
Abstract
Background
In the last decade, genomic studies have identified and replicated thousands of genetic associations with measures of health and disease and contributed to the understanding of the etiology of a variety of health conditions. Proteins are key biomarkers in clinical medicine and often drug-therapy targets. Like genomics, proteomics can advance our understanding of biology.
Methods and Results
In the setting of the Cardiovascular Health Study (CHS), a cohort study of older adults, an aptamer-based method that has high sensitivity for low-abundance proteins was used to assay 4979 proteins in frozen, stored plasma from 3188 participants (61% women, mean age 74 years). CHS provides active support, including central analysis, for seven phenotype-specific working groups (WGs). Each CHS WG is led by one or two senior investigators and includes 10 to 20 early or mid-career scientists. In this setting of mentored access, the proteomic data and analytic methods are widely shared with the WGs and investigators so that they may evaluate associations between baseline levels of circulating proteins and the incidence of a variety of health outcomes in prospective cohort analyses. We describe the design of CHS, the CHS Proteomics Study, characteristics of participants, quality control measures, and structural characteristics of the data provided to CHS WGs. We additionally highlight plans for validation and replication of novel proteomic associations.
Conclusion
The CHS Proteomics Study offers an opportunity for collaborative data sharing to improve our understanding of the etiology of a variety of health conditions in older adults.
Similar content being viewed by others


Introduction
In studies of cardiovascular disease (CVD), the emergence of various types of -omics data has transformed population science. In the last decade, genome-wide association studies have identified and replicated thousands of genetic associations with measures of human health and disease. While risk scores aggregating multiple genetic variants can improve prediction of several cardiovascular conditions [1], the individual genetic associations are perhaps most valuable for providing biological insights about disease subtypes and potential molecular targets for therapeutics [2]. For instance, genetic studies that identified associations of ANGPTL3 variants with triglyceride levels [3] have resulted in the development of two new drug therapies for lipid disorders [4].
Proteomics, the large-scale study of hundreds to thousands of proteins, provides another avenue to biologic discovery. Proteins perform most biological functions, and they are often the targets of drug therapies [5]. The human genome encodes about 20,000 proteins, many of which undergo post-translational modifications that may affect their function [5]. Recent advances in technology have improved multiplexed protein assays [5, 6]. Linking proteomics with genomics is especially valuable. For instance, multiple studies have described associations between levels of the inflammation biomarker C-reactive protein (CRP) and CVD [7, 8], but these associations are not necessarily causal. Biologically, CRP is an acute phase reactant that increases in response to interleukin (IL)-6. Mendelian randomization (MR) studies, which use genetic variants as instrumental variables for modifiable risk factors, [9] suggest that CRP itself is not in the causal pathway for CVD [10]. Thus, direct efforts to reduce the levels of CRP for CVD prevention are unlikely to yield therapeutic benefit. In contrast, MR studies suggest a causal role for IL-6 in CVD [11]. Indeed, a clinical trial evaluating canakinumab, an IL-1-beta monoclonal antibody that decreases the levels of IL-6, significantly reduced the risk of recurrent CVD events [12].
The Cardiovascular Health Study (CHS), which recently completed large-scale proteomic assays on 3,188 participants, is providing both shared data and centralized analytic support to CHS investigators. Genomic and proteomic studies are expensive, and the National Institutes of Health (NIH) appropriately insists on passive methods of data sharing [13]. Approved NIH data-sharing methods involve depositing publicly funded data for widespread access, typically on dbGaP or BioLINCC. While CHS provides regular updates to these public sites, the CHS investigators developed a novel method of broad data sharing designed to be both productive and instructive. The NIH investment in cohort studies includes not only the participants, the data, and the biospecimens but also the investigators who have a deep understanding of the study design and its conduct. Over the last two decades, CHS developed a working-group (WG) model to provide “mentored access” to data, an approach that takes advantage of the investigators’ knowledge and experience [14]. Currently, contract funding provides active support, including central analysis, for seven trait-specific WGs. Each CHS WG is led by one or two senior investigators and includes 10 to 20 early or mid-career scientists. In this paper, we describe CHS, the WG model, the methods used to measure protein levels in CHS participants, data characteristics and the quality control measures undertaken, and the active design to share the proteomic data actively and widely.
