
Abstract
The achievement of a better life for cystic fibrosis (CF) patients is mainly caused by a better management and infection control over the last three decades. Herein, we want to summarize the cornerstones for an effective management of CF patients and to give an overview of the knowledge about the fungal epidemiology in this clinical context in Europe. Data from a retrospective analysis encompassing 66,616 samples from 3235 CF patients followed-up in 9 CF centers from different European countries are shown.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cystic fibrosis (CF) is one of the most common autosomal recessive diseases in Europe. The dysfunction of epithelial electrolyte transport causes the production of viscous secretion by all exocrine glands. Due to the viscous mucus in the CF patients’ airways, recurrent bacterial infections of the respiratory tract commonly occur which play a crucial role in the progression of the disease. The colonization of the lung with bacteria, predominantly Pseudomonas aeruginosa, is thought to be the main cause of morbidity and mortality in CF. In the course of the CF disease, fungal colonization of the airways and respiratory infections play an increasing role, especially due to rising awareness of fungal growth in microbiological diagnostic, a longer life expectancy and a rising percentage of immunocompromised CF patients.
The scope of this study is to give a short and comprehensive overview on the management of CF patients and a synopsis of the cornerstones in diagnostic and therapy in general. In a second part, we elucidated published data concerning the fungal epidemiology in the CF context in Europe and retrospectively analyzed the fungal epidemiology and diversity from 9 CF centers, distributed in 9 different European countries.
Management of Patients with Cystic Fibrosis
In the last decade, CF care worldwide and specifically in Europe has evolved in many ways. Early diagnosis through neonatal screening has proliferated across Europe. The adult population of patients has been steadily increasing, and in some countries it has now numerically passed the pediatric population. New treatments are available, including mutation-specific therapies that might have the potential to change the natural history of the disease [1, 2]. More emphasis is given to clinical research, and a network of European CF centers has been established with the aim of facilitating high-quality clinical trials to translate new therapies into clinical care. Epidemiological data from many European countries have become available through the EuroCareCF project [3] and national and international registries. Evidence of disparities in allocation of resources and in clinical outcomes has also emerged.
The landscape of CF will change dramatically in the future in Europe [4]. Data obtained from the European Cystic Fibrosis Society Patient Registry revealed forecasts for 2025 in 16 European countries that are on one side promising and on the other side dramatic. The data indicated that the number of CF patients will increase by approximately 50%, mainly due to neonatal screening programs. Moreover, the number of CF adults will increase by approximately 75%, a finding that mostly results from the transition of children to adults, whereas the number of CF children will show a 20% increase. These results are crucial as people with CF have complex care needs that demand specialist with medical and allied healthcare expertise. The life expectancy has increased significantly in successive patient birth cohorts [5] as a result of more effective treatments and crucially because most patients attend CF centers in line with the demonstration that patients attending CF centers for their care have better well-being and lung function than those who do not [6, 7].
Management of the patients means nowadays implementing a multi-disciplinary team into the CF center that should have adequate resources (e.g., staffing, computer equipment) and an infrastructure (inpatient and outpatient facilities) that allows the multi-disciplinary team to provide a level of care that is in accordance with the European Cystic Fibrosis Society (ECFS) standards recommendations [8]. The CF center should have a minimum of 100 adult or pediatric patients to warrant enough experience and appropriate level of expertise.
The CF center must allow access to the center for emergency care 24 h and 7 days a week. Routine appointments for people with stable disease should be every 2–3 months. But this also should be depending on the severity of their disease. Newly diagnosed infants should be seen more frequently (initially weekly).
All patients should have an annual assessment to ensure high quality of care with the aim to prevent progress of the disease or to diagnose new CF-related diseases such as diabetes or osteoporosis. For patients treated either in the outpatient department or on the ward, standard operating procedures should exist to prevent cross-infection between patients.
One further important structure in patient management is the access to special diagnostics [8]. As aforementioned, patients with CF reach higher average of lifetime. Therefore, it is mandatory to reduce the risk of late sequelae. Annual radiologic diagnostics are performed with X-ray or computed tomography (CT) scan producing a significant amount of radiation exposure to the patient. Newer techniques such as magnetic resonance imaging (MRI) allow a comparable examination of the lung [9,10,11]. Additionally, the radiology and nuclear medicine service should still include CT scanning, liver ultrasound and dual-energy X-ray absorptiometry (DXA) bone scanning. Lung function measures must be available for outpatient department as well as for the ward. As CF patients gain more mobility, exercise testing and fitness-to-fly testing should be available, too.
