
Abstract
Porphyrins have long been proposed as key ingredients in the emergence of life yet plausible routes for forming their essential pyrrole precursor have heretofore not been identified. Here we show that the anaerobic reaction of δ-aminolevulinic acid (ALA, 1–5 mM) with the β-ketoester methyl 4-methoxyacetoacetate (2–40 mM) in water (pH 5–7) at 70–100°C for >6 h affords the porphyrinogen, which upon chemical oxidation gives the corresponding porphyrin in overall yield of up to 10%. The key intermediate is the α-methoxymethyl-substituted pyrrole, which undergoes tetramerization and macrocycle formation under kinetic control. The resulting type-I porphyrin bears four propionic acid and four carbomethoxy groups, is distinct from porphyrins (e.g., uroporphyrin or coproporphyrin) derivable from ALA alone via the extant universal biosynthetic path to tetrapyrroles, and is photoactive upon assembly into cationic micelles in aqueous solution. The simple self-organization of eight acyclic molecules into a tetrapyrrole macrocycle, from which a porphyrin is derived that is photoactive in lipid assemblies, augurs well for the spontaneous origin of catalysts and pigments essential for prebiotic metabolism and proto-photosynthesis.
Similar content being viewed by others


Introduction
The emergence of an energy-utilizing metabolism is likely to have been a critical milestone in prebiotic chemistry. Regardless of whether the earliest self-replicating systems were heterotrophic or (photo)autotrophic in nature (Lazcano and Miller 1999; Eschenmoser 2007a), the underlying metabolism undoubtedly relied on extensive redox chemistry (Canfield et al. 2006). As such, the formation of porphyrinogens, progenitors of porphyrins and diverse hydroporphyrins, would have yielded a source of extraordinarily versatile redox and photoredox catalysts (Mauzerall 1976, 1992, 1998; Eschenmoser 1986, 1988). Indeed, tetrapyrrole macrocycles are attractive if not essential in a variety of prebiotic chemistries, including proto-photosynthesis (Krasnovsky 1976; Mauzerall 1978, 1990, 1992, 1998; Mercer-Smith et al. 1984, Mercer-Smith and Mauzerall 1985; Olson and Pierson 1987). In this regard, a prebiotically plausible route to tetrapyrrole molecules has yet to be demonstrated (Miller 1986, 1987, 1998), at least in the context of robust reaction of simple species in anaerobic aqueous solution to give measurable quantities of stable product. This dearth exists despite extensive studies in the half-century since Miller’s seminal spark-discharge experiments (for a review of early work, see: Wnuk 1983; for more recent work, see: Scott 1997; Aylward and Bofinger 2005; Ksander et al. 1987; Meierhenrich et al. 2005).
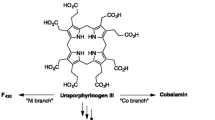
Uroporphyrin has been proposed as a likely prebiotic porphyrin (Granick 1965; Mauzerall 1978, 1992, 1998; Mercer-Smith et al. 1984; Mercer-Smith and Mauzerall 1985), given its water solubility, typical porphyrin spectral and (photo)redox properties, and ready formation by oxidation of uroporphyrinogen, the universal macrocyclic precursor (Battersby et al. 1980) in the biosynthesis of tetrapyrroles (heme, chlorophylls, corrins) (Fig. 1). Studies to mimic the biosynthesis of uroporphyrinogen (and thus form uroporphyrin) under plausible prebiotic conditions were first carried out by Mauzerall (Mauzerall and Granick 1958, Mauzerall 1960a, b), who showed that (i) porphobilinogen undergoes self-condensation in acidic solution in the absence of enzymes to give uroporphyrinogen (Mauzerall 1960b), a thermodynamically stable macrocycle (Mauzerall 1960a), (ii) under reversible conditions, the uroporphyrinogen isomer (III) that is expected in statistical abundance is observed preferentially (Mauzerall 1960a), and (iii) the oxidation of uroporphyrinogen to uroporphyrin can be achieved photochemically (Mercer-Smith and Mauzerall 1985), with autocatalytic features owing to the photochemistry of uroporphyrin (Mauzerall and Granick 1958). Thus, the seemingly complex steps of porphyrinogen formation and subsequent oxidation occur readily under simple conditions.

