
Abstract
Query Expansion is commonly used in Information Retrieval to overcome vocabulary mismatch issues, such as synonymy between the original query terms and a relevant document. In general, query expansion experiments exhibit mixed results. Overall TREC Genomics Track results are also mixed; however, results from the top performing systems provide strong evidence supporting the need for expansion. In this paper, we examine the conditions necessary for optimal query expansion performance with respect to two system design issues: IR framework and knowledge source used for expansion. We present a query expansion framework that improves Okapi baseline passage MAP performance by 185%. Using this framework, we compare and contrast the effectiveness of a variety of biomedical knowledge sources used by TREC 2006 Genomics Track participants for expansion. Based on the outcome of these experiments, we discuss the success factors required for effective query expansion with respect to various sources of term expansion, such as corpus-based cooccurrence statistics, pseudo-relevance feedback methods, and domain-specific and domain-independent ontologies and databases. Our results show that choice of document ranking algorithm is the most important factor affecting retrieval performance on this dataset. In addition, when an appropriate ranking algorithm is used, we find that query expansion with domain-specific knowledge sources provides an equally substantive gain in performance over a baseline system.
Similar content being viewed by others


1 Introduction
The Genomic era is upon us; however, popular commercial literature search engines used by biomedical researchers, such as Entrez PubMed Footnote 1 and Google Scholar, Footnote 2 provide no explicit support for genomic-focussed queries. These information needs are, broadly speaking, requests for published articles describing the specifics of how genes contribute to disease in organisms. Given the prevalence of gene and general biomedical term synonymy in the literature (Ananiadou and Nenadic 2006), it is clear that information retrieval (IR) applications have much to gain from query expansion techniques which automatically augment user queries with related terms. For example, using terminology resources, a biomedical concept such as “colorectal cancer” can be expanded with its abbreviated form “CRC”, and a gene such as “MLH1” to its other aliases “COCA2, FCC2, HNPCC, HNPCC2, MGC5172, hMLH1”. By adding these terms to the query, we explicitly address what is referred to as the vocabulary mismatch problem (Furnas et al. 1987).
In this paper, we perform a number of experiments using data from the TREC 2006 Genomics Track task. Footnote 3 This is a question answering-style task that requires the retrieval of exact answer passages in response to natural language questions. Examining TREC 2006 participant workshop papers we find a mixed bag of results for query expansion: some groups report increases in performance, for example (Si et al. 2006; Zhou et al. 2007), while others describe performance drops (Smucker 2006; Dorff et al. 2006). Despite the fact that experiments are conducted on the same set of queries and documents, there are many implementation differences between these participating systems that prevent us from establishing concrete conclusions from these experiments.
Hence, the principal contribution of this paper is the definition of a list of system design criteria that are necessary for effective query expansion in the genomic domain. We achieve this by running a set of controlled experiments that isolate specific factors affecting system performance. These factors focus on two aspects of system design: the IR ranking metric and the variety of knowledge sources available for query expansion. These query term expansion sources include corpus-based cooccurrence statistics, pseudo-relevance feedback methods, and domain-specific and -independent ontologies and databases. Our results clarify that the choice of passage ranking algorithm is the most important factor affecting retrieval performance. Another contribution of this paper is our novel ranking metric which, our results show, maximises the impact of query expansion in the genomic domain. In particular, we find that query expansion with synonyms from domain-specific terminology resources provide the most substantive gains in performance over a baseline system on our dataset.
The remainder of the paper is structured as follows. In Sect. 2, we provide an overview of related work in biomedical IR, and discuss the additional tasks explored by the TREC Genomics Track. Section 3 examines the popularity of various biomedical terminology resources used for query expansion by TREC participants. In Sect. 4, we present our novel IR framework, which includes a modified version of the Okapi ranking algorithm (Robertson et al. 1994) specifically designed to optimise results for expansion of queries containing multiple concepts. Section 5 briefly contrasts our IR framework with system descriptions published at the official TREC 2006 Genomics Track workshop. Sections 6 and 7 describe our experimental methodology and results. This discussion is concluded, in Sect. 8, with a proposed list of criteria necessary for successful query expansion for this TREC task.
2 Background
The recent deployment of high throughput analysis methods in biomedical research, such as gene expression microarrays, has facilitated a rapid increase in the rate at which experimental results are obtained and subsequently published. In the area of genomics, user queries tend to focus on genes and their corresponding proteins. More specifically, geneticists are interested in the role of genes and proteins in biological processes in the body through their interactions with other genes and their products. The “big picture” aim is the generation of hypotheses from the literature that can then be used to drive new experimental research in the area of drug discovery for diseases. The TREC Genomics Track’s motivation was to support these information searching endeavours.
Since the Genomics Track’s commencement in 2003, two important IR document collections with corresponding queries and relevance judgements (gathered from real expert information requests) have been produced: a large subset of abstracts taken from the MEDLINE bibliographic database, Footnote 4 and a collection of full-text open source documents in HTML gleaned from the HighWire website. Footnote 5 While the main focus of the TREC Genomics Track is ad hoc retrieval, text categorisation to support database curators was also investigated at the 2003–2005 forums. Database curation is the manual extraction and addition of important relevant information from the literature to a database format. This is very desirable from the point of view of clinicians and biomedical researchers, as it can significantly speed up their analysis of genomic data. However, performed manually, the process is very costly and time-consuming. The categorisation task at the Genomics Track evaluated how effective an automatic triage task would be at deciding whether a document requires additional expert review and inclusion in the database, thus saving the curators valuable time. The document subset investigated was taken from the Mouse Genome Informatics (MGI) project, Footnote 6 and the task in 2005 required systems to classify documents into one of four categories: tumour biology, embryologic gene expression, alleles of mutation and phenotypes, and Gene Ontology annotation. See the track overview papers for more on text categorisation (Hersh et al. 2004, 2005).
Like the categorisation task, full documents were not the focus of the ad hoc retrieval task until 2006. Furthermore, the ad hoc retrieval task has shifted to Question Answering (QA)-style retrieval of relevant answer passages in response to natural language queries. In 2007, entity-based QA was investigated, which requires not only a relevant answer, but also references to a certain entity type, e.g., “What mutations are responsible for Retina Blastoma?”, where the entity type is “mutation”. Obviously, an effective named-entity tagger for all 14 entities investigated by TREC is required; however, research in this area has been limited to only a few types such as mutations, and gene and protein names (Park and Kim 2006).
The experimental results discussed in this paper focus on the retrieval of relevant answer passaged from full-text articles. We do not report results for the 2007 task since entity recognition errors would make it impossible to ascertain the “true” level of effectiveness of both our query expansion framework and the knowledge resources investigated here.
Excluding the TREC datasets, the only other large scale collection for ad hoc medical IR is OHSUMED, Footnote 7 which consists of a subset of clinical MEDLINE abstracts spanning the years 1987–1991, 106 topics from clinicians, and an accompanying set of relevance judgements (Hersh et al. 1994). Footnote 8 Obviously, these queries differ from their TREC counterparts as the information requests are centred around patient conditions and treatments rather than inquiries of a genomic nature. This collection has been a testbed for many pre-TREC medical IR experiments. For example, there are a number of papers describing query expansion on this dataset. Hersh et al. (2000) explored the usefulness of expanding OHSUMED queries with related terms found in the UMLS Metathesaurus (UMLS is the Unified Medical Language System Footnote 9). Their experiments showed an overall decline in performance when expansion terms were added to original queries. However, some small portion of queries did respond well to both synonym and hierarchical expansion. A manual expansion experiment (where the user interactively adds terms to the query) also showed no significant improvement over baseline performance. Hersh et al. (2000) notes that potential improvements may have been dampened by the inclusion of MeSH indexing terms which are manually assigned when an abstract is added to MEDLINE (removing these MeSH terms reduces performance by about 10%). Related research by Aronson and Rindflesch (1997) and Srinivasan (1996) in contrast showed that both query expansion terms from automatically derived thesauri and additional terms from pseudo-relevance feedback improved baseline performance. More recently, Ruch et al. (2006) report improvements on the same collection when a pseudo-relevance feedback approach using the Rocchio algorithm (Rocchio 1971) is enhanced by selectively choosing sentences in the initial ranked list that discuss one of the following points of information, as classified by a machine learning approach: Purpose, Methods, Results or Conclusions. They show that by considering only these aspects of document argumentative structure (in particular Purpose and Conclusion tag sentences) in the feedback process, they can improve baseline performance by about 40%.
Mixed reports of query expansion effectiveness are also a characteristic of the TREC Genomics Track. The aim of this paper, as already stated, is to investigate and show that certain sources of query expansion terms do significantly improve baseline performance, with the caveat that certain IR system design issues are met. In the next section, we describe in detail some of the most popular biomedical knowledge sources used at TREC. Section 5 continues our discussion of related work, but provides specific details on TREC participant approaches.
3 Knowledge sources for genomic query expansion
In this section, we review different sources of biomedical terminology used by TREC genomic participants. These knowledges sources can be classified into two different types: hand-crafted ontological resources, and automatically generated knowledge sources derived from biomedical corpora. Since the focus of this paper is query expansion, one of our aims is to determine which word association type provides the most benefit when expanding genomic focussed queries. These word association types can be categorised into the following expansion term types:
-
Lexical variants of query terms, including plural/singular, morphological, orthographic and spelling variations. In this work we examine the impact of an automatic variation generation tool, first suggested by Buttcher et al. (2004), which segments terms at possible break points such as hyphens, and normalises common characters, such as alpha to a. A detailed explanation of this tool is provided later in this section.