In terms of the chronic lung disease and the acquisition of several different microorganisms during their lifetime, the microbiology services should have the ability to process samples from people with CF and to reliably detect Burkholderia spp., non-tuberculous mycobacteria and Aspergillus infections as well as other rare bacteria and fungi. Molecular pathogen typing and immunology for allergic bronchopulmonary aspergillosis (ABPA) monitoring should also be implemented into the structure of the CF center as ABPA can occur in up to 15% of CF patients [12].
As aforementioned, the multi-disciplinary team is the key to good quality care for a multi-organ disease like CF. Therefore, every single specialist has his own package of work and structure which has to be included into the entire concept of the patient’s care. From the long list of team members, the medical doctors, CF nurses, physiotherapists, dieticians and psychologists play the most important roles and build the core of the team. Special knowledge is needed in all subgroups of the team. The team needs the facilities, structure and standard operating procedures (SOP) available for yearly check-ups. In addition to the planned visits, patients with CF have common complications of their disease and will need additional service. For medical doctors and nurses, it is crucial that they are aware of these complications. Structures and SOPs must be implemented in the CF center for pneumothorax, hemoptysis, distal intestinal obstructive syndrome, hepatobiliary complications, CF-related diabetes, ABPA and respiratory insufficiency [13,14,15].
Furthermore, physiotherapists have their own topics regarding treatment of the CF lung diseases. CF physiotherapists should be available for regular contact and assessment of the patient for treatment, lung function testing, physical surveillance and therapy evaluation. The frequency of these will vary according to the patient’s age and clinical status, but as a minimum should happen at every routine outpatient clinic and daily during each hospitalization (including when patients are admitted under the care of other specialists and to intensive care). A more extensive assessment should take place annually [8]. Major issues for implementation in the CF care for physiotherapists are airway clearance therapy, inhalation therapy, exercise capacity testing, postural and musculoskeletal assessment and noninvasive implementation [8].
Further, team members are psychologists. They are receiving an increased number of problems to solve. One major part in this context is the mental health [16]. In a study from Quittner et al. [17], psychological symptoms were reported from 6088 patients with CF and 4102 parents. Elevated symptoms of depression were found in 10% of adolescents, 19% of adults, 37% of mothers and 31% of fathers. Elevations in anxiety were found in 22% of adolescents, 32% of adults, 48% of mothers and 36% of fathers. Overall, elevations were 2–3 times those of community samples. Participants reporting elevated anxiety were more likely to report depression (ORs: adolescents = 14.97, adults = 13.64, mothers = 15.52, fathers = 9.20). These results show a strong need for implementing a screening into the routine setting in CF centers [17].
The role of the specialist CF dietician is to take the lead in providing high-quality treatment and care to ensure optimal nutritional status, including nutritional screening and surveillance, and regular patient assessment with review of all aspects of nutrition and gastrointestinal status. The frequency and type of assessment will vary with age and clinical status [8].
In terms of CF care, the framework of microbiology is of special interest as patients suffer from chronic and acute infections due to a high number of different bacteria and fungi. A clinical microbiologist with particular knowledge of CF infection should be part of the CF multi-disciplinary team. This person may be a medically trained clinical microbiologist or an infectious disease specialist, but a clinical scientist with relevant knowledge and experience could also undertake this role. The CF clinical microbiologist should work closely with the microbiology laboratory providing diagnostic services and the other members of the CF multi-disciplinary team, as well as with the local infection control and prevention team. To provide support to the CF multi-disciplinary team for the diagnosis and treatment of infection, the CF clinical microbiologist needs to know about the range of infections and colonization in CF. In particular, he needs to be aware of the role of unusual microorganisms, the risk of cross-infection and the impact of long-term chronic infection on microbiological laboratory testing and anti-infective treatment. In addition to a good basic knowledge, the CF clinical microbiologist should have evidence of continuing professional development in CF microbiology and regularly attend specialist CF meetings and conferences [8].
The CF clinical microbiologist should ensure that the full range of microbiology laboratory tests needed for the CF center is available and that the laboratory service provided is based on currently published guidelines [18]. The laboratory should be fully accredited by a recognized national scheme for clinical microbiology and should participate in external quality assurance programs, which include CF-associated pathogens. There should be provision to send relevant samples to a reference laboratory specialized in CF microbiology when required. The laboratory should provide accurate and timely results to the CF center with an agreed system for notifying urgent and important results. The technical staff in the laboratory should have sufficient expertise and knowledge to deal with the complex microbiology of CF infections. There should be a framework for recording and investigating errors and other incidents, with evidence of how the lessons learned are used to inform a program of service improvement. The service should be regularly audited. Examples of audits are the turnaround time (i.e., the time between receipt of the sample in the laboratory and availability of the result to the CF multi-disciplinary team), the accuracy of identification and susceptibility testing, and the appropriate and prompt communication of urgent results to the CF multi-disciplinary team [8].