Method
CHS design
CHS was designed to evaluate risk factors for coronary heart disease and stroke in older adults [15]. In 1989–90, four Field Centers used random samples from Medicare eligibility lists to recruit 5201 participants. Among eligible sampled individuals at baseline, 57% were enrolled [16]. In 1992–93, an additional sample of 687 predominantly African-American participants was recruited using similar methods. The primary outcomes in CHS are myocardial infarction (MI), angina, heart failure (HF), peripheral arterial disease, stroke, transient ischemic attack, and total mortality [17,18,19,20]. The events data collection through 2015 also included information about all hospitalizations so that other events such as venous thromboembolism, hip fracture, and pneumonia could be studied. After 2015, study methods include mortality follow-up, Medicare hospitalization data, and semiannual phone calls. The multiple methods of follow-up have allowed for near-complete ascertainment of CVD events and deaths [21].
The baseline examination assessed traditional risk factors and measures of subclinical cardiovascular disease. During the 1990s, contacts alternated between in-clinic examinations and telephone calls. At each annual in-clinic visit, selected examination components were repeated, and some new components such as cranial magnetic resonance imaging were added. Processing for EDTA plasma was standardized, and samples were shipped to the University of Vermont Core Laboratory on dry ice, stored initially in -70 °C freezers, with storage at -80 °C for the last 20 years. Since 2000, participants have been contacted by telephone every 6 months for information, including events, functional status, cognitive function, and quality of life. Additional details can be found at https://chs-nhlbi.org/CHSData. Ancillary studies have provided a wealth of other data, including rare-variant exome chip data on 5028 CHS participants; genome-wide genotype data (n = 4094); whole exome sequence (WES) data (n = 2940); and whole-genome sequence (WGS) data (n = 4932). The genetic data, both GWAS [22] and sequence data [23], have undergone extensive quality control evaluations. The CHS contract and ancillary studies have also funded novel assays and produced multiple publications on a score of established biomarkers [24,25,26].
CHS WG model: mentored access to CHS data
In 2001, several CHS investigators developed the approach of “mentored access” to CHS data and recruited young scientists from across the country to work in CHS and participate in the newly established Renal WG. The Renal WG was so productive that CHS conducted novel two-day workshops for early-career scientists targeting new investigators in March 2005 and May 2007. The CHS new-investigator workshops soon spawned a set of new WGs (currently, Cardiovascular, Aging, Bone, Diabetes, Neurology, Renal and Genetics). Each WG includes one or more lead investigators, a CHS Coordinating Center (CHSCC) analyst, and other (often early-career) investigators from about 40 institutions. Typically, WG members participate in regular calls; and the central CHSCC analysts perform the data analyses for the WG investigators. Data sharing across WGs has been the norm. For instance, The Diabetes WG funded assays of 7 novel biomarkers [25], and also shared these data with the Cardiovascular [25], Bone, and Aging WGs. The CHS network of mentored access, described in a paper from the NHLBI [14], not only serves as a major national training effort in CVD epidemiology, but also promotes multiple uses of costly data to advance high-quality science and the health of older adults. As illustrated in Supplemental Fig.1, the increase in CHS publications after the workshops was driven primarily by collaborators, often members of the WGs, rather than by the original CHS investigators. The variety of available phenotypes in CHS provides a rich resource for a team-based approach to the analysis of the proteomic data.
Design of CHS Proteomics Ancillary Study
For the CHS Proteomics Study, we chose the 1992–93 examination as the baseline. All 3188 CHS participants for whom we had unthawed plasma in the 1992–93 repository were selected. In addition, to estimate the stability of protein levels through correlation of sample aptamer levels from plasma collected five years apart, unthawed plasma for a random sample of 100 participants from the 1997–98 examination was selected. These samples were shipped on dry ice to SomaLogic (Boulder, CO) for assays on the 5 k SOMAscan. Samples were randomized across the plates with the exception of the 100 paired samples. Each pair was kept on the same plate. The variety of available phenotypes in CHS provide a rich resource for a team-based approach to proteomics.
SOMAscan assays
The SOMAscan assays have been described previously [27]. Briefly, slow off-rate modified aptamers (SOMA) are oligonucleotides that bind tightly and specifically at a ratio of 1:1 with a target protein and permit the evaluation of multiple proteins on a single assay [28]. The method, which takes advantage of novel chemically modified nucleotides [28], converts the measurement of protein levels into the measurement of nucleic acid levels assessed by a DNA oligo-array plate reader. Measurement of the SOMAmer (aptamer) concentration across a 7 log10 range (100 fM to 1 uM) reflects the concentration of the protein with an average coefficient of variation of 6%. Results of these assays, reported in relative fluorescence units (RFU), are approximately proportional to plasma protein concentrations. The SomaLogic assays included 4979 proteins: including those related to inflammation, CVD, aging, metabolism/endocrine, neurology, pulmonary, renal, and others.