-
Synonyms are lexically distinct, but semantically equivalent terms; synonyms are considered, with lexical variants, to be the most effective type of query expansion term. Three ontological resources containing biomedical synonyms are explored in this paper: UMLS MetaThesaurus, MeSH and SNOMED-CT. Given the genomic focus of the TREC queries, the following gene and protein synonym sources are also investigated: HUGO (Eyre et al. 2006), UniProt (Bairoch et al. 2005), Entrez Gene (Maglott et al. 2005), and OMIM (McKusick 1998). A useful source of synonymy can also be found in abbreviation databases such as ADAM (Zhou et al. 2006a). A more detailed description of these resources is provided later in this section.
-
Ontological relationships include specialisation/generalisation associations between query term and expanded term—e.g., “liver” has part “bile ducts”. These relationships are found in knowledge resources such as the ontological resources listed above.
-
Cooccurrence relationships are automatically generated from a corpus of biomedical documents (in our case MEDLINE). These related terms represent word associations that cannot be described by any of the previous relationship types, but which are often considered intuitive due to their high frequency of occurrence with query terms in the same context; e.g., “heart disease” and “statins”, where the latter is a drug used to control the former. Two sources of cooccurrence are explored in this paper: pseudo-relevance feedback; and statistically frequent n-grams extracted from the MEDLINE collection of biomedical abstracts. We discuss these expansion sources in more detail in Sect. 4.
Of the 20 groups that participated at TREC 2006, 16 used a terminological or ontological resource for query expansion. Table 1 compares the popularity of the different biomedical resources, where MeSH was the most used ontological resource, and Entrez Gene was the most popular source of gene synonyms. The four entries in the “Other” category refer to the HUGO gene database (\({\tt Cornell\,U.\,(Twease)}\)), UniProt protein database (\({\tt Berkeley}\)), the T2K group’s gene synonym expansion tool (\({\tt Amsterdam}\)),Footnote 10 and suggested PubMed term expansions (\({\tt Fudan\,U.}\)). Most of the groups that explored abbreviation expansion use resources described in Schwartz and Hearst (2003) for mining acronyms from the TREC Genomics collection, and then storing their frequencies and corresponding longforms in a database. Ready-made abbreviation resources were also used such as AcroMed (Pustejovsky et al. 2001) and ADAM (Zhou et al. 2006a). We use the ADAM abbreviation database in our experiments. An overview of this and the other terminology resources we investigate in the paper are provided in Table 2.
In Table 2, vocabulary resources are either classified as GENE or GENERAL (more specifically, general biomedical terms). Classifying terminologies in this way is a bit misleading as many of the general resources, such as MeSH, also have gene and protein entries; however, their coverage is poor compared to a gene specialised resource such as Entrez Gene. In particular, MeSH entries cover only well-known genes such as “tp53” and “BRCA1”. Classifying resources in this way helps to clarify how they contribute to our query expansion process described in Sect. 4, since all TREC Genomics queries match one of the following predefined query templatesFootnote 11:
-
What is the role of a gene [GENE] in a disease[GENERAL]?
-
What effect does a gene [GENE] have on a biological process [GENERAL]?
-
How do genes [GENE] interact in organ [GENERAL] function?
-
How does a mutation in a gene [GENE] influence a biological process [GENERAL]?
As we can see, all italicised “concept” query terms are followed by their type, which indicates which subset of knowledge sources we will use to expand them. The aim of the experiments described in Sect. 7 is to determine which of these resources provides the most effective query expansion terms for the genomic domain.
So far we have discussed terminology expansion without commenting on the issue of sense disambiguation, that is, the automatic determination of the sense of a particular word in a particular concept. For example, given the ambiguous phrase “Big Apple”, if the context indicates that this is the fruit sense then “cooking apple” is an appropriate term; however, if we interpret it as its “city” sense then the synonym “New York City” should be added to the query.
Experiments by Sanderson (2000) have shown that unless automatic sense disambiguation methods achieve over 90% accuracy (around 30% more than current state-of-the-art systems are capable of), no increase in IR performance will be observed. Voorhees and Buckland (1994) shows that even when the senses of the query terms are correctly interpreted, query expansion using WordNetFootnote 12 only achieves performance gains when a subset of highly relevant expansion terms are manually chosen from the knowledge source. These results are indicative of domain independent query expansion experiments using ontological resources. Hence, the IR community has focussed more on corpus-based expansion methods such as pseudo relevance feedback (Ruthven and Lalmas 2003). The strength of these methods is accredited to the query collocation effect (Krovetz and Croft 1992). More specifically, in pseudo relevance feedback, query terms mutually disambiguate each other, which means that the top ranked documents, from which the query expansion terms are derived, tend to contain more domain specific relevant terms. For example, given the query “Big Apple City Tours”, we can expect that the most highly ranked documents for this topic are more likely to describe excursions in New York city than recipes for an apple pie.
This discussion shows that in domain independent query expansion, the topic of disambiguation is an important consideration. However, the experiments described in this paper are performed on a collection of domain specific documents and queries. Hence, like many other TREC Genomics participants we assume that multiple senses of a term are rare, and that no explicit sense disambiguation method is required. Our results show this to be the case, in all except one instance: the addition of abbreviated terms to the query. This result, and a proposed solution to the problem are discussed in more detail in Sect. 7.
4 A genomic query expansion framework
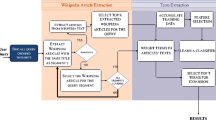
In this section, we describe the different components of our Genomic IR architecture (Fig. 1). Our IR system is a version of the Zettair engineFootnote 13 which we have specifically modified for passage retrieval and biomedical query term expansion. This system will form the basis for the experiments reported on below, wherein we vary the resources and techniques used for query expansion for genomic IR tasks.

A genomic query expansion framework
4.1 Document preprocessing
The TREC 2006 Genomics document collection consists of 162,259 full-text journal articles obtained by crawling the Highwire site. When uncompressed, the full collection is about 12.3 GB in size. After preprocessing, the whole collection becomes 7.9 GB. The collection is pre-processed as follows:
-
Paragraph Segmentation: for evaluation purposes the Genomics Track requests that the ranked answer passages must be within specified paragraph boundaries.
-
Sentence Segmentation: all sentences within paragraphs are segmented using an open source tool.Footnote 14
-
Character Replacement: Greek characters represented by gifs are replaced by textual encodings; accented characters such as “À” or “Á” are replaced by “A”; Roman numbers are replaced by Arabic numerals. These replacements are very important for capturing variations in gene names. In addition, hyphens in terms such as “Creutzfeldt-Jakob” are replaced by spaces (similar changes must be made to query terms).
-
Text Removal: all HTML tags, very short sentences, paragraphs with the heading Abbreviations, Figures, Tables, and some special characters such as hyphens, slashes and asterisks, are removed.
-
Abbreviation Resolution: all abbreviations and their corresponding long forms within the same article are detected using ADAM as a filter, and the long forms are added after the abbreviations in the original text. For example, “HIV” is replaced by “HIV (Human Immunodeficiency Virus)”.
4.2 Query parsing and expansion
Once pre-processing on the collection has been completed, and an index consisting of paragraphs (rather than full-text documents) has been created, querying can begin. As already explained, TREC simplifies the query preprocessing task by ensuring that all topics conform to the query templates discussed in Sect. 3. The following is a sample query, Topic 160 from the 2006 track, which contains two concepts: “PrnP” (a gene) and “mad cow disease” (a general biomedical term):
What is the role of PrnP in “mad cow disease”?
To use our concept-based search engine model, we first need to parse the query and identify phrases corresponding to concepts. For this task we leverage PubMed; we submit the natural language query to PubMed and use information presented in the details tab, to obtain a parsed version of the query. We also use information from the query templates to automatically differentiate between gene concept and general biomedical term concepts.Footnote 15 For example, in the above topic the two identified concepts (the first of type [GENE], and second of type [GENERAL]) are highlighted in bold font. After parsing, the external resources are used to expand these concept terms to their synonyms, ontologically related terms (generalisations and specialisations), and abbreviations. The following example shows the expanded terms derived from Entrez Gene and MeSH:
What is the role of PrnP, {“prion protein”, “p27-30”, “Creutzfeldt-Jakob disease”, “Gerstmann-Strausler-Scheinker syndrome”, “fatal familial insomnia”, “ASCR”, “CD230”, “CJD”, “GSS”, “MGC26679”, “PRIP”, “PrP”, “PrP27-30”, “PrP33-35C”, “PrPc”, “CD230 antigen”, “major prion protein”, “prion protein”, “prion protein PrP”, “prion-related protein”} in “mad cow disease” {“Encephalopathy, Bovine Spongiform”, “Bovine Spongiform Encephalopathy”, “BSE”, “BSEs”, “Encephalitis, Bovine Spongiform”, “Bovine Spongiform Encephalitis”, “Mad Cow Disease”, “Mad Cow Diseases”, “Spongiform Encephalopathy, Bovine”, “Spongiform Encephalopathy", "Spongiform Encephalitis", “Prion Diseases", “Cattle Diseases"}?
As mentioned in Sect. 3, while we don’t apply any explicit sense disambiguation method when deciding which expansion terms to add to the query, we do define rules for cases where our terminology databases returns multiple hits for a given query term. In addition, these resources may provide additional information that would not be of benefit to our task. The rules presented in Table 3 define how we resolve these issues.