Special items have to be cleared between the multi-disciplinary team and the microbiologist regarding infections of the patients [8]:
-
1.
Which respiratory samples should be taken and how should they be processed (e.g., sputum, bronchoalveolar lavage, cough swab or a pharyngeal swab)?
-
2.
Which samples should be taken for the diagnosis of an infected intravascular line?
-
3.
Diagnosis of other infections including infections of the gut (e.g., enteric viruses, when and how to test for toxigenic Clostridium difficile).
-
4.
The level of identification of microorganisms (e.g., genus, species, subtype) required in individual cases. This may include a discussion on the tests that can be performed in a local laboratory and what may need to be referred to a specialized laboratory with more advanced testing methodology (e.g., confirmation of first infection with Burkholderia spp. with accurate species identification).
-
5.
Typing methods and frequency of typing (i.e., how often the CF multi-disciplinary team should send samples for routine surveillance and when additional typing should be done due to suspicion of cross-infection).
-
6.
Measurement of antipseudomonal antibodies where appropriate.
-
7.
Provision of diagnostic testing for fungal and mycobacterial infection together with level of identification and role of typing.
-
8.
Susceptibility testing—agreement on the antibiotics and antifungals to be tested and when susceptibility testing is helpful.
-
9.
Virology services should include rapid identification of highly pathogenic viruses that may spread between patients—both familiar (e.g., influenza virus) and emerging viral pathogens (e.g., severe acute respiratory syndrome-related coronavirus, Middle East respiratory syndrome-related coronavirus).
-
10.
Which results need to be phoned urgently to the CF multi-disciplinary team (e.g., first isolation of Pseudomonas aeruginosa, new isolation of Burkholderia cepacia complex and other Burkholderia species from sputum, of methicillin-resistant Staphylococcus aureus and of Mycobacterium species)?
-
11.
Advice on infection prevention and control [8].
The CF clinical microbiologist should work close together with the other members of the CF multi-disciplinary team and in addition with the local infection control team to create a consented local infection control and prevention policy. Procedures should be in line with expert national and international guidelines [8]. This policy should include different strategies (Table 1).
Epidemiology of Fungal Colonization of the Airways in CF Patients
Over the last years, fungal colonization of the respiratory tract from CF patients raised attention due to better evaluation of the fungal diversity in the CF airways [19, 20]. This awareness is also caused by the increasing number of immunocompromised CF patients (e.g, after lung transplantation) and the often fatal outcome in case of invasive infections by rare fungal species.
The aim of this section is to summarize published data concerning the fungal epidemiology in CF in Europe and to give a short overview on some spots in Europe. Furthermore we will also present collected data from different European centers with the intention, to sensitize more clinicians and microbiologists to this issue.
The role of certain fungal species for the progress of the lung disease is still under discussion, as well as the prophylactic use of antifungal therapy during lung transplantation and the exclusion of CF patients when their respiratory tract is colonized by specific molds.
Yeasts and molds are often cultured from respiratory tract of CF patients. Despite the colonization in over 50% with Candida species (with a majority of Candida albicans), the clinical role of these yeasts is still not clarified.
Concerning the filamentous fungi, Aspergillus fumigatus has the highest impact on CF patients. The respiratory tract of up to 78.8% of CF patients is colonized by some Aspergillus species [21]. About 12–25% of CF patients suffer from an ABPA [22,23,24]. Localized infections are seen in CF patients with abnormalities in their airways like aspergilloma and, if they are immunocompromised, they are at severe risk for invasive aspergillosis.
Scedosporium and Lomentospora species (formerly called Pseudallescheria for some of them) are responsible for numerous infections in immunocompromised patients or healthy individuals after near-drowning accidents. The high-resistance profile of these fungi to antifungal agents often complicates the therapeutic approach. Further colonization with Scedosporium or Lomentospora species has shown a fatal clinical outcome after lung transplantation in some cases.
Exophiala dermatitidis can be cultivated from respiratory specimens from up to 17% of CF patients [25]. This black yeast appears as a chronic colonizing organism in CF patients; its clinical role is not fully understood, but an immunological reaction of the host organism, a contribution to inflammation and clinical deterioration has been shown [26].
Trichosporon mycotoxinivorans has been described as a chronic colonizer of the CF airways, but also as a highly pathogenic yeast in a couple of cases [27,28,29,30]. Until now, it is not clearly understood which factors trigger the variable effects of this yeast species on the CF lung.