Recent reports by our group and others describe not only the analytic and clinical validity of the aptamer-based assay method [29], but also high correlations between SOMAscan levels and conventional laboratory measures of IL-6, IL-8, IL-16, MMP-3, CRP, and troponins [30]. Nevertheless, in view of concerns about the specificity of some aptamers, we plan to use “orthogonal” techniques to assess analyte specificity and quantification. For selected markers, we plan to (1) use the aptamers themselves as affinity reagents to capture and identify the protein targets that are binding using liquid-chromatography-tandem mass spectrometry (LC–MS/MS) based affinity proteomics [29] and (2) develop quantitative LC–MS/MS-based assays by using anti-peptide antibodies to enrich unique tryptic peptides derived from the protein targets identified as binding to the aptamers. These analyses are done in a targeted manner, thereby increasing both the specificity and sensitivity of the assays [31, 32]. Known amounts of synthetic, stable-isotopically labeled versions of the target peptides are captured simultaneously with the native peptides and used to quantify the amounts of the latter. This method known as stable isotope standards and capture by anti-peptide antibodies (SISCAPA) or immunoMRM is more specific than antibody or aptamer-based approaches and suffers fewer interferences, although it can be less sensitive Liquid Chromatography-Mass Spectroscopy (LC–MS) [31,32,33].
Protein quantitative trait loci (pQTLs)
This study leverages genetic data from CHS and other studies to identify protein quantitative trait loci (pQTLs) both to aid in protein identification, and to assess whether novel biomarkers may belong to causal pathways. Briefly, whole-genome sequencing data on CHS participants from TOPMed were used [23]. Log-transformed and standardized SOMAscan values were residualized on age, sex, race, and principal components (PCs) of ancestry 1–10 as determined by GENetic EStimation and Inference in Structured samples (GENESIS), and the resulting residuals were normalized. To evaluate the association between these values and genetic variants, the fastGWA model was used, implemented in GCTA software package (version 1.93.2beta/gcta64) [34]. Linear mixed effects models were adjusted for age, sex, race, the estimated genetic relationship matrix, and PCs 1–10, with repeat adjustment implemented to reduce type I error and improve statistical power. After implementing a procedure to handle overlap, the variant with the lowest P value in the resulting region was labeled as the sentinel variant. Any sentinel variant within 1 Mb of the TSS for the cognate gene of a protein were considered ‘cis’.
The proteomics consortium led by Butterworth, the senior author on the Nature genomic atlas of the proteome [35], includes more than 18,000 participants with both genetic and SOMAscan data, and these data are available to validate pQTLs, to aid in the selection of genetic variants, and to provide independent estimates of effect sizes for Mendelian randomization analyses.
Data analysis
Analytic methods for studies evaluating individual candidate proteins are generally straightforward. For an unbiased search of the available proteomic space, however, there are three general approaches [36]: 1) testing all proteins individually with multiple comparison correction for statistical significance [37]; 2) data reduction strategies such as principal components [38] or network analysis [39,40,41]; and 3) variable (protein) selection strategies [36, 42] such as the adaptive elastic net [43]. Some WGs may leverage proteomic data to develop prediction models for clinical traits, in which case we will use the C-statistic to evaluate discrimination and risk-stratification tables to evaluate calibration and accuracy [44, 45]. For proteins with adequate genetic instruments, we will also perform MR analyses to examine the supporting evidence for potential causal associations between protein levels and clinical outcomes [46, 47]. The CHS Proteomics study has good power for association with a variety of outcomes. Investigators from the Framingham Heart Study (FHS) and the Jackson Heart Study (JHS), both of which have SomaLogic data and have subcontracts for replication and collaborative meta-analyses [48, 49], are active collaborators. Additionally, we are actively seeking collaboration with other national and international studies that have proteomic data to improve power.
Study organization
The CHS Proteomics Study relies on the CHS WG model but has added an analysis committee and provides modest central analytic support. In the setting of the COVID-19 pandemic, weekly videoconferences calls provide mentored access to the data and methods. During the first year, scientists have provided a series of talks: CHS design, proteomics study design, outline of analysis plans, SomaLogic assay methods, quality control, statistics for proteomics, analytic pipelines, penalized regression, mediation analysis, network analysis, and Mendelian randomization. During the same period, early-career young investigators from all 7 WG have brought at least one manuscript proposal to the group for review. A pipeline for proteomic analysis has been developed and made available to WG members and publicly available through GitHub (https://github.com/UWCHRU/OmicsPipeline). The analysis committee has provided recommendations and support for network analysis and Mendelian randomization. In addition to central analytic support, study data and analytic methods are available to WG analysts and to individual CHS investigators who are encouraged to participate in the regular study videoconferences calls to share results and analysis issues so that solutions can be widely disseminated. This group will also help make plans for the use of mass spectrometry for protein validation and quantification and the development of new inexpensive enzyme-linked immunosorbent assay (ELISA) assays for a few key proteins that might be essential to moving findings forward in other clinical studies, clinical trials, or clinical care.