As well as expanding with synonyms, hypernyms, hyponyms and abbreviations, we use a “term variant” generation tool to generate all the possible variants for both original query terms and expanded terms. Our segmentation rules are similar to those used by (Buttcher et al. 2004). We describe our rules as follows:
Given a gene name, we define a split point to be: (1) any hyphen or punctuation occurring in the name; (2) any change from lower case to upper case, or (3) any change from a character to a number (or vice versa) or a Greek character (e.g. “alpha”). A word is split according to all its split points, and all variants are generated by concatenating all these split parts, optionally with a space inserted. Greek characters are also mapped to English variants, e.g. alpha is mapped to “a”.
For example, for the query term “Sec61alpha”, we would generate the following lexical variants which are also commonly used forms of this term in the collection: “Sec 61alpha”, “Sec61 alpha”, “Sec 61 alpha”, “Sec 61a”, “Sec61 a”, “Sec 61 a”, “Sec61a”.
In query phrases, we replace any hyphens (“-”), slashes (“/”) and asterisks (“*”) with a space. For example, “subunit 1 BRCA1 BRCA2 containing complex” would be generated as a variant of “subunit 1 BRCA1/BRCA2-containing complex”.
4.3 Concept-based query normalisation
Our paragraph ranking method is based on the Okapi model (Robertson et al. 1994). Many participant systems at the TREC Genomics Track uses the Okapi method for ranking paragraphs with respect to their similarity to the query. However, there are two fundamental problems with using this model on TREC Genomics queries.
The first problem regards Okapi not differentiating between concept terms and general query terms in the query. For example, consider two paragraph, one containing the terms “mad cow disease” and “PrnP”, and the other containing the terms “role” and “PrnP”. Clearly the first paragraph containing the two biological concepts is more relevant; however, Okapi does not make the required distinction. The second problem occurs because TREC 2006 topics contain more than one concept term. It is possible that a short paragraph that discusses only one concept will be ranked higher than a longer paragraph which mentions two concepts. Again this is an undesirable outcome.
To overcome these problems, a Conceptual IR model was proposed in Zhou et al. (2006b). In this paper, we use another method called concept-based query normalisation, first introduced in Stokes et al. (2007a), which is a modified version of the geographic query normalisation model presented in Li et al. (2006).
The first problem mentioned above is solved by dividing query terms into two types: concept terms t c (e.g., “mad cow disease”), and non-concept terms t n (e.g., “role”).Footnote 16 More specifically, we define non-concept terms as query terms that do not have an entry in one of our genomic expansion resources; while concept terms, on the other hand, do. Consequently, non-concept query terms are not expanded. So given a query with both concept and non-concept terms, the similarity between a query Q and a paragraph P p is measured as follows:
where nsim(Q, P p ) is the non-concept query similarity score and csim(Q, P p ) is the concept similarity score. The non-concept similarity score is defined by:
where Q n is the aggregation of all non-concept terms in the query, and
where k 1 and b are usually set to 1.2 and 0.75 respectively, and k 3 can be taken to be ∞. Variable W p is the length of the paragraph p in bytes; avgW p is the average paragraph length in the entire collection; N is the total number of paragraphs in the collection; f t is the number of paragraphs in which term t occurs; and f {p,q},t is the frequency of term t in either a paragraph p or query q. The concept similarity score is given by:
where Q c is the aggregation of all concepts in the query, C is one concept in Q c , and t ci is a query or expanded term belonging to concept C; the t ci are listed in descending order according to their Okapi similarity scores \(sim_{t_{c1}}, \ldots , sim_{t_{cN}}.\) Without this geometric progression normalisation, a document with more synonyms belonging to the same concept will be favoured against another document which has fewer synonyms. The similarity score sim t of a concept term t is calculated as:
where r p,t and r q,t are defined in nsim(Q, P p ), and w′ t is defined as:
Note that (1) is an adjustment of the calculation for the weight w ′ t of an expanded term t appearing in the query: for expanded term t, its own term frequency f t and the corresponding original query term’s frequency f t_q are compared, and the larger value used—this ensures the term contributes an appropriately normalised “concept weight”.
To solve the second problem, we use the following rules to ensure that for two paragraphs \({\tt P\_{1}}\) and \({\tt P\_{2}}\) , where one contains more unique query concepts than the other, the number of concepts \({\tt ConceptNum(P\_{i})}\) will override the Okapi score \({\tt Score(P\_{i})}\) and assign a higher rank to the paragraph with more unique query concepts:
4.4 Relevant n-gram feedback
So far we have described how hand annotated external resources can be used for expansion. In this work, we also investigate an N-gram feedback method. The following is an overview of how this pseudo relevance feedback step contributes to the retrieval process:
-
1.
Retrieve the first 1,000 paragraphs which include at least one instance of each concept in the query.
-
2.
From this subset of paragraphs, find all unigrams, bigrams and trigrams by using a (in-house) tokenisation tool. All stop words are excluded.
-
3.
Among these n-grams, calculate their \(TF \times IDF\) scores, and find the top 20 with the highest scores. Add these into the query as additional expansion terms, and re-run the passage retrieval step.
4.5 Passage extraction and re-ranking
As already mentioned the 2006 Genomics Track defined a new question answering-type task that requires short full-sentence answers to be retrieved in response to a particular query. However, before answer passages can be generated, we first retrieve the top 1,000 ranked paragraphs for each topic, and use the following simple rules to reduce these paragraphs to answer spans.
The passage extraction method can be best described by an example. Consider a paragraph consisting of a sequence of sentences 〈(s 1, i), (s 2, i), (s 3, r), (s 4, r), (s 5, i), (s 6, r), (s 7, i), (s 8, i), (s 9, r), (s 10, i) 〉, where r is a relevant sentence (that is, mentions at least one query term) and i is an irrelevant one. Our passage extraction method removes all the irrelevant sentences from both ends of the paragraph and splits a paragraph if there are two or more consecutive irrelevant sentences within this span. Hence, it would produce the following two passages from this paragraph: 〈(s 3, r), (s 4, r), (s 5, i), (s 6, r)〉 and 〈(s 9, r) 〉.
After the passage extraction technique has been applied for a particular topic, we re-rank passages by re-indexing them, and re-querying the topic against this new index, using global statistics obtained from the original indexed collection, i.e. using term frequency f t and the average paragraph length avgW p .
4.6 Document context combination
In earlier experiments, we found that even when passage extraction and re-ranking resulted in a significant increase in Passage level MAP, we would still see a significant drop in the Document and Aspect level MAP for the same run. Zhou et al. (2006b) also witnessed this negative impact on Document and Aspect level MAPs when applying their passage reduction technique. To overcome this problem, we employed the following linear document context combination method:
-
1.
Use passage extraction and re-ranking to find the top 1,000 passages for each topic.
-
2.
Divide these 1,000 passages into different concept level groups according to the number of concepts they include. This is straightforward since our concept-based retrieval model has already ranked all the paragraphs according to (firstly) their concept numbers, and then similarity scores.
-
3.
Within each group, re-rank the passages by combining their similarity scores with their containing documents’ similarity scores.
The following rules tell us how to combine the two scores: for a passage i which has a similarity score P i and whose containing document has a similarity score D i , the final combination score S i is calculated as:
where P max and D max are the maximum similarity scores of all the 1,000 passages and their containing documents, and α controls the weight of importance of the document similarity score, which was set to 0.5 in our experiments.
5 Query expansion frameworks at the TREC Genomics Track
In Sect. 2, we briefly discussed related work on query expansion for medical IR. Early work in this area tended to focus on the information needs of clinicians (that is, medical professionals interacting directly with patients). Despite the differences in query view point between TREC and this work, we still find that many of the same query expansion techniques and resources have been used by Genomics Track participants.
In this section, our aim is to briefly summarise the most popular approaches to IR system design employed at TREC 2006 Genomics, and contrast these with our framework presented in the previous section. While the Genomics Track system papers also describe a broad spectrum of solutions to collection preprocessing and passage reduction, we will limit our discussion here to IR frameworks. The pre- and post-processing methods described in the previous section can be viewed as a good merge of the best solutions for these tasks.
One of the key aspects of our system is the implementation of a concept-based passage ranking algorithm. A similar idea was explored by the University of Chicago, Illinois (Zhou et al. 2007) who also recognised the significance of re-ranking documents with respect to the number of unique concepts they mention. However, we use a term weighting normalisation technique to ensure that the weights of expansion terms do not “drown out” the contribution of original query terms in the ranking metric; while they explicitly modify the weight of expansion term types based on their strength of association with the query term. For example, the weight associated with a hypernym (specialisation) expansion term is reduced by 5%. Zhou et al.’s post-submission results are the highest reported effectiveness scores on the 2006 data. These results are compared with our best performing expansion run in Sect. 8. Other modified Okapi approaches described in TREC system reports do not come near the performance gains achieved by Zhou et al. and those presented later in our paper. These included submissions by Abdou and Savoy (2006) and Huang et al. (2006).
The Indri retrieval engineFootnote 17 (part of the Lemur project) was also a popular choice of IR framework at 2006 Genomics workshop. Advantages of this framework include the integration of language modelling and inference networks. Indri also supports structured querying (using an extended set of operators) which facilitates more sophisticated ranking and filtering of candidate documents during the retrieval process.