Retrospective Analysis of the Data from the 9 Participating CF Centers
For data collection, collaborators from the 9 participating European CF centers were asked to provide retrospective data concerning CF patients, fungal examination and count of species detected within the time period of January 2011 to December 2016. Due to heterogeneity of the datasets provided by each center, we can only describe these data and cannot compare in detail each dataset (different isolation methods, data counting, patient cohort and patient management). To get an idea of the rate of colonized patients from each of the nine centers, we built up the mean for the patients tested positive for selected fungal species per year and related this to the mean count of patients per year for each center.
Datasets collected from 9 European CF centers (all from different countries in Europe) were thus analyzed. Overall, 66,616 samples from 3235 CF patients were included in this retrospective analysis. The range between the participating centers was from 110 samples (60 patients) to 5300 samples (680 patients) per year (in mean) (Table 2). The age distribution was quite identical; the range was 0–79 years with a mean age of about 17–28 years. In Poland, the participating center was a children hospital, which explains the mean age of 12 years for the patients followed-up in this hospital. Most of the analyzed samples originated from the respiratory tract (Table 2).
All participating centers performed homogenization of the respiratory samples using a mucolytic agent before culturing. They all included mycological culture media for the detection of yeasts and molds. The use of a Scedosporium selective medium (e.g., SceSel+-Agar) was mentioned by 4/9 laboratories (Table 3) [31]. The incubation time was variable between 7 and 21 days. Identification tools were mainly matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry for yeasts, and morphological macroscopic and microscopic criteria for molds, followed in doubtful cases by sequence analysis of the internal transcribed spacer (ITS) regions of ribosomal RNA genes mostly.
More than 36% (in mean) of the samples from CF patients were culture positive for fungi (yeasts and/or molds), which highlight the importance of a mycological examination of these samples.
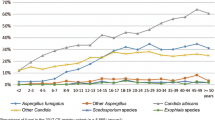
Candida albicans was the most prevalent yeast in every center, with a range (in mean) of 33.8% up to 77.9%. In a lower and diverse frequency, other Candida species were detected in each center (for detail see Table 3).
Other yeast-like fungi, e.g., Exophiala dermatitidis were reported only in 7/9 centers, with a prevalence rate ranging from 0.2 to 18.3% colonized patients (in mean). Culturing Trichosporon species (including the potential CF pathogenic species Trichosporon mycotoxinivorans) was reported only rarely in 7/9 centers (0.2–3.5% in mean).
The main genus of cultured filamentous fungi was Aspergillus, with dominance of Aspergillus fumigatus in most centers. The range in mean differed between 3.9% up to 42.4% for A. fumigatus, in the participating children hospital the mean of positive patient per year being 13.6% (Table 3).
Concerning Scedosporium and Lomentospora species, there were also geographic variations in their distribution: the mean percentages of positive patients differed from 12.1% in Italy to 0.1% in the Czech Republic center. In the majority, the prevalence rate was between 2 and 5% for these species.
Patients with a colonization of the respiratory tract with Rasamsonia (Geosmithia) argillacea were only rarely seen in this study (Table 3).
The limitations of this survey were the heterogeneity in the datasets from participating centers. This is explained by differences in isolation methods, data counting, patient cohort and patient management. Even the diversity in the study periods between the participating centers as well as in climatic features between centers has probably influenced the results. The data shown here are only from one CF center/region for each country and possibly not representative for a whole country.
Every participating laboratory in this study used a mucolytic pretreatment of respiratory samples from CF patients. The benefit of this was highlighted by Masoud-Landgraf et al. [21] who showed a higher cultivation rate for rare fungi.
All microbiological laboratories used mycological culture media for the detection of yeasts and molds. The impact of these media has been shown by different studies. Particularly in CF patients, the faster growth of associated bacteria often masks the presence of fungi which are more slowly growing. Therefore, the use of mycological culture media is mandatory for the detection of fungi from respiratory secretions from CF patients [21]. For some CF-related fungal species, even more specific media have shown benefit for the diagnosis, particularly when A. fumigatus is present in the sample. Examples for this is the SceSel+ agar for an enhanced cultivation rate of Scedosporium/Lomentospora species or the erythritol-chloramphenicol agar (ECA) medium for Exophiala spp. [25, 31,32,33]. Nevertheless, it must be kept in mind that in a clinical setting, the use of more specific media is associated with a higher cost for the laboratory and is time-consuming.
Beside improved isolation techniques, the knowledge about fungal pathogens is mandatory. Laboratories should know the limits of their skills and especially for special diagnostic questions, e.g., colonization with rare bacteria or filamentous fungi, they should utilize the assistance of specialized laboratories in this field [34]. If an in vitro antifungal susceptibility testing is necessary for the filamentous fungi that are detected, this should be done in reference centers or laboratories that are familiar with this.
In our retrospective study, we saw great variations in fungal diversity and quantity. Each center showed a unique profile with an own local climate, regional biocenosis and even a different hygiene management in the wards or in the outpatient center of their clinic.