Dataset creation and quality control analyses
In the results, we describe characteristics of the dataset received from SomaLogic and analytic decisions based on the quality of study samples, the characteristics of the CHS participants, the numbers of incident events, and the stability of protein levels over a five-year period. For selected biomarkers, we compare protein concentrations at the 1992–93 examination cycle based on ELISA assays and SomaLogic-based proteomics measurements and describe the plan for multiple testing. Finally, we calculate the median RFU over all proteins for each CHS participant and describe the presence of outliers in the dataset.
Result
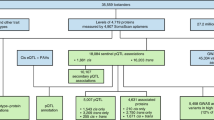
Results were returned from SomaLogic for all 3288 samples (3188 participants plus 100 repeat measures) on 4979 aptamers. Our analysis excluded three participants with samples flagged by SomaLogic for poor quality, yielding a final analysis sample of 3185 participants, 100 of whom had samples from both the 1992–93 and 1997–98 CHS examinations (Fig. 1). There were no missing proteomic data for those 3185 participants. Calibrators included in the SOMAscan assays had a median intra-assay coefficient of variation of 3.4 (10%—90%: 1.6 – 7.6), and quality control samples had a median inter-assay coefficient of variation of 4.4 (10%—90%: 2.6 – 10.1). We excluded from the analysis dataset aptamers marked as “deprecated” (indicating a retired aptamer) and those marked as “non-human.” The final analysis sample included 3285 proteomes of 4979 aptamers from 3185 unique CHS participants. The 731 aptamers flagged for potential quality concerns remained in the analysis sample, and users are alerted to interpret results with caution. Aside from analyses of long-term stability, all the following analyses used the 4979 aptamers from the 1992–93 examination on 3185 CHS participants.

Flowchart of Inclusion in Cardiovascular Health Study proteomics analyses CHS = Cardiovascular Health Study QC = Quality Control
Characteristics of participants included in our analysis are described in Table 1, along with characteristics of those participants who attended the 1992–93 examination but were not included in the analysis sample. Previous ancillary studies, which had partially depleted unthawed plasma, account for the differences. At baseline, 8% percent of those included in the analysis sample had a history of myocardial infarction (MI), 4% stroke, and 4% heart failure (HF). During 25 years of follow-up for adjudicated events, these 3185 participants had 555 incident MIs, 577 incident strokes, 1064 incident HF events, and 2776 deaths, of which 963 were classified as cardiovascular deaths.
For all analyses, aptamer levels were log transformed and standardized to have a mean of 0 and a standard deviation of 1. Among the 100 participants for whom plasma was sampled from both the 1992–93 and 1997–98 examinations, the median intraclass correlation coefficient across all proteins was 0.66 (IQR, 0.46 – 0.81). Within each individual, the median RFU of all 4979 aptamers served as a marker of total plasma protein level. Among the 100 participants with two assays five years apart, the intra-individual correlation between the median of all an individual’s RFU values was 0.61 (Supplemental Fig.2).
Figure 2 illustrates the relationships of biomarkers assayed by SomaLogic with those previously assayed in CHS using ELISA-based methods at the same examination. The pairwise correlation between the SomaLogic aptamers and ELISA-based methods (both log transformed) ranged from 0.65 to 0.96: the correlations for C-reactive protein, Cystatin C, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) were all > 0.90.

Comparison of log-transformed and standardized SomaLogic aptamer levels and CHS-assayed biomarkers for (A) C-reactive protein (n = 3,158), (B) Cystatin C (n = 3,182), (C) Factor VII (n = 3,129), (D) Fibrinogen (n = 3,175), and (E) NT-proBNP (n = 2,837)
To determine a statistical significance threshold to account appropriately for multiple testing, principal components (PC) were calculated from the standardized log protein RFUs in the analysis dataset in order to estimate the amount of independent information available within the protein data. The first PC explained 31% of the variance, while the first five collectively explained 45% of the variance. A total of 1566 PCs was required to explain 95% of the variance in the 4979 standardized log protein RFUs (Supplemental Fig.3). We propose to use 1566 as a proxy for the number of independent tests performed per analysis, so that the empirically-informed Bonferroni-corrected 0.05 significance level of 0.05/1566 is 3.2 × 10–05 [50].