An example of the flexibility of the Indri operators can be found in the University of Wisconsin system report (Goldberg et al. 2006) which defines their ranking strategy using the Indri operators as follows: first retrieve all documents using the \( {\tt \#band}\) constraint that contains at least one reference to all query concepts represented by either an original term or any synonyms as defined by the \( {\tt \#syn}\) operand. This subset of the collection is then ranked using the \( {\tt \#combine}\) operand, which considers the frequency of occurrence of any of the original and expanded terms in its ranking score.
It is not surprising that this ranking strategy performed poorly, given that the first filtering step often results in a ranked list length that is considerable shorter than the required 1,000 passages for the TREC evaluation metrics, which can lead to lower effectiveness scores.
CMU (Si et al. 2006) make more effective use of the Indri’s structured querying language as follows: each original term and its expanded terms are combined using the weighted synonym operator \( {\tt \#wsyn}\), where different weights are assigned to expanded terms depending on their word type (aliases, synonyms, acronyms, function word) and expansion source. These different \( {\tt \#wsyn}\) expression are then combined into a single score for a document using the \( {\tt \#weight}\) belief operator, which ensures that highly reliable operators (in this case acronyms from AcroMed) will contribute more to the final document rank.
CMU also use Indri’s language modelling IR capabilities. Indri’s default mode combines language modelling and Dirichlet smoothing. Variations on the Language Modelling (LM) approach to IR were explored by many research groups at the Genomics Track. The LM approach to IR is based on the observation that we can estimate the relevance of a document with respect to a query by computing the probability of generating each query term from the document model. The product of these probabilities is then used to rank the document. Smoothing methods are used to tackle the problem of zero query term probabilities in this calculation. Smoothing addresses the data sparseness problem by assigning small amounts of probability mass to all unseen query terms in the document language model. A more detailed explanation of the language modelling paradigm for IR can be found in Croft and Lafferty (2003).
Data sparseness is an even greater problem when the language model is generated for a passage rather than a full-text document. CMU (Si et al. 2006) use a modified version of the Dirichlet smoothing method which incorporates query term probability evidence from the passage, document and collection level for calculating passage relevance. The University of Guelph (Song et al. 2006) use two different smoothing techniques, Good-Turning estimates and curve-fitting functions, and a combination of various language models similar to the CMU approach: passage, document, journal and collection models. They state that borrowing information from outside the passage model helps to further differentiate the contribution of unseen terms.
The University of Illinois at Urbana-Champaign (Jiang et al. 2006) investigate the performance of a Kullback-Leibler (KL) divergence retrieval framework, another language modelling approach, and use vocabulary retrieved from pseudo-relevance feedback to improve the estimation of the query model. UMass (Smucker 2006) use a similar approach called relevance modelling (Lavrenko and Croft 2001) to incorporate pseudo-relevance feedback information into the language modelling framework. They also investigated query-biased pseudo-relevance feedback, which generates a document model from the terms surrounding the query term occurrences in a document.
Of these four LM approaches, CMU performs best followed by Urbana-Champaign, Guelph, and UMass. One reason for CMU’s strong performance may be its use of terminology resources during query expansion, while UMass and Urbana-Champaign use relevance feedback. The Guelph result is impressive given that no explicit query expansion is used. In Sect. 7, we compare our baseline (no-expansion) concept normalisation framework with the Guelph LM results, as it is one of the few reported examples where a baseline system beats the median Genomics Track values for effectiveness.
All of the systems discussed so far are classed as automatic runs, i.e. no human intervention contributed to final ranked list of results. However, a number of groups submitted interactive runs. The most effective example of an interactive run was reported in Demner-Fushman et al. (2006). This group collected manually expanded queries from a computational biologist and a medical librarian for one of their official runs. This interactive run is one of the top ranked systems at TREC 2006. It achieved the highest passage-based Mean Average Precision score of all official TREC 2006 runs (Hersh et al. 2006). While their effectiveness scores are impressive, our results in Sect. 8 demonstrate that even domain experts can be outperformed by a competent automatic query expansion system.
6 Experimental methodology
As already stated, this work focusses on the TREC 2006 Genomics passage-level retrieval task. The TREC collection for this task consists of 162,259 full-text documents from 49 journals published electronically via the Highwire Press website.Footnote 18 With the collection comes 28 topics expressed as natural language questions, formatted with respect to seven general topic templates (see Sect. 3). A list of these queries and their corresponding topic numbers, which are referenced in Sects. 7 and 8, can be found in the track overview paper (Hersh et al. 2006).
As with other TREC tasks, participating systems must submit the first 1,000 retrieved passages for each topic (Hersh et al. 2006). Passages in this task are defined as text sequences that cannot cross paragraph boundaries (delimited by HTML tags), and are subsets of the original paragraphs in which they occur. As is the custom at TREC, human judges were used to decide the relevance of passages in the pooled participating system results. These judges also defined exact passage boundaries, and assigned topic tags called aspects from a control vocabulary of MeSH terms to each relevant answer retrieved.
System results are evaluated with respect to three distinct versions of the Mean Average Precision (MAP) score calculated at different levels of answer granularity: Document, Passage and Aspect. Traditionally the MAP score is defined as follows: first, the average of all the precision values at each recall point on a topic’s document ranked list is calculated; then, the mean of all the topic average precision scores is determined.
Since the retrieval task at the Genomics Track is a question answering-style task, a metric that is sensitive to the length of the answer retrieved was developed. Passage MAP is similar to document MAP except average precision is calculated as the fraction of characters in the system passage overlapping with the gold standard answer, divided by the total number of characters in every passage retrieved up to that point in the ranked list. Hence, a system is penalised for all additional characters retrieved that are not members of the human evaluated answer passages.
For the TREC 2007 Track, Passage MAP was modified in response to 2006 participant reports that the score could be increased by simply halving passages without any consideration to content (Smucker 2006). For example, consider a topic with one relevant passage, 100 characters long, and two ranked lists. The first list ranked this relevant passage at position 1, but also retrieves 50 extra characters so the AP for the topic is 0.25 (or 100/400). The second list, retrieves 75 correct and 125 irrelevant characters at position 1, and at rank 4 it retrieves the remaining correct 25 characters and 500 characters in total up to this point in the ranked list. This results in an overall MAP score for this topic of 0.29 (or (75/200 + 100/500)/2)). It is clear from this contrived example that the second ranked list will achieve a higher score than the first, which contradicts our intuitive understanding of how a MAP score should work. The improved Passage2 MAP scores, on the other hand, calculates MAP as if each character in each passage were a ranked document. Using this modified version, the first ranked list is guaranteed to obtain a higher MAP score at the passage level.
Despite these enhancements, Passage2 MAP (like passage MAP) is a very harsh metric for evaluating system performance. Given the utility of the task, it is questionable whether users would be considerably hampered if they were presented with additional text surrounding a relevant answer. In fact it has been shown that, in general, users prefer paragraph size chunks to exact answers in question answering applications (Lin et al. 2003). Hence, as an alternative to Passage-level MAP, we define a Paragraph MAP score which calculates the fraction of paragraphs retrieved that contain a correct passage, divided by the total number of paragraphs retrieved. As before, the average of these scores at each recall point is the final score for that topic. We use this metric to evaluate the effectiveness of different system parameters (knowledge sources and expansion framework), but also report a set of Passage2 MAP scores for our final, best run in order to evaluate the effectiveness of our passage reduction method described in Sect. 4.
The final metric defined by the track is used to measure to what extent a particular passage captures all the necessary information required in the answer. Judges were asked to assign at least one MeSH heading to all relevant passages. Aspect average precision is then calculated as the number of aspects (MeSH headings) captured by all the relevant documents up to this recall point in the ranked list for a particular query. Relevant passages that do not contribute any new aspect to the aspects retrieved by higher ranked passages are removed from the ranking. Aspect MAP is defined as the mean of these average topic precision scores. In essence, it captures the completeness of an answer, but doesn’t reward redundant answer removal as repeated aspects are not penalised.
7 Experimental results
This section reports on the findings of our experimental evaluation of three important factors in effective query expansion. Firstly, we discuss baseline performance without query expansion by comparing our modified-Okapi ranking algorithm with other baseline approaches reported at TREC. We then investigate the effectiveness of different corpus-based and manual-derived expansion resources that have been used by TREC participants. As many of the ontological resources provide more than one type of semantic relationship, we also look at which relationship types, as defined in Sect. 3, provide the most gain. All of these results are then used in Sect. 8 to derive a list of optimal conditions for successful query expansion in the genomic domain. A paired Wilcoxon signed-rank test at the 0.05 confidence level was used in all our experiments to determine significance. When a significant result is reported in a table we use the following symbol \(\dag .\)
7.1 Comparing baseline IR frameworks
The objective of this experiment is to show that the improvements in effectiveness reported later in this section are achieved on a strong baseline system; that is, a system which uses no external resources for query expansion. Table 4 presents Paragraph, Aspect and Document MAP scores for a baseline Okapi approach (\( {\tt Okapi}\)), our concept-based normalisation baseline approach (\( {\tt Baseline}\)) as discussed in Sect. 5, and a baseline Language Modelling (LM) run with the official run name \( {\tt UofG0}\) (see Sect. 5 for more details). This LM approach was chosen for comparison purposes since it is one of the few non-expansion runs that outperforms the median MAP scores at the 2006 track. This comparison confirms that our baseline system is competitive. This helps to motivate the significance of our results, since reports of incremental increases in effectiveness on weak baselines are not always repeatable on stronger baseline systems. Table 4 shows that the \( {\tt Baseline}\) run is significantly better than the \( {\tt Okapi}\) run across all MAPs. The significance scores reported for the LM run are based on a comparison with the \( {\tt Baseline}\) run rather than \( {\tt Okpai}\). We can see that \( {\tt UofG0}\) is significantly worse than \( {\tt Baseline}\) for Aspect and Paragraph MAP.
