNA. Knockdown efficiency was examined in L-02 cells (Fig. S6A–B). After METTL14 interference, the iAs-induced insulin signaling impairment was significantly reversed (Fig. 8A–C), and glucose uptake was correspondingly increased (Fig. 8D). Similar results were also observed during IGF2BP2 downregulation upon siIGF2BP2 transfection (Fig. 8E–H). Altogether, METTL14 and IGF2BP2 appeared to play essential roles in iAs-induced hepatic IR.

Inhibition of m6A methylation attenuates iAs-induced hepatic IR. Before treatment with iAs, L-02 cells were transfected with siMETTL14 or siIGF2BP2. Cells were also stimulated with insulin for 10 min before protein isolation. (A–C) The effect of siMETTL14 on AKT and GSK3β phosphorylation levels in iAs-treated L-02 cells. (E–G) The effect of siIGF2BP2 on AKT and GSK3β phosphorylation levels in iAs-treated L-02 cells. (D and H) The effect of siMETTL14 or siIGF2BP2 on glucose uptake levels in iAs-treated L-02 cells. *P < 0.05 and **P < 0.01 versus the control group, #P < 0.05 and ##P < 0.01 versus the iAs-treated group. The data represent the mean ± standard error of the mean (SEM), n = 3. One-way ANOVA was used to determine statistical significance
Discussion
The NLRP3 inflammasome is closely associated with inflammatory metabolic diseases such as NAFLD/NASH. We previously demonstrated that the NLRP3 inflammasome was involved in iAs-induced hepatic IR, NASH, and HSC activation (Qiu et al. 2018, Jia et al. 2020, Tao et al. 2020). Here, we demonstrated an association between NLRP3 inflammasome activation and early onset iAs-induced hepatic IR. In addition to NLRP3, other inflammasomes are also involved in iAs-induced inflammatory responses, such as NLRC4 and AIM2 (Maier et al. 2014, Zhang et al. 2016); therefore, we assessed the expression these proteins. Except for the significant upregulation of NLRP3 expression levels, NLRC4 expression was slightly decreased, while AIM2 levels remained almost unchanged. Thus, iAs promoted inflammatory responses mainly by regulating NLRP3 inflammasome activation in hepatocytes. Nevertheless, the effects of iAs on the transcription and post-transcriptional modification of NLRP3 remain elusive.
AS3MT is a critical enzyme during iAs methyl metabolism and may be a link between iAs and IR (Douillet et al. 2017, Huang et al. 2018b). However, associated mechanisms are unknown. We observed that AS3MT interference significantly restored insulin signaling activity, enhanced the glucose uptake capacity of L-02 cells, and effectively alleviated iAs-induced IR. Furthermore, AS3MT interacted with NLRP3, at the 201–375 amino acid position of AS3MT and the 211–550 position of NLRP3, and promoted NLRP3 inflammasome activation. Also, AS3MT interference significantly inhibited the iAs-induced translocation of METL14 and IGF2BP2 to the nucleus. Furthermore, AS3MT was identified as a key modulator of m6A methylation-mediated NLRP3 inflammasome activation. Specifically, AS3MT interacted with NLRP3 to enhance the m6A methylation of NLRP3, thereby facilitating iAs-induced hepatic IR.
However, contrary to our results, Huang et al. reported that AS3MT knockout mice showed a more severe IR phenotype after iAs treatment (Huang et al. 2018b). However, as few studies on AS3MT mechanisms in iAs-induced liver metabolism disorders have been conducted, more studies are required in the future. Notably, significant differences in methylation levels (both the degree and pattern) after iAs exposure exist between individuals and lead to substantial differences in susceptibility to iAs exposure. Key genetic factors are genetic polymorphisms, which include single nucleotide polymorphisms (SNPs) (Das et al. 2016; Balakrishnan et al. 2017; Hsieh et al. 2017; De Loma et al. 2018; Lin et al. 2018). Therefore, changes in AS3MT SNPs after iAs exposure and their associated functions in iAs-induced disease should be given more attention in future studies.