As part of initial quality control, we examined the protein data for evidence of batch effects. A plot of PC1 against PC2 did not display any batch effects by plate, nor did a plot of t-distributed stochastic neighbor embedding [51] (Supplemental Fig.4). Similarly, there was no apparent pattern among the first five PCs based upon self-identified race or sex (Supplemental Figs. 5 and 6). To identify whether samples with high median RFU values clustered by laboratory handling, we calculated the median RFU for all proteins from each sample and found that no plate had systematically higher or lower median RFU (Fig. 3).

Boxplots of median relative fluorescence unit for each participant sample, stratified by plate.RFU = relative fluorescence unit
Discussion
The CHS Proteomics Study offers high quality data on the relative concentrations of 4979 proteins in 3185 participants and will be an important source of collaborative research on the etiology of cardiovascular disease and other health conditions of older adults. The WG structure in CHS allows for widespread distribution and use of these data, and the project offers opportunities for replication and integration of genomics and MR into ongoing projects. The results of these analyses will help inform our understanding of a variety of diseases and may inform biologic targets for drug development.
Out of 3188 total samples, only three samples were flagged for poor quality and removed from analysis dataset, and the proteomic data did not appear to show any evidence of batch effects or undue influence of outlier values. For analysis of the raw RFUs, we have recommended log-transformation and standardization to a mean of 0 and SD of 1, and the number of PCs required to explain 95% of the variation in the data may be used empirically to inform adjustment for multiple comparisons rather than a conservative Bonferroni approach that assumes 4979 independent tests. The finding that relative protein concentrations for a number of proteins were strongly correlated with concentrations measured by ELISA-based methods in CHS suggests that the aptamer-based method is comparable to other available methods of measuring protein concentrations [30].
Because many longitudinal hypotheses in this study involve the associations of baseline protein levels and the incidence of a variety of clinical events, the long-term biological stability of these biomarkers is important. The median long-term correlation for the 4,979 proteins was 0.66 (IQR, 0.46 – 0.81), which is similar to what has been previously observed for clinical biomarkers measured by standard assays. For instance, in CHS, the between-visit intra-individual correlation (ICC) from traditional assay methods across 3–4 years for fibrinogen was 0.62 (95% CI, 0.60–064); for factor VII, 0.77 (95% CI, 0.76 to 0.78); and for cystatin C, 0.71 (95% CI, 0.69 to 0.72). For comparison with other risk factors in CHS, the ICC for total cholesterol between the 1992–93 and 1997–98 exams was 0.47 (95% CI, 0.44–0.49) and the ICC for systolic blood pressure over the same period was 0.65 (95% CI, 0.63–0.67). In other words, the within-individual ICCs for these protein log RFUs are comparable to the ICCs for several traditional CVD risk factors.
The specificity of some of the SomaLogic aptamers has been questioned. For instance, an aptamer for GDF11 was also reported to bind to the protein GDF8 [52, 53]. The early GDF11 reports appear to be a misidentification related to high homology between these two proteins. Genetic evidence can assist in protein validation (if genetic variation does not influence aptamer binding). For selected key findings, MS-based validation of the protein identity is essential to rigorous design; and the CHS Proteomics Study includes modest resources for unambiguous identification and quantification [29].
Many proteins exhibit strong genetic associations [48]. Using an aptamer-based assay in 3301 participants, for instance, Butterworth and colleagues identified 1927 genetic associations (p < 1.5E-11) with 1478 of 3622 proteins to produce a genomic atlas of the human plasma proteome [35]. Protein QTLs (pQTLs) replicated well. The median protein-level variation explained by pQTLs was 5.8% (interquartile range, 2.6 to 12.4%), and pQTLs explained more than 20% of the variation of 193 proteins. The pQTL findings have already aided in drug-target prioritization and identifying new indications for approved drugs [35]. The pQTL data in this genomic atlas will assist in assuring the specificity of selected aptamers and will also provide strong instruments for Mendelian randomization.
CHS is well designed and rigorously conducted study that has extensive longitudinal data, including events data and serial measures of physical and cognitive function. Loss to follow-up has been minimal. Unthawed plasma stored at -80° C makes protein degradation unlikely. Moreover, the extensive genetic data in CHS make possible MR analyses, which, under certain assumptions, can provide supportive evidence for a potential causal role. Major strengths of the study are the research team with expertise in protein biomarker research and population-based studies and the collaboration with FHS and JHS investigators. Additionally, CHS used Medicare files to sample a population-based cohort of older adults to whom the results may be generalized. Nevertheless, we recognize inherent limitations in an observational cohort study, including the possibilities of measurement error, residual confounding, unmeasured confounding, and the difficulties of causal inference. Although CHS has data on a rich set of covariates, confounding is difficult to exclude, and a cautious interpretation of the findings is appropriate. While CHS was able to obtain data on the SomaLogic 5 K platform, FHS and JHS currently only have data on the 1 K and 1.3 K platforms. Additional assays are planned or in progress. The platform differences, as well as differences in some participant characteristics among the studies, may limit the ability of FHS and JHS to replicate some findings in CHS.