The final result presented in Table 4 shows how critical phrase-based querying is in the genomic domain. The \({\tt No\_Phrase}\) run (which treats each constituent word of a phrase as an independent query term) shows a 13.8% drop in performance when compared to the \( {\tt Baseline}\) run. This result is consistent with domain-independent IR experiments reported by Pickens and Croft (2000). Consequently, all subsequent runs reported in this section use phrase-based querying.
7.2 Gene and biomedical expansion with lexical variation
In Sect. 3, we outlined four distinct query expansion term types: lexical variation (e.g., “HIV-1”, “HIV 1”, “HIV1”), synonymy (e.g., “sonic hedgehog gene”, “HHG1”), ontologically related words (e.g., “lupus” is a type of “autoimmune disorder”), and cooccurring terms (e.g., “cancer”, “chemotherapy”). In the following subsection, we will explore the effect of these expansion term types on retrieval. In this subsection, our experiments focus on the strongest relationship type: term equivalence through lexical variation. In Sect. 4, we described a term variation generation tool that splits words at appropriate breakpoints such as a hyphen or change in case. Our intention in this section was also to explore the value of variants in UMLS Specialist Lexicon which lists plural forms and alternative spellings for biomedical terms. Unfortunately, our experiments show that for the 2006 queries no benefit was gained from expansion with the Specialist Lexicon. While alternative spellings are sure to have high impact on appropriate queries (for example, a query containing “estrogen” will benefit from the term “oestrogen”), there were no 2006 query terms that contained an alternative spelling in this lexicon. We note that the addition of morphological variations, in contrast, would have been appropriate for some queries, e.g., the nominalisations of verbs (“mutate” to “mutation”). However, one of the failings of the lexicon is that no explicit links between these variants exist, hence fuzzy term matching is needed to make these inferences. This would be an interesting avenue for future research, but is not pursued any further here.
Table 5 shows the positive impact of the lexical variation generation tool, where Paragraph MAP increases by 16.5%, which is statistically significant. Improvements in Document and Aspect MAP scores are also observed. Later in this section, we report on the impact of the generation tool after query expansion has been applied, that is, after both original query terms and expanded terms are expanded using this tool.
7.3 Gene and biomedical synonym expansion
In this subsection, we consider expanding original queries with synonyms, i.e., terms that can be swapped without change of meaning in a given context. Hence, like lexical variation, query expansion has much to gain from this term relationship type.
The following experiments are divided into two parts: first we compare sources of gene synonyms, followed by the expansion of general biomedical concepts (such as disease names) with another selection of terminology resources. Detailed descriptions of these knowledge bases are provided in Sect. 3.
In Table 6, the rows from \( {\tt E\_Gene}\) (Entrez Gene) to \( {\tt OMIM}\) show MAP performance increases after gene synonyms from these resources have been used. Entrez Gene marginally outperforms the other resources with a nearly statistically significant increase in effectiveness over the baseline for its Paragraph and Aspect MAP scores, which amounts to a 20.9% improvement over baseline Paragraph MAP.
The second half of Table 6, shows the performance gains when general biomedical terms in the query are expanded. We observe that the impact of these synonyms is much lower than for gene synonyms. A manual examination of the gold standard passages shows that there are fewer examples of general biomedical terms from the original query being referred to by their synonyms, where as the opposite is true for gene names. Hence, adding gene synonyms has more potential for impact than for other biomedical term-type expansion.
As explained in Sect. 3, UMLS is a concatenation of ontological and terminology resources. In our experiments we use a subset of these: MeSH, SNOMED Clinical Terms (\( {\tt SNOMEDCT}\)), the NCBI thesaurus, and UMLS’s own homegrown resource the MetaThesaurus (\( {\tt MTH}\)). Considering the standalone resources, \( {\tt SNOMEDCT}\) provides the greatest improvement over the baseline. However, the combined resource run (\( {\tt UMLS}\)) marginally outperforms the baseline at the Paragraph, Aspect and Document level. Like gene synonym expansion this improvement is not statistically significant. From these results we also see that the inclusion of UMLS’s own thesaurus, \( {\tt MTH}\), provides little or no value to the experiment, as its coverage of synonymous terms for the 2006 queries is poor. SNOMED is perhaps the surprise winner in this category of ontologies, as it outperforms its more popular competitor MeSH. SNOMED was only released in 2004 (available as part of UMLS) and so reports on the usefulness of this resource are limited in both the IR and NLP communities. Given the lack of contribution from \( {\tt MTH}\), the \( {\tt UMLS}\) run is basically a combination of \( {\tt MeSH}\) and \( {\tt SNOMEDCT}\). This run performs as well as individual runs for \( {\tt SNOMEDCT}\) and \( {\tt MeSH}\), indicating a high degree of overlap between these two resources.
In Sect. 3, we discussed ambiguity and its observed negative effect on IR performance. As already stated, we perform no explicit disambiguation. Our general rule for biomedical term expansion is to expand with the first ranked concept returned by the knowledge resource, where in the case of genes the first ranked human reference to the gene is the expansion seed. The results of our synonym expansion experiments reported here provide strong evidence that, in a terminology-specific domain such as genomics, ambiguity is negligible. More specifically, negative effects are drowned out by the large boosts in performance that the correct synonymous expansion terms provide.
7.4 Expansion with abbreviation databases
Another special instance of synonymy is abbreviation. In domain-specific documents, particularly in the Sciences, abbreviations are used prolificly as a shorthand version of important concepts that are frequently repeated in a particular publication. As already stated, there are three options for addressing abbreviations: automatically generate a collection of longform-shortform pairs (see Schwartz and Hearst 2003); use a static resource (in our case we use the ADAM database); or resolve all abbreviations in the document collection to their longforms. Table 7 shows that resolving abbreviations (\( {\tt Rsolve}\)) in the document collection is more effective than adding shortforms to the query from a static resource (ADAM).
From a manual analysis of the results it appears that abbreviation expansion is one instance where ambiguity becomes an issue. For example, looking at the gold standard passages for topic 161 (What is the role of IDE in Alzheimer’s disease?), abbreviation “AD” is a commonly used reference to “Alzheimer’s disease”. Hence, the addition of “AD” to the query should boost performance; however, according to the ADAM database, “AD” can refer to 35 unique longform concepts, such as “after discharge”, “autosomal dominant”, “autistic disorder”. In contrast, replacing abbreviations with longforms in the collection (\( {\tt Rsolve}\)) is a low-risk expansion strategy that provides an impressive boost in Paragraph MAP (28.6% over the baseline). This improvement even exceeds the contribution made from gene synonym expansion reported in the previous subsection (20.9%). Later in this section, we discuss the results of an extension to this ADAM experiment, whereby abbreviations for expansion terms are also added to the query. This experiment provides additional evidence that abbreviations expansion can be a dangerous pursuit.
7.5 Gene and biomedical hierarchical term expansion
In this section we evaluate the effect of ontological relationships, specialisations and generalisations, on retrieval performance. The following extracted sentence from a gold standard passage is a typical example of how these ontological relationships can benefit passage ranking:
Huntington’s disease (HD)1 is an autosomal-dominant neurodegenerative disorder caused by a CAG expansion in the huntingtin gene (htt) (1), and is characterized by involuntary movements, personality changes, dementia, and early death.
More specifically, since “Huntington’s disease” is a type of “neurodegenerative disorder” and a form of “dementia”, when these hierarchically related terms are added to the query, they can provide a positive ranking boost for the passage. However, gold standard passages where original query terms (such as a disease) are referred to solely by their more general or specific terms are rarer. Hence, we don’t expect expansion with hierarchical terms to have as significant an effect on paragraph retrieval performance as synonyms do. Results shown in Table 8 confirm this; however, in all instances, except for MeSH parents (that is, MeSH_Gener which consists of generalised terms), expansion with hierarchial terms has a negative effect on Paragraph, Aspect and Document MAP. In some cases this under-performance is statistically significant compared to the baseline. The worse performing resources are the MetaThesaurus (MTH) and the combined resource UMLS.
A close examination of the individual topic performance for MeSH hierarchical expansion shows that there are no topics where specialised terms outperformed baseline performance (\({\tt MeSH_Spec}\) vs. \( {\tt Baseline}\)). On the other hand, there are three topics which show improved Paragraph MAP scores after generalised terms have been added to the query (\({\tt MeSH\_Gener}\)). For example, topic 163 on “colon cancer” benefits from the generalised term “colorectal cancer”. Drops in performance are rare for \({\tt MeSH\_Gener}\); topic 181 is one such example where the generalised form of “mutation”, according to MeSH, is “variation” which is perhaps too broad and ambiguous to be beneficial. While our normalisation technique, described in Sect. 4, ensures that multiple references to the same concepts are not given undue weight by our ranking metric, it might also pay to weight hierarchical terms more conservatively than other expansion terms types. We leave this for future investigation.