RNA m6A modifications have essential roles in metabolic liver diseases, such as NAFLD/NASH and hepatocellular carcinoma (Zhao et al. 2020b). Nevertheless, the role of m6A in metabolic disorders caused by environmental pollutants such as arsenic remains unclear. In this study, iAs treatment promotes METTL14 and IGF2BP2 translocation into the nucleus and upregulated m6A enrichment in hepatocytes, leading to early onset hepatic IR. Additionally, iAs-induced NLRP3 inflammasome activation was dependent on METTL14-mediated m6A methylation, and NLRP3 mRNA stability was enhanced by m6A methylation through METTL14 and reader IGF2BP2.
Previously, the effects of iAs on m6A methylation mainly focused on keratinocytes, with limited research on the liver. Zhao et al. reported that iAs upregulated m6A methylation enrichment in keratinocytes (Zhao et al. 2019, Zhao et al. 2020a). However, Cui et al. showed that chronic low-level arsenic exposure upregulated FTO stability and downregulated m6A enrichment in keratinocytes (Cui et al. 2021). Consistent with Zhao et al., we showed that iAs treatment upregulated m6A enrichment in hepatocytes by enhancing METTL14 and IGF2BP2 translocation into the nucleus. However, in keratinocytes, iAs upregulated methyltransferase expression and promoted m6A methylation enrichment, while we showed that iAs upregulated m6A methylation enrichment by promoting MEETL14 translocation into hepatocyte nuclei.
In summary, after iAs treatment, AS3MT interacted with NLRP3 and enhanced the m6A methylation of NLRP3 mRNA, thereby promoting NLRP3 inflammasome activation, and ultimately leading to early onset of iAs-induced hepatic IR. Together, these results may open new angles for therapeutic interventions in arsenicosis and identified a novel post-transcriptional function of AS3MT in promoting arsenicosis.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Abbreviations
- iAs :
-
Inorganic arsenic
- m 6 A :
-
N6-methyladenosine
- IR :
-
Insulin resistance
- T2D :
-
Type 2 diabetes
- NAFLD :
-
Non-alcoholic fatty liver disease
- AS3MT :
-
Arsenite methyltransferase
- SAM :
-
S-adenosylmethionine
- SNP :
-
Single nucleotide polymorphism
- NLRP3 :
-
NOD-like receptor protein 3
- METTL14 :
-
Methyltransferase-like 14
- METTL3 :
-
Methyltransferase-like 3
- IGF2BPs :
-
Insulin-like growth factor 2 mRNA-binding proteins
- YTHDFs :
-
YTH domain-containing family proteins
References
Allen, A. M. and B. A. Neuschwander-Tetri (2021). "The importance of glycemic equipoise in NASH." Hepatology.
Argos M, Kalra T, Rathouz PJ, Chen Y, Pierce B, Parvez F, Islam T, Ahmed A, Rakibuz-Zaman M, Hasan R, Sarwar G, Slavkovich V, van Geen A, Graziano J, Ahsan H. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): a prospective cohort study. Lancet. 2010;376(9737):252–8.
Balakrishnan P, Vaidya D, Franceschini N, Voruganti VS, Gribble MO, Haack K, Laston S, Umans JG, Francesconi KA, Goessler W, North KE, Lee E, Yracheta J, Best LG, MacCluer JW, Kent J, Cole SA, Navas-Acien A. Association of Cardiometabolic Genes with Arsenic Metabolism Biomarkers in American Indian Communities: The Strong Heart Family Study (SHFS). Environ Health Perspect. 2017;125(1):15–22.
Castriota F, Rieswijk L, Dahlberg S, La Merrill MA, Steinmaus C, Smith MT, Wang JC. A State-of-the-Science Review of Arsenic’s Effects on Glucose Homeostasis in Experimental Models. Environ Health Perspect. 2020;128(1):16001.
Chen X, Guo X, He P, Nie J, Yan X, Zhu J, Zhang L, Mao G, Wu H, Liu Z, Aga D, Xu P, Smith M, Ren X. Interactive Influence of N6AMT1 and As3MT Genetic Variations on Arsenic Metabolism in the Population of Inner Mongolia, China. Toxicol Sci. 2017;155(1):124–34.