Perhaps the most innovative aspect of this study is the reliance on the collaborative team science of the CHS WGs, where early-career investigators receive mentored access to the data and methods. The proteomics study has also linked the WGs with central analytic support. The central analysis committee makes recommendations about best practices. The video calls provide opportunities to identify problems, disseminate “best practices,” present or review analytic methods, develop manuscript proposals, discuss analytic approaches, review interim results, establish collaborations, decide on replication plans, and coordinate efforts across WGs and investigators. Typically, the manuscripts are led by early-career investigator “champions” who have the time, energy, and enthusiasm to bring them to fruition. In an effort to pursue the best science efficiently, this approach attempts to maximize the scientific and educational value of these publicly-funded research resources. While the data will be made publicly available on dbGaP and BioLINCC, we encourage national and international early-career investigators to consider joining a CHS WG (https://chs-nhlbi.org for CHS and https://chs-nhlbi.org/WorkingGroups for the WGs).
References
Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–24. https://doi.org/10.1038/s41588-018-0183-z.
Sinner MF, Tucker NR, Lunetta KL, et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. 2014;130(15):1225–35. https://doi.org/10.1161/CIRCULATIONAHA.114.009892.
Romeo S, Pennacchio LA, Fu Y, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39(4):513–6. https://doi.org/10.1038/ng1984.
Graham MJ, Lee RG, Brandt TA, et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N Engl J Med. 2017;377(3):222–32. https://doi.org/10.1056/NEJMoa1701329.
Ebhardt HA, Root A, Sander C, Aebersold R. Applications of targeted proteomics in systems biology and translational medicine. Proteomics. 2015;15(18):3193–208. https://doi.org/10.1002/pmic.201500004.
Smith JG, Gerszten RE. Emerging Affinity-Based Proteomic Technologies for Large-Scale Plasma Profiling in Cardiovascular Disease. Circulation. 2017;135(17):1651–64. https://doi.org/10.1161/CIRCULATIONAHA.116.025446.
Tracy RP, Lemaitre RN, Psaty BM, et al. Relationship of C-reactive protein to risk of cardiovascular disease in the elderly. Results from the Cardiovascular Health Study and the Rural Health Promotion Project. Arterioscler Thromb Vasc Biol. 1997;17(6):1121–7. https://doi.org/10.1161/01.atv.17.6.1121.
Zakai NA, Katz R, Jenny NS, et al. Inflammation and hemostasis biomarkers and cardiovascular risk in the elderly: the Cardiovascular Health Study. J Thromb Haemost. 2007;5(6):1128–35. https://doi.org/10.1111/j.1538-7836.2007.02528.x.
Smith GD, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Prins BP, Abbasi A, Wong A, et al. Investigating the Causal Relationship of C-Reactive Protein with 32 Complex Somatic and Psychiatric Outcomes: A Large-Scale Cross-Consortium Mendelian Randomization Study. PLoS Med. 2016;13(6): e1001976. https://doi.org/10.1371/journal.pmed.1001976.
Interleukin-6 Receptor Mendelian Randomisation Analysis C, Swerdlow DI, Holmes MV, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379(9822):1214–24.https://doi.org/10.1016/S0140-6736(12)60110-X
Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119–31. https://doi.org/10.1056/NEJMoa1707914.
NHLBI. NHLBI policy for data sharing from clinical trials and epidemiological studies.
Eblen MFR, Olson JL, Pearson K, Pool LR, Puggal M, Wu C, Wagner RM. Social network analysis comparing researcher collaborations in two cardiovascular cohort studies. Res Eval. 2012;21(5):392–405.
Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1(3):263–76. https://doi.org/10.1016/1047-2797(91)90005-w.
Tell GS, Fried LP, Hermanson B, Manolio TA, Newman AB, Borhani NO. Recruitment of adults 65 years and older as participants in the Cardiovascular Health Study. Ann Epidemiol. 1993;3(4):358–66. https://doi.org/10.1016/1047-2797(93)90062-9.
Ives DG, Fitzpatrick AL, Bild DE, et al. Surveillance and ascertainment of cardiovascular events. Cardiovas Health Study Ann Epidemiol. 1995;5(4):278–85. https://doi.org/10.1016/1047-2797(94)00093-9.