7.6 Expansion with related and cooccurring terms
Up to this point, our experiments have focussed on the effectiveness of query expansion using ontological resources. We turn our attention now to the final expansion term type: cooccurring terms. Two sources of cooccurring terms are examined here: those derived from MEDLINE and those acquired from pseudo-relevance feedback. Our feedback method was described in Sect. 4. Our MEDLINE cooccurring terms are uni-, bi- and tri-grams, extracted from all 8 million abstracts. After all n-grams containing a stopword are removed, as well as n-grams with frequency less than two, the following breakdown of n-grams and their corresponding MEDLINE abstract ids are obtained and stored in a database: 127 million uni-gram, 30 million bi-grams, and 5 million tri-grams. Then, for a given general biomedical query term we calculate the top 20 strongest (statistically) associated phrases using a log-likelihood association metric (Dunning 1993), where the window size for a cooccurrence pair is the length of an abstract. These cooccurring terms are then added to the query in the same fashion as ontologically related terms are.
Table 9 shows the results from our expansion experiments using cooccurring terms. While most approaches to relevance feedback use uni-grams from top ranked documents, we also compare this run (\({\tt RelF\_uni}\)) with a bi- and tri-gram run (\({\tt RelF\_bi-tri}\)). For each of these we see a non-statistically significant improvement over the baseline across Paragraph and Document MAPs, where the use of bi and tri-grams result in marginally better performance. In contrast, MEDLINE cooccurring terms, whose addition to the query is similar in concept to query expansion using an automatically derived thesaurus (Srinivasan 1996), consistently underperformed compared to the baseline run. For Paragraph MAP, the drop in performance is 22.4%. One possible explanation for the strength of Relevance Feedback is the query collocation effect, where top ranked documents in many instances will contain two or more query concepts which mutually disambiguate each other. This increases the likelihood that feedback terms will be appropriate query expansion terms. MEDLINE cooccurring terms, on the other hand, are calculated without knowledge of additional query terms, and in many cases cooccurring terms relate to multiple distinct senses of the term.
7.7 Combining query expansion term sources
In this subsection, we combine the most effective sources of expanded terms as determined by the experiments reported in our paper so far. Our criteria for including an expansion source is: all three MAP scores must improve baseline performance. Table 10 compares baseline performance with this optimal combination of query terms from the following positive runs: \({\tt E\_Gene}\), \({\tt UniProt}\), \({\tt OMIM}\), \({\tt SNOMEDCT}\) and \({\tt Gener\_MeSH}\). We can see that the \( {\tt All\_Source}\) run achieved a 45.3% Paragraph MAP increase in performance over the baseline with similar large gains over Aspect (40.1%) and Document MAP (20.6%).
In Sects. 7.2 and 7.4, we explored the effectiveness of adding lexical variants and abbreviations as expansion terms. These expansion terms were derived with respect to the original query terms only. We repeated these experiments on our optimal expanded queries to measure the effect of adding variants and abbreviations for expansion terms as well. Table 11 shows that expansion term variants \( {\tt All+V}\) provides a smaller benefit to paragraph MAP (4.54% increase) than they did when applied to original query terms. However, for abbreviation expansion using the ADAM database, performance significantly drops (\( {\tt All+V+Adam}\)). This confirms our previous observation that the addition of abbreviations to queries can be more harmful than good, due to the effects of ambiguous shortforms (Stokes et al. 2007b). Again we see that the most effective method of dealing with abbreviations is to resolve them in the collection before retrieval is performed, where our \( {\tt All+V+Rsolve}\) run consistently increases MAP scores (by up to 5.45%) when compared with \( {\tt All+V}\).
This concludes our experiments on issues regarding expansion resources. The next subsection focusses on the final step in the retrieval process: the reduction of candidate paragraphs to exact answer passages.
7.8 Passage reduction
Previously we have described our approach to reducing paragraphs to answer passages. In brief, our method searches for the longest span of query terms (occurring in consecutive sentences) in each of our retrieved candidate paragraphs for a particular topic (see Sect. 4 for more details).
Table 12 compares the performance of our best expansion run (\( {\tt All+V+Rsolve}\), abbreviated to \( {\tt AVR}\) in this table) when passage reduction (\( {\tt AVR+PSG}\)) and passage reduction with document context is considered in the answer re-ranking process (\( {\tt AVR+PSG+Ctxt}\)). Runs up to this point have been evaluated with our paragraph MAP score. As already stated, paragraph MAP does not penalise a run for returning additional text surrounding a correct answer passage. Since the focus of the experiments in this section is to evaluate our passage reduction approach, we will evaluate our runs with the official TREC Passage2 MAP metric.
The results in Table 12 show that while passage reduction has a positive effect on Passage2 MAP, slight drops in Aspect and Document MAP occur (\( {\tt AVR+PSG}\)). Similar drops were reported by other TREC participants (Zhou et al. 2006b, 2007). In Sect. 4, we proposed a new passage re-ranking method that as well as considering the relevance of the passage to the query, also incorporates the relevance score of the document containing that passage. Our results show that this run (\( {\tt AVR+PSG+Ctxt}\)) counteracts the tradeoff between Passage2 MAP increases and Aspect/Document MAP decreases. In addition, document context further increases our Passage2 and Document MAP scores.
8 Discussion
The objective of this section is threefold. First, we present a detailed topic analysis of our optimal run \( {\tt AVR+PSG+Ctxt}\) which we now refer to as \( {\tt NICTA\_Best}\). Second, we motivate the contribution of this work by presenting it in the context of other reported results on the TREC 2006 data. And thirdly, we collate the findings of our experiments, and present a list of optimal criteria necessary for successful query expansion in the genomic domain.
8.1 Topic-based discussion of results
In Sect. 7, we drew conclusions regarding system effectiveness from mean Average Precision scores across all 28 topics in the TREC 2006 Genomics evaluation. In this subsection, we provide a deeper understanding of the strengths and weaknesses of our best run in comparison with our baseline concept retrieval system (described in Sect. 4) by analysing performance on a topic by topic basis.
In Fig. 2, we see that \( {\tt NICTA\_Best}\) outperforms the baseline system on all but 4 topics: 169, 181, 183 and 185. This observation concurs with our statistical significance tests, thus confirming that the increase in performance is contributed to by the majority of topics rather than a few very high scoring ones. We now consider each of the under-performing topics, and attempt to explain their sub-optimal performance compared to the baseline:
-
Topic 169 (How does adenomatous polyposis coli (APC) affect actin assembly?): Looking at the top ranked passages returned by the expanded query, we see a number of long, irrelevant chunks of text from the Reference Sections of papers. Like many other TREC participants we took the decision to index references, since human judges have added many such instances to the gold standard passages. So it is possible that, while the content of a document is irrelevant, it may contain a relevant reference to a paper that has been judged to contain an answer to the query.
-
Topic 181 (How do mutations in the Huntingtin gene affect Huntington’s disease?): The expansion of the Huntingtin gene to “HD” causes problems for this topic as there are a number of references in the text to a paper containing this abbreviation in its title (“Behavioral abnormalities and selective neuronal loss in HD”). This paper is represented 4 times in the top 20 retrieved passages (as it is referenced by multiple documents), but has not been judged relevant. This problem is similar in spirit to topic 169. However, it also highlights the need for some redundancy removal on the ranked list: that is, lower ranking passages that look distinctly similar to a higher ranked one should be removed from the answer list. The performance of this topic drops by around 60% compared to the baseline, which is the biggest drop of the four under-performing topics.
-
Topic 183 (How do mutations in the NM23 gene affect tracheal development?): Here we see a slight drop in performance by the \( {\tt NICTA\_Best}\) run as a number of irrelevant passages are retrieved that contain references to the following expansion terms: NM23-H1, NM23/AWD, ASP/NM23-M1, NM23-M1, AWD. These gene expansion terms are correct; however, we must assume that these passage were not judged relevant as they made no reference to the concept “tracheal development”.
-
Topic 185 (How do mutations in the hypocretin receptor 2 gene affect narcolepsy?): The performance of this topic drops slightly because one high ranked passage contains the gene expansion term “orexin receptor”, but is not considered relevant.

Passage2 AP scores per topic for two system runs: \({\tt Baseline}\) and \({\tt NICTA\_Best}\)
In summary then, it appears that the indexing of reference sections in papers is a bigger problem than the addition of irrelevant expansion terms due to ambiguity.
8.2 Performance comparison with TREC participants
In this subsection, we motivate the significance of our results with respect to official TREC 2006 MAP scores, and the highest reported post-submission scores, achieved by the University of Illinois, Chicago (UIC). Table 13 shows that the \( {\tt NICTA\_Best}\) run has the highest MAP score at each level of granularity (Passage2, Paragraph, Aspect and Document), except for the now defunct Passage MAP score. A brief description of the UIC and National Library of Medicine (NLM) approach (the only interactive run presented here) can be found in Sect. 5. The \( {\tt THU2}\) run, submitted by Tsinghua University, does not have a corresponding participant workshop paper, so it is unclear which techniques they have used to achieve these impressive results. The median values of each MAP score, for the official TREC results, are also reported (\( {\tt TREC\_MEDIAN}\)). Since we don’t have access to the ranked lists of the \( {\tt UIC\_SIGIR}\) run (Zhou et al. 2007), the Passage2 and Paragraph values are missing. Overall, our \( {\tt NICTA\_Best}\) run achieves a 185% increase in performance (Passage2 MAP, 0.137 vs. 0.048) over a baseline Okapi approach (\( {\tt Okapi}\)).