Candida Species
In a cross-sectional analysis of the European Epidemiologic Registry of Cystic Fibrosis, positive sputum cultures for Aspergillus spp. and Candida spp. were associated with 5–10% lower forced expiratory volume in one second (FEV1) at all ages [35]. No decline in FEV1 was found in a single-center study in the UK [36]. It can be assumed that there is an ecological niche for Candida spp. in the respiratory tract due to recurrent antibiotic treatment courses against bacterial pathogens, e.g., Pseudomonas spp. and other non-fermenting rods, or to corticosteroid treatments. If so, it is finally the consequence of bacterial lung deterioration, through antibiotic treatment, that the recovery of yeasts from sputum samples is higher in patients with a lower FEV1. In a study from Norway, it was shown that children with CF had the highest prevalence of C. albicans, especially when treated with amoxicillin, azithromycin or third-generation cephalosporins [37].
Among Candida species, prevalence rates in Germany differed from 38 to 78% for C. albicans and 12% for Candida dubliniensis or 9% for Candida glabrata, to 3% for Candida parapsilosis and 2% for Candida lusitaniae or only 1% for Candida krusei [38, 39]. In Austria, they found C. albicans (74%), followed by C. dubliniensis (17%), C. parapsilosis (12%), C. glabrata (7%), C. lusitaniae (6%) and C. krusei (1%) [21]. In a retrospective study from South Italy, the prevalence rate was C. albicans 19.1%, followed by C. glabrata (0.5%) [40]. In a culture and molecular approach to detect fungal species in CF patients from Northern Ireland, the cultivation rates were 44% for C. albicans and 27% for C. dubliniensis, whereas they were only 3.9% for C. parapsilosis and 1.3% for C. glabrata [41].
In our data setting, the mainly cultured fungal species in respiratory samples were Candida spp., with a majority of C. albicans. As already seen in other studies, C. dubliniensis, C. parapsilosis and C. glabrata were following with wide variations in their respective frequency from one center to another (Table 3).
Despite the widespread use of azole antifungals, we could not detect any rising count of C. glabrata or C. krusei, which may be selected under azole treatment because of their reduced susceptibility.
Aspergillus fumigatus
As mentioned before, A. fumigatus is the main clinically relevant filamentous fungus for CF patients. It may be responsible for various diseases, including asthma, bronchitis and aspergilloma, and may also cause invasive pulmonary infections in immunocompromised patients. According to previous published data, its prevalence rate ranges from 16 to 56.7% [19]. In recent studies, we found for single centers in Europe colonization rates of 29% in Germany, 78.8% in Austria, 43.5–88.9% in the UK, 8.6–52.6% in France, 9.6% in Italy and 5.2% in Northern Ireland [21, 39,40,41].
This wide range of colonization rate was also seen in our setting, the rate differed between 3.4% to up to 42.4% of positive patients (in mean, Table 3). Further, this rate did not correlate with the colonization rate of C. albicans, which may support the hypothesis that this colonization status depends more on diagnostic or geographic/climate differences than on different antifungal regimens.
Concerning the major clinical manifestation ABPA and A. fumigatus bronchitis, the data from the ECFS show an age dependency: In young childhood, it is below 5% and increases up to 10.3% in adolescents [22].
Scedosporium and Lomentospora Species
For Scedosporium and Lomentospora species in patients with CF, the prevalence rate in a multicenter prospective study, Germany varied from 0.0 to 10.5%, despite the use of the selective culture medium SceSel+ [42]. In other European countries, it was reported a prevalence rate for Scedosporium sp. of 3.9% in Northern Ireland, of 10.6% in Austria (Lomentospora sp. 0.9%) and of 8.6% in France [21, 39, 41, 43]. In France, geographic discrepancies and the dependence from human-impacted areas for these species have been clearly shown [44, 45].
These regional variations in the epidemiology were also seen in our analysis: Prevalence rates differed from 0.1 to 12.1% (in mean) between the participating centers (Table 3).
Exophiala (Wangiella) dermatitidis
The black yeast Exophiala dermatitidis is one of the fungi for which differences in cultivation methods, especially the time of culturing (> 10 days) and the use of a selective medium, may influence the detection rate [32, 33]. Despite this, we found in Europe different published frequencies: in Sweden up to 19%, Northern Ireland 3.9%, Germany 3 to 9%, Austria 9% and France 6% [19, 21, 25, 33, 39, 41, 46, 47]. Data from our retrospective analysis showed colonization rates (in mean) between 0.2 to 18.3% (Table 3).
Trichosporon spp.