Price TR, Psaty B, O’Leary D, Burke G, Gardin J. Assessment of cerebrovascular disease in the Cardiovascular Health Study. Ann Epidemiol. 1993;3(5):504–7. https://doi.org/10.1016/1047-2797(93)90105-d.
Gottdiener JS, Arnold AM, Aurigemma GP, et al. Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol. 2000;35(6):1628–37. https://doi.org/10.1016/s0735-1097(00)00582-9.
Longstreth WT Jr, Bernick C, Fitzpatrick A, et al. Frequency and predictors of stroke death in 5,888 participants in the Cardiovascular Health Study. Neurology. 2001;56(3):368–75. https://doi.org/10.1212/wnl.56.3.368.
Psaty BM, Delaney JA, Arnold AM, et al. Study of Cardiovascular Health Outcomes in the Era of Claims Data: The Cardiovascular Health Study. Circulation. 2016;133(2):156–64. https://doi.org/10.1161/CIRCULATIONAHA.115.018610.
Psaty BM, O’Donnell CJ, Gudnason V, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2(1):73–80. https://doi.org/10.1161/CIRCGENETICS.108.829747.
Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845).https://doi.org/10.1038/s41586-021-03205-y
Kizer JR, Arnold AM, Benkeser D, et al. Total and high-molecular-weight adiponectin and risk of incident diabetes in older people. Diabetes Care. 2012;35(2):415–23. https://doi.org/10.2337/dc11-1519.
Agarwal I, Glazer NL, Barasch E, et al. Fibrosis-related biomarkers and incident cardiovascular disease in older adults: the cardiovascular health study. Circ Arrhythm Electrophysiol. 2014;7(4):583–9. https://doi.org/10.1161/CIRCEP.114.001610.
Parikh RH, Seliger SL, Christenson R, Gottdiener JS, Psaty BM, deFilippi CR. Soluble ST2 for Prediction of Heart Failure and Cardiovascular Death in an Elderly, Community-Dwelling Population. J Am Heart Assoc. 2016;5(8).https://doi.org/10.1161/JAHA.115.003188
Mehan MR, Ostroff R, Wilcox SK, et al. Highly multiplexed proteomic platform for biomarker discovery, diagnostics, and therapeutics. Adv Exp Med Biol. 2013;735:283–300. https://doi.org/10.1007/978-1-4614-4118-2_20.
Gold L, Ayers D, Bertino J, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE. 2010;5(12):e15004. https://doi.org/10.1371/journal.pone.0015004.
Ngo D, Sinha S, Shen D, et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation. 2016;134(4):270–85. https://doi.org/10.1161/CIRCULATIONAHA.116.021803.
Fitzgibbons TP, Edwards YJK, Shaw P, et al. Activation of Inflammatory and Pro-Thrombotic Pathways in Acute Stress Cardiomyopathy. Front Cardiovasc Med. 2017;4:49. https://doi.org/10.3389/fcvm.2017.00049.
Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J Proteome Res. 2004;3(2):235–44. https://doi.org/10.1021/pr034086h.
Kuhn E, Whiteaker JR, Mani DR, et al. 2012 Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics. 2012;11(6):M111 013854.https://doi.org/10.1074/mcp.M111.013854
Kuhn E, Carr SA. Multiplexed Immunoaffinity Enrichment of Peptides with Anti-peptide Antibodies and Quantification by Stable Isotope Dilution Multiple Reaction Monitoring Mass Spectrometry. Methods Mol Biol. 2016;1410:135–67. https://doi.org/10.1007/978-1-4939-3524-6_9.
Jiang L, Zheng Z, Qi T, et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat Genet. 2019;51(12):1749–55. https://doi.org/10.1038/s41588-019-0530-8.
Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–9. https://doi.org/10.1038/s41586-018-0175-2.
Chadeau-Hyam M, Campanella G, Jombart T, et al. Deciphering the complex: methodological overview of statistical models to derive OMICS-based biomarkers. Environ Mol Mutagen. 2013;54(7):542–57. https://doi.org/10.1002/em.21797.
Benjamini YHY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Richard A, Johnson DWW. Applied Multivariate Statistical Analysis. Englewood Cliffs, NJ: Prentice Hall; 1992.
Shojaie A, Michailidis G. Analysis of gene sets based on the underlying regulatory network. J Comput Biol. 2009;16(3):407–26. https://doi.org/10.1089/cmb.2008.0081.
Shojaie A, Michailidis G. Network enrichment analysis in complex experiments. Stat Appl Genet Mol Biol. 2010;9:Article22.https://doi.org/10.2202/1544-6115.1483
Shojaie AMG. Penalized Principal Component Regression on Graphs for Analysis of Subnetworks. Adv Neural Inf Process Syst. 2010;23:2155–63.