8.3 Criteria for successful query expansion
As stated at the beginning, and throughout this paper, our aim here is to suggest some necessary criteria for optimising the effectiveness of query expansion for genomic-based search. In Sect. 7, our experiments isolated and investigated factors affecting system performance. These experiments focussed around two specific aspects of the retrieval framework presented in Sect. 4: knowledge resource and relationship type used in expansion; and IR ranking algorithm.
Before we formally state our criteria for effective query expansion, it is worth pointing out that there are many other parameters that significantly affect performance. For example, our decision to index references in journal papers was based on the observation that cited papers appear in gold standard passages for the task. More specifically, of the 3,451 gold standard passages, 458 (13.3%) of them are references.
Similarly, other researchers have shown that small changes in tokenisation of the collection (a preprocessing step) can significantly improve performance (Trieschnigg et al. 2006). To the best of our ability, we have tried to optimise these additional factors to ensure that performance losses by, for example, the addition of specialised terms to the query, can be solely attributed to this term type rather than an unfortunate side-effect of, say, our tokenisation strategy.
Figure 3 summarises the findings of our experiments in Sect. 7, where clearly our concept normalisation ranking method (\( {\tt Con}\)) provides more performance gains than any of the individual expansion term types, and slightly more than the optimal combination of expansion terms (\( {\tt ALLEXP}\)).

Graph comparing the relative improvement in Passage2 MAP performance achieved by various system parameters (factors). These factors are: Concept Normalisation (\({\tt Con}\)), Phrase Retrieval (\( {\tt Phr}\)), Lexical Variation (\( {\tt VAR}\)), Gene Synonyms (\( {\tt GeSyn}\)), General term synonyms (\( {\tt GTSyn}\)), Generalised ontological terms (\( {\tt Gnr}\)), Abbreviation resolution in collection (\( {\tt Res}\)), Adam abbreviations (\( {\tt Adm}\)), Relevance feedback (\( {\tt Rel}\)), Passage reduction (\( {\tt Psg}\)). The factor \( {\tt ALLEXP}\) is a combination of all the following expansion factors: \( {\tt VAR}\), \( {\tt GeSyn}\), \( {\tt GTSyn}\), \( {\tt Gnr}\) and \( {\tt Res}\)
So the principal conclusions of this paper are as follows:
-
Without doubt the single biggest contributing factor to the success of our experiments can be attributed to the modified Okapi ranking algorithm we have developed, which is based around concept re-ranking, and the normalisation of expansion terms. The former ensures that a passage that contains multiple distinct query concepts will be ranked higher than a passage that contains multiple instances of the same query concept. The latter, the normalisation technique, ensures that expansion terms contribute less to the ranking process than original query terms do.
-
Phrase-based querying is essential, and becomes more critical as additional expansion terms are added.
-
In general, expansion terms gleaned from ontologies are more effective than those provided by corpus-based analysis methods, such as pseudo relevance feedback and cooccurrence statistics derived from MEDLINE. Our results show that while relevance feedback achieves small non-statistical gains, MEDLINE cooccurring terms significantly decrease performance.
-
Gene synonym expansion provides larger performance improvements than general biomedical synonym term expansion. Entrez Gene provides the largest improvement for gene expansion, while UMLS (a combination of SNOMED_CT, MeSH and the Metathesaurus) is the best option for general biomedical terms.
-
Ontological relationships such as parents and children of the original query term produce little or no improvement to the retrieval process. However, adding parent terms from MeSH showed some minor improvement, where a limited number of topics performed better and all other topics experienced a neutral rather than negative effect, making it a useful expansion term type. Other sources of generalised terms (SNOMED_CT, the MetaThesaurus and UMLS) did not exhibit this characteristic.
-
Abbreviation expansion is an ineffective form of query expansion, especially when shortforms of expanded terms are also added (see Sect. 7.4). This is due to the additional ambiguity that shortforms with multiple semantically distinct longform concepts contribute. We show that abbreviation resolution in the collection eliminates the need for abbreviation query expansion, and produces a statistically significant improvement over the baseline.
-
Since none of the expansion term resources examined differ greatly in their effect on performance, we can conclude that the differences in expansion effectiveness reported at TREC are caused by the use of an inappropriate IR framework rather than from a poor source of the ontological expansion terms.
-
Passage Reduction helps improve Passage2 MAP scores. However, as reported by other participants this increase can negatively impact Document MAP. We have shown that considering document context in the ranking algorithm can alleviate this trade-off.
9 Conclusions
The aim of this paper was to explore the criteria necessary for successful query expansion in the genomic domain. We presented a set of controlled experiments that isolated and evaluated two important aspects of system design: passage ranking strategy, and knowledge resources for query expansion. Our experiments showed that the single biggest factor affecting the accuracy of the retrieval process is the ranking metric used.
We presented a novel concept normalisation ranking strategy that addresses two problems with the standard Okapi ranking algorithm: first, the concept re-ranking strategy ensures that documents containing multiple unique concepts are ranked higher than documents that make reference to the same concept multiple times; and secondly, the query-term normalisation strategy ensures that expansion terms for the same concept are not given undue influence by the ranking metric.
Our results also conclusively show that query expansion has a positive effect on genomic retrieval performance; with the added caveat that expansion terms should be gleaned for manually-derived domain specific resources rather than automatically-generated corpus-derived terms. Our results also demonstrate that expansion with synonyms and lexical variants is much more effective than with hierarchical terms from ontologies. The only exceptions to the “synonym efficacy rule” are abbreviations, whose utility is questionable due to their innate ambiguity (that is, shortforms may refer to multiple concepts). Our experiments establish that resolving abbreviations in the collection (replacing them with their longforms) provides a more effective alternative to using abbreviations to expand the query.
An interesting direction for future work would be to investigate the importance of weighting expansion terms according to their strength of semantic association with the original query term. This idea is similar in vein to work on weighted query terms by Aronson and Rindflesch (1997) and Zhou et al. (2007), and work by Liu and Chu (2005) on thesaurus guided relevance feedback. In addition, we have only examined expansion in the context of two ranking metrics: Okapi and our own modified version of this metric. It would be interesting to repeat these experiments on other IR models, such as Language Modelling approaches, which have shown some promise at the TREC 2006 Genomics track.
In conclusion then, this paper demonstrates that query expansion has a positive effect on retrieval performance in the genomic domain, when the basic criteria, regarding system design issues, are adhered to as outlined in this paper.
Notes
Entrez PubMed: http://www.ncbi.nlm.nih.gov/sites/entrez.
Google Scholar: http://scholar.google.com.au/.
Some papers report results on retrieval from a subset of the OHSUMED collection containing all MEDLINE citations that have an abstract (that is 233,455 records from 348,566, e.g.), and for the 101 queries which have at least one positive relevance judgement (Ruch et al. 2006).
T2K provides an online service which collects information from GenBank, OMIM and MEDLINE data http://www.bioinformatics.org/textknowledge/synonym.php.
Query templates are officially referred to as generic topic templates (GTT); see Hersh et al. (2006) for more details.
WordNet is a domain independent machine-readable, manually-derived thesaurus developed by researchers at Princeton (Fellbaum 1998).
Since no manual intervention occurred in the query parsing process, we consider our runs to be automatic. For example, if PubMed’s parsing tool returned an incorrect parse, we did not manually modify the query. This PubMed tool was also used in the official automatic runs submitted by Zhou et al. (2006b).
For readability purposes, we refer to both a single term unit and a multi-term unit (that is, a phrase) as a term, since the latter is treated in the same manner as the former in the following equations.
More information on the TREC dataset can be found at: http://ir.ohsu.edu/genomics/2006data.html.
References
Abdou, S., & Savoy, J. (2006). Report on the TREC 2006 Genomics experiment. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Ananiadou, S., & Nenadic, G. (2006). Automatic terminology management in biomedicine. In S. Ananiadou & J. McNaught (Eds.), Text mining for biology and biomedicine (pp. 67–98). Norwell, MA: Artech House Books.
Aronson, A. R., & Rindflesch, T. C. (1997). Query expansion using the UMLS metathesaurus. In Proceedings of the 1997 Annual AMIA Symposium, Nashville, TN (pp. 485–489).
Bairoch, A., Apweiler, R., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., Gasteiger, E., Huang, H., Lopez, R., Magrane, M., Martin, M. J., Natale D. A., O’Donovan, C., Redaschi, N., & Yeh, L. S. (2005). The Universal Protein Resource (UniProt). Nucleic Acids Research, 33, D154–D159.
Buttcher, S., Clarke, C. L. A., & Cormack, G. V. (2004). Domain-specific synonym expansion and validation for biomedical information retrieval. In E. M. Voorhees & L. P. Buckland (Eds.), The Thirteenth Text REtrieval Conference (TREC 2004) Proceedings, 16–19 November. Gaithersburg, MD: NIST.
Croft, W. B., & Lafferty, J. (2003). Language modeling for information retrieval. Norwell, MA: Kluwer Academic Publishers.
Demner-Fushman, D., Humphrey, S. M., Ide, N. C., Loane, R. F., Ruch, P., Ruiz, M. E., Smith, L. H., Tanabe, L. K., Wilbur W. J., & Aronsona, A. R. (2006). Finding relevant passages in scientific articles: Fusion of automatic approaches vs. an interactive team effort. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Dorff, K., Wood, M., & Campagne, F. (2006). Twease at TREC 2006: Breaking and fixing BM25 scoring with query expansion, a biologically inspired double mutant recovery experiment. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Dunning, T. (1993). Accurate methods for the statistics of surprise and coincidence. Computational Linguistics, 19(1), 61–74.