Data concerning the epidemiology of T. mycotoxinivorans are scarce. The first fulminant reported case in a CF patient was published in 2009 [27]. In a German study, the positive patient rate was below 0.6% and kept stable over a 5-year period [39]. In Northern Ireland, the rate for Trichosporon sp. was 1.3% [41]. In our survey, only rarely T. mycotoxinivorans was detected and no emerging counts were observed.
Rasamsonia (Geosmithia) argillacea
The role of this filamentous fungus, which is often misidentified as Penicillium sp. or Paecilomyces sp., is still unclear. Rarely, this fungus is cultivated in CF patients, and usually no acute clinical deterioration is observed [48]. In published data, the prevalence rate varies from 0.3% in Germany to 4.4% in Austria [21, 39]. In our analysis of the 9 CF centers, this species was rarely found in only 6 centers.
Other Species
Several other fungal species have been identified from the respiratory tract of CF patients in participating centers: non-fumigatus Aspergillus species, Paecilomyces sp., Fusarium sp., Cryptococcus sp. etc. These species can be clinically relevant in single cases, but in a general view up to the current knowledge, they are not CF-related pathogens and we have no suspicion for an epidemiological arising of these rare species.
Relation Between Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Genotype and Infections
The great variability from one center to another in the detected fungal species and their respective frequencies may be due to differences in the populations that are followed-up in each center (children or adults) and in the procedures used for mycological examination, or as above mentioned to climatic differences or to differences in hygiene management. Nevertheless, one may also consider the influence of the CFTR genotype. Due to our heterogeneous dataset, we cannot, however, conclude any correlation between CFTR mutations and a specific risk for a fungal infection. Data from the European Cystic Fibrosis Society Patient Registry for European CF patients have shown a large majority for the F508del mutation with more than 80% [49]. Nevertheless, the distribution and frequency of the other mutations within the CFTR gene are highly variable.
Alteration in CFTR function correlates with infection with a variety of respiratory pathogens. This has already been shown for patients with residual CFTR function and infection with P. aeruginosa and A. fumigatus [50]. The reduced clearance of Aspergillus spores in the CF airways emphasizes the relation between CFTR mutation and fungal infections [51]. Further, during treatment with ivacaftor for patients with the G551D mutation, a reduction of 53% for Aspergillus colonization rate has been observed [52]. With the new CFTR modulators, we will probably have a new epidemiology in a short time and further studies are urgently needed to identify CF patients at risk for fungal infections.
Conclusion
Published data concerning the epidemiology and clinical relevance of fungal species are rare. Here, we tried to give a short summary on published data from European countries and to show local data from different European CF centers. As mentioned before, the heterogeneity in diagnostic, treatment, environment and hygiene management for CF patients in each clinic or laboratory influences the detection and occurrence of fungal species. Nevertheless, 66,616 samples from 3235 CF patients were included in this retrospective analysis and 23,696 fungi were identified.
So each CF-specialized clinic should know its respective frequencies of occurrence for fungal species. This might lead to a deeper knowledge of the clinical impact of some fungal species and to improved diagnostic and treatment options for CF patients. Further correlations between CFTR genotype, bacterial colonization and fungal species colonizing the airways will help us to identify more rapidly patients at high risk for suffering from allergic diseases or invasive fungal infections.
References
Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2:527–38.
Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–72.
McCormick J, Mehta G, Olesen HV, et al. Comparative demographics of the European cystic fibrosis population: a cross-sectional database analysis. Lancet. 2010;375:1007–13.
Burgel PR, Bellis G, Olesen HV, et al. Future trends in cystic fibrosis demography in 34 European countries. Eur Respir J. 2015;46:133–41.
Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J. 2007;29:522–6.
Johnson C, Butler SM, Konstan MW, Morgan W, Wohl ME. Factors influencing outcomes in cystic fibrosis: a center-based analysis. Chest. 2003;123:20–7.
Mahadeva R, Webb K, Westerbeek RC, et al. Clinical outcome in relation to care in centers specialising in cystic fibrosis: cross sectional study. BMJ. 1998;316:1771–5.
Conway S, Balfour-Lynn IM, De Rijcke K, et al. European Cystic Fibrosis Society standards of care: framework for the cystic fibrosis center. J Cyst Fibros. 2014;13(Suppl 1):S3–22.
Eichinger M, Heussel CP, Kauczor HU, Tiddens H, Puderbach M. Computed tomography and magnetic resonance imaging in cystic fibrosis lung disease. J Magn Reson Imaging. 2010;32:1370–8.
Renz DM, Scholz O, Bottcher J, et al. Comparison between magnetic resonance imaging and computed tomography of the lung in patients with cystic fibrosis with regard to clinical, laboratory, and pulmonary functional parameters. Investig Radiol. 2015;50:733–42.