Cui YH, Yang S, Wei J, Shea CR, Zhong W, Wang F, Shah P, Kibriya MG, Cui X, Ahsan H, He C, He YY. Autophagy of the m(6)A mRNA demethylase FTO is impaired by low-level arsenic exposure to promote tumorigenesis. Nat Commun. 2021;12(1):2183.
Das N, Giri A, Chakraborty S, Bhattacharjee P. Association of single nucleotide polymorphism with arsenic-induced skin lesions and genetic damage in exposed population of West Bengal, India. Mutat Res. 2016;809:50–6.
De Loma J, Skröder H, Raqib R, Vahter M, Broberg K. Arsenite methyltransferase (AS3MT) polymorphisms and arsenic methylation in children in rural Bangladesh. Toxicol Appl Pharmacol. 2018;357:80–7.
Douillet C, Huang MC, Saunders RJ, Dover EN, Zhang C, Stýblo M. Knockout of arsenic (+3 oxidation state) methyltransferase is associated with adverse metabolic phenotype in mice: the role of sex and arsenic exposure. Arch Toxicol. 2017;91(7):2617–27.
Ferguson, D. and B. N. Finck (2021). "Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus." Nat Rev Endocrinol.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361(6409):1346–9.
Gao N, Yao X, Jiang L, Yang L, Qiu T, Wang Z, Pei P, Yang G, Liu X, Sun X. Taurine improves low-level inorganic arsenic-induced insulin resistance by activating PPARγ-mTORC2 signalling and inhibiting hepatic autophagy. J Cell Physiol. 2019;234(4):5143–52.
Grau-Perez M, Kuo CC, Gribble MO, Balakrishnan P, Jones Spratlen M, Vaidya D, Francesconi KA, Goessler W, Guallar E, Silbergeld EK, Umans JG, Best LG, Lee ET, Howard BV, Cole SA, Navas-Acien A. Association of Low-Moderate Arsenic Exposure and Arsenic Metabolism with Incident Diabetes and Insulin Resistance in the Strong Heart Family Study. Environ Health Perspect. 2017;125(12):127004.
Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 2018;122(12):1722–40.
Hsieh RL, Su CT, Shiue HS, Chen WJ, Huang SR, Lin YC, Lin MI, Mu SC, Chen RJ, Hsueh YM. Relation of polymorphism of arsenic metabolism genes to arsenic methylation capacity and developmental delay in preschool children in Taiwan. Toxicol Appl Pharmacol. 2017;321:37–47.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, Hu YC, Huttelmaier S, Skibbe JR, Su R, Deng X, Dong L, Sun M, Li C, Nachtergaele S, Wang Y, Hu C, Ferchen K, Greis KD, Jiang X, Wei M, Qu L, Guan JL, He C, Yang J, Chen J. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018a;20(3):285–95.
Huang MC, Douillet C, Dover EN, Zhang C, Beck R, Tejan-Sie A, Krupenko SA, Stýblo M. Metabolic Phenotype of Wild-Type and As3mt-Knockout C57BL/6J Mice Exposed to Inorganic Arsenic: The Role of Dietary Fat and Folate Intake. Environ Health Perspect. 2018;126(12):127003.
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buée L, Heneka MT. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–73.
Jia X, Qiu T, Yao X, Jiang L, Wang N, Wei S, Tao Y, Pei P, Wang Z, Zhang J, Zhu Y, Yang G, Liu X, Liu S, Sun X. Arsenic induces hepatic insulin resistance via mtROS-NLRP3 inflammasome pathway. J Hazard Mater. 2020;399:123034.
Lin YC, Chen WJ, Huang CY, Shiue HS, Su CT, Ao PL, Pu YS, Hsueh YM. Polymorphisms of Arsenic (+3 Oxidation State) Methyltransferase and Arsenic Methylation Capacity Affect the Risk of Bladder Cancer. Toxicol Sci. 2018;164(1):328–38.
Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D, He C. N (6)-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science. 2020;367(6477):580–6.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, Huang JT, Chen SM, Xu ZG, Leng XH, Yu XC, Cao J, Zhang Z, Liu J, Lengyel E, He C. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20(9):1074–83.
Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184(10):2537–64.