Eyre, T. A., Ducluzeau, F., Sneddon, T. P., Povey, S., Bruford, E. A., & Lush, M. J. (2006). The HUGO gene nomenclature database. Nucleic Acids Research, 34(Database issue), D319–21.
Fellbaum, C. (1998). Wordnet: An Electronic Lexical Database. Cambridge, MA: MIT Press.
Furnas, G. W., Landauer, T. K., Gomez, L. M., & Dumais, S. (1987). The vocabulary problem in human-system communication. CACM, 30(11), 964–971.
Goldberg, A., Andrzejewski, D., Van Gael, J., Settles, B., & Zhu, X. (2006). Ranking biomedical passages for relevance and diversity: University of Wisconsin at TREC Genomics 2006. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Hersh, W., Cohen, A., Roberts, P., & Rekapalli, H. (2006). TREC 2006 Genomics Track overview. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Hersh, W., Cohen, A., Yang, J., Bhupatiraju, R. T., Roberts, P., & Hearst, M. (2005). TREC 2005 Genomics Track overview. In E. M. Voorhees & L. P. Buckland (Eds.), The Fourteenth Text REtrieval Conference (TREC 2005) Proceedings, 15–18 November, Gaithersburg, MD. NIST.
Hersh, W., Price, S., & Donohoe, L. (2000). Assessing thesaurus-based query expansion using the UMLS metathesaurus. In Proceedings of the 2000 Annual AMIA Fall Symposium.
Hersh, W. R., Buckley, C., Leone, T. J., & Hickam, D. H. (1994). OHSUMED: An interactive retrieval evaluation and new large test collection for research. In W. B. Croft & C. J. van Rijsbergen (Eds.), SIGIR ’94: Proceedings of the 17th Annual International ACM SIGIR Conference on Research and Development in Information Retrieval, 3–6 July, Dublin, Ireland (pp. 192–201). New York: ACM.
Hersh, W. R., Bhuptiraju, R. T., Cohen, A. M., Ross, L., Kraemer, D. F., & Johnson, P. (2004). TREC 2004 Genomics Track overview. In E. M. Voorhees & L. P. Buckland (Eds.), The Thirteenth Text REtrieval Conference (TREC 2004) Proceedings, 16–19 November, Gaithersburg, MD. NIST.
Huang, X., Hu, B., & Rohian, H. (2006). York University at TREC 2006: Genomics Track. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Jiang, J., He, X., & Zhai, C. (2006). Robust pseudo feedback estimation and HMM passage extraction: UIUC at TREC 2006 Genomics Track. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Krovetz, R., & Croft, W. B. (1992). Lexical ambiguity and information retrieval. Information Systems, 10(2), 115–141.
Lavrenko, V., & Croft, W. B. (2001). Relevance based language models. In W. B. Croft, D. J. Harper, D. H. Kraft, & J. Zobel (Eds.), SIGIR’01: Proceedings of the 24th Annual International ACM SIGIR Conference on Research and Development in Information Retrieval, 9–13 September, New Orleans, LA, USA (pp. 120–127). New York: ACM.
Li, Y., Moffat, A., Stokes, N., & Cavedon, L. (2006). Exploring probabilistic toponym resolution for geographical information retrieval. In R. Purves & C. Jones (Eds.), Workshop on Geographic Information Retrieval, SIGIR 2006, 6–11 August, Seattle, WA. New York: ACM.
Lin, J., Quan, D., Sinha, V., Bakshi, K., Huynh, D., Katz, B., & Karger, D. (2003). What makes a good answer? The role of context in question answering. In The Ninth IFIP TC13 International Conference on Human-Computer Interaction (INTERACT 2003), 1–5 September, Zurich, Switzerland (pp. 392–399).
Liu, Z., & Chu, W. (2005). Knowledge-based query expansion to support scenario-specific retrieval of medical free text. In ACM-SAC Information Access and Retrieval Track, Santa Fe, NM (pp. 1076–1083). New York: ACM.
Maglott, D. R., Ostell, J., Pruitt, K. D., & Tatusova, T. A. (2005). Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Research, 33(Database Issue), 54–58.
McKusick, V. A. (1998). Mendelian inheritance in man. A catalog of human genes and genetic disorders (12th ed.). Baltimore: Johns Hopkins University Press.
Park, J. C., & Kim, J. (2006). Named entity recognition. In S. Ananiadou & J. McNaught (Eds.), Text mining for biology and biomedicine (pp. 121–142). Norwell, MA: Artech House Books.
Pickens, J., & Croft, W. B. (2000). An exploratory analysis of phrases in text retrieval. In RIAO ’00: Proceedings of RIAO (Recherche d’Information assiste par Ordinateur), Paris (pp. 1179–1195).
Pustejovsky, J., Castano, J., Cochran, B., Kotecki, M., & Morrell, M. (2001). Automatic extraction of acronym-meaning pairs from MEDLINE databases. Medinfo, 10, 371–375.
Robertson, S. E., Walker, S., Jones, S., Hancock-Beaulieu, M., & Gatford, M. (1994). Okapi at TREC-3. In E. M. Voorhees & L. P. Buckland (Eds.), (1994). The Third Text REtrieval Conference (TREC 3) Proceedings, 2–4 November, Gaithersburg, MD. NIST.
Rocchio, J. (1971). Relevance feedback in information retrieval. In G. Salton (Ed.), The SMART Retrieval System—Experiments in Automatic Document Processing (pp. 313–323). Englewood Cliffs, NJ: Prentice Hall.
Ruch, P., Tbahriti, I., Gobeill, J., & Aronson, A. R. (2006). Argumentative feedback: A linguistically-motivated term expansion for information retrieval. In Annual Meeting of the Association for Computation Linguistics (ACL), 17–81 July, Sydney, Australia (pp. 675–682). Morristown, NJ: ACL.
Ruthven, I., & Lalmas, M. (2003). A survey on the use of relevance feedback for information access systems. Knowledge Engineering Review, 18(2), 95–145.
Sanderson, M. (2000). Retrieving with good sense. Information Retrieval, 2(1), 49–69.
Schwartz, A. S., & Hearst, M. A. (2003). A simple algorithm for identifying abbreviation definitions in biomedical text. In Pacific Symposium on Biocomputing 2003 (Vol. 8, pp. 451–462).
Si, L., Lu, J., & Callan, J. (2006). Combining multiple resources, evidence and criteria for genomic information retrieval. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Smucker, M. (2006). UMass Genomics 2006: Query-biased pseudo relevance feedback. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Song, F., Vasak, J., & Wang, W. (2006). Passage retrieval by shrinkage of language models. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Srinivasan, P. (1996). Query expansion and MEDLINE. Journal of the American Medical Informatics Association, 13, 157–167.
Stokes, N., Li, Y., Cavedon, L., Huang, E., Rong, J., & Zobel, J. (2007a). Entity-based relevance feedback for genomic list answer retrieval. In E. M. Voorhees & L. P. Buckland (Eds.), The Sixteenth Text REtrieval Conference (TREC 2007) Proceedings, 5–9 November, Gaithersburg, MD. NIST.
Stokes, N., Li, Y., Cavedon, L., & Zobel, J. (2007b). Exploring abbreviation expansion for genomic information retrieval. In Australasian Language Technology Workshop 2007, 10–11 December, Melbourne, Australia (pp. 100–108).
Trieschnigg, D., Kraaij, W., & de Jong, F. (2007). The influence of basic tokenization on biomedical document retrieval. W. Kraaij, A. P. de Vries, C. L. Clarke, N. Fuhr & N. Kando (Eds.), (2007). SIGIR ’07: Proceedings of the 30th Annual International ACM SIGIR Conference on Research and Development in Information Retrieval, 23–27 July, Amsterdam, The Netherlands (pp. 803–804). New York: ACM.
Voorhees, E. M., & Buckland, L. P. (Eds.). (1994). The Third Text REtrieval Conference (TREC 3) Proceedings, 2–4 November, Gaithersburg, MD. NIST.
Zhou, W., Torvik, V. I., & Smalheiser, N. R. (2006a). ADAM: Another database of abbreviations in MEDLINE. Bioinformatics, 22(22), 2813–2818.
Zhou, W., Yu, C., Smalheiser, N., Torvik, V., & Hong, J. (2007). Knowledge-intensive conceptual retrieval and passage extraction of biomedical literature. In W. Kraaij, A. P. de Vries, C. L. Clarke, N. Fuhr & N. Kando (Eds.), SIGIR ’07: Proceedings of the 30th Annual International ACM SIGIR Conference on Research and Development in Information Retrieval, 23–27 July, Amsterdam, The Netherlands (pp. 655–662). New York: ACM.
Zhou, W., Yu, C., Torvik, V., & Smalheiser, N. (2006b). A concept-based framework for passage retrieval in genomics. In E. M. Voorhees & L. P. Buckland (Eds.), The Fifteenth Text REtrieval Conference (TREC 2006) Proceedings, 14–17 November, Gaithersburg, MD. NIST.
Acknowledgements
NICTA is funded by the Australian Government as represented by the Department of Broadband, Communications and the Digital Economy and the Australian Research Council through the ICT Centre of Excellence program. The authors would also like to thank the reviewers for their helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stokes, N., Li, Y., Cavedon, L. et al. Exploring criteria for successful query expansion in the genomic domain. Inf Retrieval 12, 17–50 (2009). https://doi.org/10.1007/s10791-008-9073-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10791-008-9073-9