Wielputz MO, Puderbach M, Kopp-Schneider A, et al. Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med. 2014;189:956–65.
Geller DE, Kaplowitz H, Light MJ, Colin AA. Allergic bronchopulmonary aspergillosis in cystic fibrosis: reported prevalence, regional distribution, and patient characteristics. Scientific advisory group, investigators, and coordinators of the epidemiologic study of cystic fibrosis. Chest. 1999;116:639–46.
FitzPatrick MEB, Bilton D, Perrin F, Westaby D, Simmonds NJ. 245 A 10-year retrospective study of cystic fibrosis patients with distal intestinal obstruction syndrome (DIOS). J Cyst Fibros. 2013;12:S111.
Houwen RH, van der Doef HP, Sermet I, et al. Defining DIOS and constipation in cystic fibrosis with a multicenter study on the incidence, characteristics, and treatment of DIOS. J Pediatr Gastroenterol Nutr. 2010;50:38–42.
Plant BJ, Goss CH, Plant WD, Bell SC. Management of comorbidities in older patients with cystic fibrosis. Lancet Respir Med. 2013;1:164–74.
Quittner AL, Abbott J, Georgiopoulos AM, et al. International Committee on Mental Health in Cystic Fibrosis: cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus statements for screening and treating depression and anxiety. Thorax. 2016;71:26–34.
Quittner AL, Goldbeck L, Abbott J, et al. Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: results of The International Depression Epidemiological Study across nine countries. Thorax. 2014;69:1090–7.
Zhou J, Garber E, Desai M, Saiman L. Compliance of clinical microbiology laboratories in the United States with current recommendations for processing respiratory tract specimens from patients with cystic fibrosis. J Clin Microbiol. 2006;44:1547–9.
Pihet M, Carrère J, Cimon B, et al. Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis—a review. Med Mycol. 2009;47:387–97.
Liu JC, Modha DE, Gaillard EA. What is the clinical significance of filamentous fungi positive sputum cultures in patients with cystic fibrosis? J Cyst Fibros. 2013;12:187–93.
Masoud-Landgraf L, Badura A, Eber E, et al. Modified culture method detects a high diversity of fungal species in cystic fibrosis patients. Med Mycol. 2014;52:179–86.
Armstead J, Morris J, Denning DW. Multi-country estimate of different manifestations of aspergillosis in cystic fibrosis. PLoS ONE. 2014;9:e98502.
Mastella G, Rainisio M, Harms HK, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis. A European epidemiological study. Epidemiologic Registry of Cystic Fibrosis. Eur Respir J. 2000;16:464–71.
Maturu VN, Agarwal R. Prevalence of Aspergillus sensitization and allergic bronchopulmonary aspergillosis in cystic fibrosis: systematic review and meta-analysis. Clin Exp Allergy. 2015;45:1765–78.
Kondori N, Gilljam M, Lindblad A, et al. High rate of Exophiala dermatitidis recovery in the airways of patients with cystic fibrosis is associated with pancreatic insufficiency. J Clin Microbiol. 2011;49:1004–9.
Kondori N, Lindblad A, Welinder-Olsson C, Wenneras C, Gilljam M. Development of IgG antibodies to Exophiala dermatitidis is associated with inflammatory responses in patients with cystic fibrosis. J Cyst Fibros. 2014;13:391–9.
Hickey PW, Sutton DA, Fothergill AW, et al. Trichosporon mycotoxinivorans, a novel respiratory pathogen in patients with cystic fibrosis. J Clin Microbiol. 2009;47:3091–7.
Hirschi S, Letscher-Bru V, Pottecher J, et al. Disseminated Trichosporon mycotoxinivorans, Aspergillus fumigatus, and Scedosporium apiospermum coinfection after lung and liver transplantation in a cystic fibrosis patient. J Clin Microbiol. 2012;50:4168–70.
Martinez Muniz FB, Martinez Redondo M, Prados Sanchez C, Garcia Rodriguez J. Chronic lung infection caused by Trichosporon mycotoxinivorans and Trichosporon mucoides in an immunocompetent cystic fibrosis patient. Arch Bronconeumol. 2016;52:400.
Shah AV, McColley SA, Weil D, Zheng X. Trichosporon mycotoxinivorans infection in patients with cystic fibrosis. J Clin Microbiol. 2014;52:2242–4.
Rainer J, Kaltseis J, de Hoog SG, Summerbell RC. Efficacy of a selective isolation procedure for members of the Pseudallescheria boydii complex. Antonie Van Leeuwenhoek. 2008;93:315–22.
De Hoog GS, Haase G. Nutritional physiology and selective isolation of Exophiala dermatitidis. Antonie Van Leeuwenhoek. 1993;64:17–26.