Universal biosynthesis of tetrapyrrole macrocycles
By contrast, examination of earlier steps, namely the formation of porphobilinogen from its universal biosynthetic precursor, ALA, has heretofore failed to identify prebiotically plausible conditions for this deceptively simple dimerization despite extensive studies over the ensuing half-century (for overviews, see Butler and George 1992; Stauffer et al. 2001; Chaperon et al. 2003). ALA in solution readily undergoes self-condensation to form the dihydropyrazine and pseudo-porphobilinogen rather than the required porphobilinogen (Fig. 2) (Granick and Mauzerall 1958; Butler and George 1992). While dihydropyrazine formation presumably is reversible under anaerobic conditions (facile oxidation to the pyrazine occurs in air), the dominant pathway to pseudo-porphobilinogen stems from the greater reactivity of the methylene at the C5 versus C3 site (Butler and George 1992, 1993, 1994). A route to a pyrrole, different from porphobilinogen yet that exhibits the functional features of porphobilinogen, would offer a chemical model for the genesis of tetrapyrrole macrocycles under plausible prebiotic conditions. The requisite functional features of such a pyrrole include (1) self-constitution from simple starting materials under simple conditions, (2) some degree of water-solubility, and (3) self-condensation to give the porphyrinogen. In this paper, we explore one example of such a route.

Self-condensation of ALA without enzymes affords the dihydropyrazine and pseudo-porphobilinogen rather than porphobilinogen
Methods
Handling of ALA
The stability of ALA in aqueous solution has been much studied in the past 15 years owing to the use of ALA as an in vivo precursor for photodynamic therapy. The studies encompass the effects of pH, concentration, oxygen, and temperature (Novo et al. 1996; Elfsson et al. 1998; Bunke et al. 2000; de Blois et al 2002; Hunter et al. 2005; for a review, see Donnelly et al. 2007). ALA is stable for years upon storage in solid form as the hydrochloride salt, and at least months upon dissolution in water given that the pH of the resulting aqueous stock solution is quite low (pH ~2.3). The ALA samples were obtained as the hydrochloride salt from one of several commercial sources (Aldrich, Sigma, or Alfa Aesar) and examined by 1H NMR spectroscopy to confirm integrity prior to use. The stock solutions of ALA (0.1 or 0.5 M in distilled water) were stored in frozen form at −15°C when not in use. Aliquots of the ALA stock solution were added on the open benchtop to frozen solutions containing all other components so as to minimize exposure of ALA in neutral solution to molecular oxygen. The reactions were carried out under anaerobic conditions either through use of Schlenk-line procedures (5–100 mL reactions) or an enclosed reaction chamber (~1 mL reactions) for investigating multiple reactions in parallel (see ESM for further information).
Solutions
The buffer solutions for the experiments were prepared as concentrated stock solutions (2M for sodium acetate, 0.5 M for sodium phosphate). The pH values stated herein were measured with a pH meter for the target solution (typically 100 mM buffer) at room temperature (21°C), and are typically within ±0.1 pH unit for the target solution (see ESM for further information). The expected change in pH with temperature is slight, given that the change in pKa value over the range examined (21–95°C) is expected to be <0.2 for the acetate buffer (Olofsson 1984; Goldberg et al. 2002) and the phosphate buffer (Bates 1962).
The micellar solutions were prepared by dissolving the desired quantity of the surfactant in aqueous sodium phosphate (50 mM, pH 7.0). In each case the quantity of surfactant including sodium dodecyl sulfate (10 mM), cetyl trimethylammonium bromide (10 mM), or Triton X-100 (2% w/v) was above the critical micelle concentration.