R T. Regression shrinkage and selection via the lasso. J Roy Stat Soc Series B (Methodological). 1996;58:267–88.
Zou H, Zhang HH. On the Adaptive Elastic-Net with a Diverging Number of Parameters. Ann Stat. 2009;37(4):1733–51. https://doi.org/10.1214/08-AOS625.
Janes H, Pepe MS, Gu W. Assessing the value of risk predictions by using risk stratification tables. Ann Intern Med. 2008;149(10):751–60. https://doi.org/10.7326/0003-4819-149-10-200811180-00009.
Kerr KF, Wang Z, Janes H, McClelland RL, Psaty BM, Pepe MS. Net reclassification indices for evaluating risk prediction instruments: a critical review. Epidemiology. 2014;25(1):114–21. https://doi.org/10.1097/EDE.0000000000000018.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey SG. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63. https://doi.org/10.1002/sim.3034.
Burgess S, Thompson SG. Improving bias and coverage in instrumental variable analysis with weak instruments for continuous and binary outcomes. Stat Med. 2012;31(15):1582–600. https://doi.org/10.1002/sim.4498.
Benson MD, Yang Q, Ngo D, et al. Genetic Architecture of the Cardiovascular Risk Proteome. Circulation. 2018;137(11):1158–72. https://doi.org/10.1161/CIRCULATIONAHA.117.029536.
Ngo D, Wen D, Gao Y, et al. Circulating testican-2 is a podocyte-derived marker of kidney health. Proc Natl Acad Sci U S A. 2020;117(40):25026–35. https://doi.org/10.1073/pnas.2009606117.
Davis JR, Fresard L, Knowles DA, et al. An Efficient Multiple-Testing Adjustment for eQTL Studies that Accounts for Linkage Disequilibrium between Variants. Am J Hum Genet. 2016;98(1):216–24. https://doi.org/10.1016/j.ajhg.2015.11.021.
van der Maaten L, Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–605.
Loffredo FS, Steinhauser ML, Jay SM, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153(4):828–39. https://doi.org/10.1016/j.cell.2013.04.015.
Schafer MJ, Atkinson EJ, Vanderboom PM, et al. Quantification of GDF11 and Myostatin in Human Aging and Cardiovascular Disease. Cell Metab. 2016;23(6):1207–15. https://doi.org/10.1016/j.cmet.2016.05.023.
Acknowledgements
This research was supported by contracts 75N92021D00006, HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086 and R01HL132320, and grants U01HL080295, U01HL130114, and HL144483 from the National Heart, Lung, and Blood Institute (NHLBI), with additional contribution from RF1AG063507 from the national Institute of Aging, and from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS‐NHLBI.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH). ND and GDS work in a Unit supported by the Medical Research Council (MC_UU_00011/1) and the University of Bristol. JZ is supported by the Academy of Medical Sciences (AMS) Springboard Award, the Wellcome Trust, the Government Department of Business, Energy and Industrial Strategy (BEIS), the British Heart Foundation and Diabetes UK (SBF006\1117).
Author information
Authors and Affiliations
Contributions
BP, RT and RG conceived of, designed, and obtained funding for the study. TA, CM, and BP wrote the draft manuscript, which all authors reviewed and revised. TA, CM, JAB, JCB, CS, TB, NB, PB, AE, BM, DN, AS, NS, KW, and JZ contributed to statistical design and analysis. SC, CD, ME, HF, JF, RG, SH, DK, JK, RL, WL, HM, KM, AN, MO, RV, GDS, ND, DS, NS, RT, and BP contributed to design, implementation, and ongoing study analyses and projects. All Authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
Psaty serves on the Steering Committee for the Yale Open Data Access Project funded by Johnson & Johnson. Elkind receives study drug in kind from the BMS-Pfizer Alliance for Eliquis® and funding from Roche for a NIH-funded stroke prevention trial; royalties from UpToDate for chapters on stroke; and serves as an unpaid Officer of the American Heart Association. Floyd has consulted for Shionogi Inc. Kizer has stock ownership in Abbott, Bristol Myers Squibb, Johnson & Johnson, Medtronic, Merck and Pfizer. Odden is a consultant for Cricket Health, Inc.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Austin, T.R., McHugh, C.P., Brody, J.A. et al. Proteomics and Population Biology in the Cardiovascular Health Study (CHS): design of a study with mentored access and active data sharing. Eur J Epidemiol 37, 755–765 (2022). https://doi.org/10.1007/s10654-022-00888-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10654-022-00888-z