Haase G, Skopnik H, Groten T, Kusenbach G, Posselt HG. Long-term fungal cultures from sputum of patients with cystic fibrosis. Mycoses. 1991;34:373–6.
Hogardt M, Ulrich J, Riehn-Kopp H, Tummler B. EuroCareCF quality assessment of diagnostic microbiology of cystic fibrosis isolates. J Clin Microbiol. 2009;47:3435–8.
Navarro J, Rainisio M, Harms HK, et al. Factors associated with poor pulmonary function: cross-sectional analysis of data from the ERCF. European Epidemiologic Registry of Cystic Fibrosis. Eur Respir J. 2001;18:298–305.
Baxter CG, Moore CB, Jones AM, Webb AK, Denning DW. IgE-mediated immune responses and airway detection of Aspergillus and Candida in adult cystic fibrosis. Chest. 2013;143:1351–7.
Gammelsrud KW, Sandven P, Hoiby EA, et al. Colonization by Candida in children with cancer, children with cystic fibrosis, and healthy controls. Clin Microbiol Infect. 2011;17:1875–81.
Valenza G, Tappe D, Turnwald D, et al. Prevalence and antimicrobial susceptibility of microorganisms isolated from sputa of patients with cystic fibrosis. J Cyst Fibros. 2008;7:123–7.
Ziesing S, Suerbaum S, Sedlacek L. Fungal epidemiology and diversity in cystic fibrosis patients over a 5-year period in a national reference center. Med Mycol. 2016;54:781–6.
Montagna MT, Barbuti G, Paglionico F, et al. Retrospective analysis of microorganisms isolated from cystic fibrosis patients in Southern Italy, 2002–2010. J Prev Med Hyg. 2011;52:209–14.
Nagano Y, Elborn JS, Millar BC, et al. Comparison of techniques to examine the diversity of fungi in adult patients with cystic fibrosis. Med Mycol. 2010;48(Suppl 1):S166–76.
Sedlacek L, Graf B, Schwarz C, et al. Prevalence of Scedosporium species and Lomentospora prolificans in patients with cystic fibrosis in a multicenter trial by use of a selective medium. J Cyst Fibros. 2015;14:237–41.
Cimon B, Carrère J, Vinatier JF, et al. Clinical significance of Scedosporium apiospermum in patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis. 2000;19:53–6.
Rougeron A, Schuliar G, Leto J, et al. Human-impacted areas of France are environmental reservoirs of the Pseudallescheria boydii/Scedosporium apiospermum species complex. Environ Microbiol. 2015;17:1039–48.
Zouhair R, Rougeron A, Razafimandimby B, et al. Distribution of the different species of the Pseudallescheria boydii/Scedosporium apiospermum complex in French patients with cystic fibrosis. Med Mycol. 2013;51:603–13.
Horré R, Schaal KP, Siekmeier R, et al. Isolation of fungi, especially Exophiala dermatitidis, in patients suffering from cystic fibrosis. A prospective study. Respiration. 2004;71:360–6.
Lebecque P, Leonard A, Huang D, et al. Exophiala (Wangiella) dermatitidis and cystic fibrosis—prevalence and risk factors. Med Mycol. 2010;48(Suppl 1):S4–9.
Matos T, Cerar T, Praprotnik M, Krivec U, Pirs M. First recovery of Rasamsonia argillacea species complex isolated in adolescent patient with cystic fibrosis in Slovenia—case report and review of literature. Mycoses. 2015;58:506–10.
De Boeck K, Zolin A, Cuppens H, Olesen HV, Viviani L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros. 2014;13:403–9.
Green DM, McDougal KE, Blackman SM, et al. Mutations that permit residual CFTR function delay acquisition of multiple respiratory pathogens in CF patients. Respir Res. 2010;11:140.
Chaudhary N, Datta K, Askin FB, Staab JF, Marr KA. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to Aspergillus and resultant pulmonary inflammation. Am J Respir Crit Care Med. 2012;185:301–10.
Heltshe SL, Mayer-Hamblett N, Burns JL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. 2015;60:703–12.
Author information
Authors and Affiliations
Contributions
CS, JPB, WB, VC, HD, EGGP, RC, EF, YG, NK, TM, ER, SZ, LS were involved in the manuscript design, data selection and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
None of the authors has any potential financial or non-financial conflicts of interests related to this manuscript.
Rights and permissions
About this article

Cite this article
Schwarz, C., Bouchara, JP., Buzina, W. et al. Organization of Patient Management and Fungal Epidemiology in Cystic Fibrosis. Mycopathologia 183, 7–19 (2018). https://doi.org/10.1007/s11046-017-0205-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11046-017-0205-x