Quantitation of Porphyrinogen Formation
The oxidation was carried out with slight modification from the procedure described by Mauzerall and Granick (1958). The following is the procedure employed for reactions at 5 mM ALA. An aliquot (50 μL) of the reaction mixture was removed, and on the open benchtop exposed to air, the aliquot was added into 250 μL of water. The resulting sample was then treated with 10 μL of a freshly prepared solution of I2 (50 mM) in absolute ethanol. The sample was gently mixed by hand for a few seconds, and then transferred in its entirety to 2.7 mL of DMF in an absorption cuvette. The resulting solution was treated with 100 μL of Na2S2O3 (100 mM in water). (For reactions that were carried out with phosphate buffer, 300 μL of water was then added to the cuvette to dissolve any inorganic precipitates.) The absorption spectrum was then recorded. The molar absorption coefficient employed was 300,000 M−1cm−1 on the basis of reported data including 284,000 M−1cm−1 for the type-I porphyrin 2,7,12,17-tetracarboethoxy-3,8,13,18-tetramethylporphyrin in CHCl3 (Kleinspehn et al. 1966). Values for the type II-IV regioisomers were 287,000, 251,000, and 247,000 M−1cm−1, respectively (Kleinspehn et al. 1966). The yield of porphyrin was assessed by the intensity of the Soret peak after correcting for the baseline (Lindsey et al. 1987). This procedure differs from the Mauzerall and Granick procedure in (i) use of ethanolic rather than aqueous iodine (Abe and Konaka 1989), and (ii) use of DMF as a solvent for absorption spectroscopy. The higher solubility of I2 in ethanol (670 mM; Hildebrand et al. 1950) than that in water (1.3 mM; Hartley and Campbell 1908) facilitates use of a concentrated stock solution. The use of DMF enables solubilization of the resulting porphyrin, which exhibits lower aqueous solubility than uroporphyrins (vide infra). Note that for the reaction with 5 mM ALA, the stoichiometric requirement for I2 (on the basis of 100% yield of porphyrinogen) corresponds to 7.5 μL of the 50 mM stock solution.
Reaction Temperatures
The temperature was assessed using a certified NIST-traceable thermometer. In each case, the reported reaction temperatures refer to the temperature of the contents inside the reaction flask, which often was as much as 10°C lower than the temperature of the external heat bath.
ESI-MS Analysis
High resolution exact mass measurements were carried out using electrospray ionization (ESI) on an Agilent Technologies 6210 LC-TOF mass spectrometer. Samples were analyzed via a 5 μL flow injection at 300 μL/min in a water:methanol mixture (25:75 v/v) containing 0.1% formic acid. The mass spectrometer was operated in positive-ion mode. Porphyrins in very crude samples have been analyzed under similar conditions (Danton and Lim 2006).
Deuterium Exchange Studies
Deuteriation of β-ketoesters was examined using neat CD3OD at 22°C (relaxation time = 3 s). Because deuterium exchange at the 2-position in D2O was too rapid to follow the exchange, CD3OD was used instead. In D2O, no deuterium exchange was observed at the 4-position with either methyl acetoacetate or methyl 4-methoxyacetoacetate (up to 24 h). An exemplary procedure is as follows: A sample of β-ketoester (10 μL) was placed in an NMR tube, and treated with CD3OD (600 μL) immediately before the measurement (t = 0). The resulting concentration of β-ketoester was on the order of 0.1 M. The 1H NMR spectrum was recorded at 22°C over time, and the resonances from the 2- and 4-positions were integrated. The deuterium exchange processes were followed relative to the non-exchanging protons (methyl ester). Only one enol form, wherein the double bond is located between the 2-and 3-positions, was observed. Over the course of the exchange study, the molar ratio of enol to keto form was 0.07 to 0.20 for methyl acetoacetate, and 0.07 to 0.22 for methyl 4-methoxyacetoacetate. The process of exhaustive deuteriation at the 2-position involves two steps: (1) the deuteriation of CH2 to form CHD, and (2) the deuteriation of CHD to form CD2. The formation of CHD was observed by the appearance of a triplet (~0.02 ppm shifted upfield compared to the resonance from CH2). The kinetics of these two steps can be calculated separately; however, we have ignored secondary deuterium kinetic isotope effects, given that the effects are considered to be small. Additionally, it was very difficult to integrate separately the resonance from CH2 and CDH, which have similar chemical shifts under the experimental condition (300 MHz). Thus, the half-life obtained represents the sum of the two steps.
Fluorescence Analysis
The fluorescence spectra were obtained at room temperature with excitation and emission slit widths of 1.5 nm, and 1 nm/sec sweep rates. The spectra were corrected for excitation intensity and instrument response on emission. The samples were excited at the peak of the Soret band with samples having absorbance values <0.12 in 1-cm pathlength cuvettes. The emission was integrated in each case over the range 550–790 nm.