Maier NK, Crown D, Liu J, Leppla SH, Moayeri M. Arsenic trioxide and other arsenical compounds inhibit the NLRP1, NLRP3, and NAIP5/NLRC4 inflammasomes. J Immunol. 2014;192(2):763–70.
Meng J, Lu Z, Liu H, Zhang L, Zhang S, Chen Y, Rao MK, Huang Y. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods. 2014;69(3):274–81.
Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, Suk WA. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect. 2013;121(3):295–302.
Nombela P, Miguel-López B, Blanco S. The role of m(6)A, m(5)C and Ψ RNA modifications in cancer: Novel therapeutic opportunities. Mol Cancer. 2021;20(1):18.
Perry MR, Wyllie S, Raab A, Feldmann J, Fairlamb AH. Chronic exposure to arsenic in drinking water can lead to resistance to antimonial drugs in a mouse model of visceral leishmaniasis. Proc Natl Acad Sci U S A. 2013;110(49):19932–7.
Qin F., B. Cai, J. Zhao, L. Zhang, Y. Zheng, B. Liu and C. Gao (2021). "Methyltransferase-Like Protein 14 Attenuates Mitochondrial Antiviral Signaling Protein Expression to Negatively Regulate Antiviral Immunity via N(6) -methyladenosine Modification." Adv Sci (Weinh): e2100606
Qiu T, Pei P, Yao X, Jiang L, Wei S, Wang Z, Bai J, Yang G, Gao N, Yang L, Qi S, Yan R, Liu X, Sun X. Taurine attenuates arsenic-induced pyroptosis and nonalcoholic steatohepatitis by inhibiting the autophagic-inflammasomal pathway. Cell Death Dis. 2018;9(10):946.
Rodrigues, T. S., K. S. G. de Sá, A. Y. Ishimoto, A. Becerra, S. Oliveira, L. Almeida, A. V. Gonçalves, D. B. Perucello, W. A. Andrade, R. Castro, F. P. Veras, J. E. Toller-Kawahisa, D. C. Nascimento, M. H. F. de Lima, C. M. S. Silva, D. B. Caetite, R. B. Martins, I. A. Castro, M. C. Pontelli, F. C. de Barros, N. B. do Amaral, M. C. Giannini, L. P. Bonjorno, M. I. F. Lopes, R. C. Santana, F. C. Vilar, M. Auxiliadora-Martins, R. Luppino-Assad, S. C. L. de Almeida, F. R. de Oliveira, S. S. Batah, L. Siyuan, M. N. Benatti, T. M. Cunha, J. C. Alves-Filho, F. Q. Cunha, L. D. Cunha, F. G. Frantz, T. Kohlsdorf, A. T. Fabro, E. Arruda, R. D. R. de Oliveira, P. Louzada-Junior and D. S. Zamboni (2021). "Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients." J Exp Med 218(3).
Rodriguez KF, Ungewitter EK, Crespo-Mejias Y, Liu C, Nicol B, Kissling GE, Yao HH. Effects of in Utero Exposure to Arsenic during the Second Half of Gestation on Reproductive End Points and Metabolic Parameters in Female CD-1 Mice. Environ Health Perspect. 2016;124(3):336–43.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–32.
Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42(4):245–54.
Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol. 2000;74(6):289–99.
Tao Y, Qiu T, Yao X, Jiang L, Wang N, Jia X, Wei S, Wang Z, Pei P, Zhang J, Zhu Y, Yang G, Liu X, Liu S, Sun X. Autophagic-CTSB-inflammasome axis modulates hepatic stellate cells activation in arsenic-induced liver fibrosis. Chemosphere. 2020;242:124959.
Tao Y, Qiu T, Yao X, Jiang L, Wang N, Jiang J, Jia X, Wei S, Zhang J, Zhu Y, Tian W, Yang G, Liu X, Liu S, Ding Y, Sun X. IRE1α/NOX4 signaling pathway mediates ROS-dependent activation of hepatic stellate cells in NaAsO(2) -induced liver fibrosis. J Cell Physiol. 2021;236(2):1469–80.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–88.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19(1):88.