Synthesis of 4-(2-Carboxyethyl)-3-methoxycarbonyl-2-methylpyrrole (5)
Following established procedures (Mauzerall and Granick 1956, Butler and George 1993), samples of methyl acetoacetate (95.5 mg, 0.82 mmol) and δ-aminolevulinic acid hydrochloride (138 mg, 0.82 mmol) were added to a solution of 1.6 mL of water and 1.6 mL of acetate buffer (2 M, composed of equal amounts of sodium acetate and acetic acid) in a conical vial. The reaction mixture was heated at 90°C with stirring exposed to air for 1 h. The reaction mixture was allowed to cool, whereupon white crystals formed. The mixture was acidified by addition of 20 drops of 2N HCl. After standing overnight at 0–5°C, the mixture was filtered. The filtered material was washed with water and dried under vacuum over KOH pellets to give white crystals (73.7 mg, 42%): mp 178–179°C; 1H NMR (400 MHz, DMSO-d 6 ) δ 2.35 (s, 3H), 2.42–2.43 (m, 2H), 2.76–2.80 (m, 2H), 3.67 (s, 3H), 6.39–6.41 (m, 1H), 10.90–11.02 (br, 1H), 11.90–12.00 (br, 1H); 13C NMR (100 MHz, DMSO-d 6 ) δ 13.5, 22.2, 34.6, 50.1, 108.6, 114.4, 123.5, 135.5, 165.5, 174.3; ESI-MS obsd m/z 212.0915 for the protonated quasimolecular ion, corresponding to 211.08422 for the neutral species, calcd 211.08446 (C10H13NO4).
Synthesis of 4-Methylpyrrole-3-carboxylic acid (6)
A sample (0.766 g, 5.00 mmol) of known ethyl 4-methylpyrrole-3-carboxylate (Cheng et al. 1976) was treated with an ethanolic solution of KOH (20% in EtOH, 5 mL). The resulting mixture was refluxed for 2 h. Water (5 mL) was added and the volatile material (mostly EtOH) was removed by rotary evaporation under reduced pressure. The resulting solution was acidified with concentrated HCl (to pH ~2) yielding a light yellow precipitate, which was filtered, washed with a small amount of cold water and dried under vacuum (0.443 g, 71%, purity ~95%): mp 190–193°C (dec.), lit. mp 190–191°C (Rapoport and Bordner 1964); 1H NMR (400 MHz, DMSO-d 6 ) δ 2.14 (s, 3H), 6.54–6.56 (m, 1H), 7.24–7.26 (m, 1H), 10.90–11.10 (br, 1H), 11.48 (s, 1H); 13C NMR (100 MHz, DMSO-d 6 ) d 11.7, 113.9, 117.6, 119.3, 124.3, 166.3; ESI-MS obsd m/z 126.05473 for the protonated quasimolecular ion, corresponding to 125.04746 for the neutral species, calcd 125.04768 (C6H7NO2).
Quantitative Assay for Pyrrole Formation
The reaction of ALA and methyl acetoacetate was carried out as done for ALA and methyl 4-methoxyacetoacetate (1) in the porphyrinogen-forming reactions with the following changes: (i) the reaction volume was typically 1 mL; and (ii) the temperature was as stated below. Reactions were run in triplicate. At the end of the reaction period, a sample of the internal standard (6) was added to each of the reaction vessels. The amount of standard added corresponded to a yield of 10%, 20%, or 30% for the acetate-buffered reactions (pH 5.0), or 25%, 50%, 75%, or 100% for the phosphate-buffered samples (pH 7.0). After adding the internal standard, each reaction mixture was frozen and lyophilized. The resulting residue in each case was treated with 0.6 mL of DMSO-d 6 (99.9% isotopic abundance) and the resulting suspension was vortexed. A small amount of residue was undissolved. The mixture was decanted by pipette into an NMR tube for 1H NMR analysis. The samples obtained from the reaction of ALA and methyl acetoacetate afforded clean spectra particularly in the region for analysis. The residue was dissolved in D2O and analyzed by 1H NMR spectroscopy to verify that no significant quantity of pyrrole had been left behind upon extraction.
The quantitation of pyrrole formation was established through use of mock reaction mixtures prepared from authentic samples of the target pyrrole 5 and the internal standard 6. The 1H NMR spectrum was relatively clean, and the spectral region of 5–10 ppm was devoid of peaks with the exception of the two multiplets for 6 and the peak (6.37 ppm) of 5. The peak intensities determined in the mock samples were used to correct the raw data obtained in the experimental samples.
Results
A Porphyrinogen Directly from Acyclic Materials
The route that we investigated entails the condensation of ALA with a 4-substituted β-ketoester (methyl 4-methoxyacetoacetate, 1) as shown in Fig. 3. The nature of the 4-substituent of the β-ketoester is critical, as this substituent becomes the leaving group at the α-methyl site of the pyrrole (2) for self-condensation to give the porphyrinogen, yet must not interfere with the pyrrole-forming process. Moreover, a key feature is that the β-ketoester 1 cannot self-condense, unlike that of ALA. This approach builds on the well-known reaction of aminoketones, including ALA, with β-dicarbonyl compounds to give the corresponding pyrroles (Corwin 1950; Mauzerall and Granick 1956). In this Knorr-like reaction, initial formation of the conjugated enamine (an enaminone) focuses the subsequent aldol reaction at the central methylene unit (C2). A related approach where ALA is reacted with a 4-unsubstituted β-ketoester (e.g., acetoacetate) followed by oxidation of the resulting α-methylpyrrole group was briefly mentioned by Mauzerall as a possible prebiotic route to porphyrins (Mauzerall 1990, 1998). To our knowledge, the only report of a synthetic pyrrole-forming reaction with a 4-substituted β-ketoester employed ethyl 4-phthalimidoacetoacetate with ALA, but without information concerning conditions, yield data, or chemical reactivity of the resulting pyrrole (Franck and Stratmann 1981).

De novo formation of α-methoxymethyl-substituted pyrrole 2, which self-condenses to give the porphyrinogen 3. Subsequent oxidation gives the porphyrin 4
β-Ketoesters including 1 (Comer and Murphy 2003) are known to react with primary amines in organic solvents to give the enaminone (Greenhill 1977). Here, we quantitated the relative reactivity in neutral protic media of the sites both within 1 and versus those of the unsubstituted methyl acetoacetate. A 1H NMR study in D2O of 1 and methyl acetoacetate at 22°C showed rapid and quantitative exchange at the C2-position with no exchange at the C4-position over the 24 h timecourse. In neat CD3OD at 22°C, a steady decrease over a few hours was observed in the intensity of the resonances from the protons at the 2-position, while the 4-position again remained intact over the duration of the experiment (up to 24 h). The half-life for complete deuterium exchange at the 2-position for 1 (t 1/2 = 211 min) and methyl acetoacetate (t 1/2 = 6.9 min) indicate that 1 has reactivity typical for β-ketoesters albeit slower than that of the unsubstituted analogue. Thus, β-ketoester 1 is expected to react at position C2 rather than C4.
The reaction of ALA (5 mM) with 1 (40 mM) in water (pH 5, 100 mM sodium acetate buffer) was carried out under anaerobic conditions at 90°C. The reaction mixture was faintly cloudy and only slightly discolored over the course of 24 h. A sample taken from the reaction after 3 h and examined by electrospray ionization mass spectrometry (ESI-MS) showed peaks characteristic of the two starting materials, the α-methoxymethyl-substituted pyrrole 2, α-methoxymethyl-substituted oligopyrromethane intermediates (dipyrromethane, tripyrrane, and bilane), and the porphyrinogen 3 (see ESM for spectra and conditions). The structures of the intermediates and the masses are displayed in Table 1. The good agreement in each case between the observed mass and the calculated mass (ΔM <5 ppm) provides confirmation of the elemental composition of the assigned specie. No oligopyrromethane species larger than the bilane were observed.

At 24 h the peaks due to the pyrrole and oligopyrromethanes had disappeared yet that for the porphyrinogen remained, with no peak observed for the corresponding porphyrin (Fig. 4a). The porphyrinogen and porphyrin differ by six mass units owing to saturation of the former macrocycle. Removal of an aliquot from the reaction mixture and treatment with I2 (aqueous ethanol), a relatively clean method for oxidizing porphyrinogens (Mauzerall and Granick 1958; Lin and Timkovich 1994), afforded the corresponding porphyrin 4 in 7.5% yield as determined by absorption spectroscopy. The ESI-MS spectrum following oxidation and purification exhibited a peak characteristic of the porphyrin (Fig. 4b). We note that a handful of pyrroles bearing α-alkoxymethyl groups have been used previously to prepare porphyrins, but in each case the critical feature for pyrrole self-condensation, the α-alkoxymethyl group, has been installed via separate synthetic steps on intact pyrroles (Hayes et al 1958; Eisner and Gore 1958; Kay 1962; Kobayashi et al. 1978; Kinoshita et al. 1992; Jeandon and Callot 1993; Thyrann and Lightner 1996 and references therein) rather than formed de novo as demonstrated here.