Wang X, Wang J, Tsui YM, Shi C, Wang Y, Zhang X, Yan Q, Chen M, Jiang C, Yuan YF, Wong CM, Liu M, Feng ZY, Chen H, Ng IOL, Jiang L, Guan XY. RALYL increases hepatocellular carcinoma stemness by sustaining the mRNA stability of TGF-β2. Nat Commun. 2021;12(1):1518.
Xie L, Hu WY, Hu DP, Shi G, Li Y, Yang J, Prins GS. Effects of Inorganic Arsenic on Human Prostate Stem-Progenitor Cell Transformation, Autophagic Flux Blockade, and NRF2 Pathway Activation. Environ Health Perspect. 2020;128(6):67008.
Yi YC, Chen XY, Zhang J, Zhu JS. Novel insights into the interplay between m(6)A modification and noncoding RNAs in cancer. Mol Cancer. 2020;19(1):121.
Zhang H, Ge Y, He P, Chen X, Carina A, Qiu Y, Aga DS, Ren X. Interactive Effects of N6AMT1 and As3MT in Arsenic Biomethylation. Toxicol Sci. 2015;146(2):354–62.
Zhang M, Qi Y, Li H, Cui J, Dai L, Frank JA, Chen J, Xu W, Chen G. AIM2 inflammasome mediates Arsenic-induced secretion of IL-1 β and IL-18. Oncoimmunology. 2016;5(6):e1160182.
Zhang Q, Hou Y, Wang D, Xu Y, Wang H, Liu J, Xia L, Li Y, Tang N, Zheng Q, Sun G. Interactions of arsenic metabolism with arsenic exposure and individual factors on diabetes occurrence: Baseline findings from Arsenic and Non-Communicable disease cohort (AsNCD) in China. Environ Pollut. 2020;265(Pt A):114968.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42.
Zhao T, Li X, Sun D, Zhang Z. Oxidative stress: One potential factor for arsenite-induced increase of N(6)-methyladenosine in human keratinocytes. Environ Toxicol Pharmacol. 2019;69:95–103.
Zhao T, Sun D, Zhao M, Lai Y, Liu Y, Zhang Z. N(6)-methyladenosine mediates arsenite-induced human keratinocyte transformation by suppressing p53 activation. Environ Pollut. 2020;259:113908.
Zhao Z, Meng J, Su R, Zhang J, Chen J, Ma X, Xia Q. Epitranscriptomics in liver disease: Basic concepts and therapeutic potential. J Hepatol. 2020b;73(3):664–79.
Zong X, Xiao X, Shen B, Jiang Q, Wang H, Lu Z, Wang F, Jin M, Min J, Wang F, Wang Y. The N6-methyladenosine RNA-binding protein YTHDF1 modulates the translation of TRAF6 to mediate the intestinal immune response. Nucleic Acids Res. 2021;49(10):5537–52.
Acknowledgements
The HEK293T cells were kindly gifted by Dr. Jiayang Zhang (Advanced Institute for Medical Sciences of Dalian Medical University).
Funding
This study was supported by the National Natural Science Foundation of China (NSFC, 81872566, 81602881); Liaoning Province People's Livelihood Science and Technology Plan Project (2021JH2/10300048); 2021 Liaoning BaiQianWan Talents Program.
Author information
Authors and Affiliations
Contributions
T. Q., X. Y., and X. S. Conceptualization. T. Q., C. W., Q. H., and W. Y. Methodology. T. Q., J. Z., and Y. S. Formal analysis and Data curation. T. Q. and N. W. Writing—original draft. T. Q., N. W., L. J., X. L., and G. Y. Writing—review & editing. X. S. is the guarantor of the article.
Corresponding author
Ethics declarations
Ethics approval
The Animal Ethics Committee of Dalian Medical University approved all animal care and study procedures (AEE18065).
Consent to participate
Not applicable.
Consent for publication
All authors agree to publish the manuscript.
Competing interests
The authors declare that there are no conflicts of interest in the present work.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Qiu, T., Wu, C., Yao, X. et al. AS3MT facilitates NLRP3 inflammasome activation by m6A modification during arsenic-induced hepatic insulin resistance. Cell Biol Toxicol 39, 2165–2181 (2023). https://doi.org/10.1007/s10565-022-09703-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-022-09703-7