ESI-MS analysis of tetrapyrrole macrocycles. a Crude reaction mixture after 24 h at 90°C shows the sodium-cationized quasimolecular ion of the porphyrinogen 3 [calculated 859.26445 for (C40H44N4O16 + Na+)]. b Purified product following oxidation shows the protonated quasimolecular ion of the porphyrin 4 [calculated 831.23555 for (C40H38N4O16 + H+)]
Reaction Robustness
A series of experiments was carried out to define the reaction parameters. Each of the reactions in sets (i)-(vi) below was carried out using the following conditions with variations therefrom as specified: 5 mM ALA and 40 mM 1 (8 equiv) in water at pH 5 (100 mM sodium acetate buffer) for 27 h at 90°C under an anaerobic atmosphere.
-
(i)
The reaction at pH 4 or 5 (100 mM sodium acetate buffer) gave 3.3 or 7.5% yield, while pH 5, 6, 7, or 8 (100 mM sodium phosphate buffer) gave 4.4, 9.0, 4.8 or 0%, respectively.
-
(ii)
The reaction at pH 5 with sodium acetate buffer concentrations of 20, 100, or 500 mM gave a yield of 7.2, 7.5, or 10.1%, respectively.
-
(iii)
The reaction with ALA (5 mM) and 16, 8, 4, 2, or 1 equiv of 1 gave a yield of 8.3, 7.5, 7.4, 5.3, or 3.4%, respectively.
-
(iv)
The reaction with 1 mM ALA with 16, 8, 4, or 2 equiv of 1 gave a yield of 1.3, 1.0, 0.6, or ~0.2%, respectively.
-
(v)
The reaction at 20 or 50 mM ALA and 8 equiv of 1 gave extensive white precipitates and considerably lower yields than at 5 mM.
-
(vi)
The reaction under aerobic conditions for 24 h at 90°C gave the porphyrin in 1.0% yield (without use of any added chemical oxidant).
The pH-dependence of the yield is readily interpreted in terms of the individual steps on the reaction pathway. The decrease in yield at lower pH likely stems from protonation of ALA to give the non-nucleophilic ammonium salt, thereby suppressing enaminone formation. In this regard, the pKa of the ammonium group of ALA has been reported to be 8.2 (Granick and Mauzerall 1958), 8.9 (Butler and George 1992), 8.03 (Novo et al. 1996), and 8.05 (Elfsson et al. 1998). The decline in yield at higher pH likely stems from (i) the requirement for protonation in the dehydration step to give the pyrrole, and (ii) the requirement for protonation of the methoxy group as part of the pyrrole self-condensation process leading to the porphyrinogen. Indeed, the Knorr pyrrole reaction is known to proceed most efficaciously at near-neutral pH (Haley and Maitland 1951). Thus, the highest yield at intermediate pH values reflects a compromise between the competing pH requirements of the individual steps.
The use of a stoichiometric excess of the β-ketoester 1 versus ALA is possible because the β-ketoester cannot self-condense, whereas ALA does so to give the dihydropyrazine. An excess of 1 versus ALA should suppress the dimerization of the latter to give the dihydropyrazine. In fact, the yield increased from 3.4 to 8.3% upon an increase in stoichiometric ratio from 1:1 to 16:1. On the other hand, the use of higher overall concentrations of 1 and ALA resulted in the formation of white precipitates of unknown composition.
The reaction also was carried out at different temperatures with periodic monitoring to determine the timecourse. The results for reactions at ~60–90°C are shown in Fig. 5a. As the temperature was increased, the rate and yield also increased, reaching a maximum yield of ~0.3, ~1.4, ~5 or ~6–7% at 62, 73, 80 or 90°C, respectively. At 90°C the reaction was half-complete at 6–9 h and leveled off at 18 h. The reaction at reflux (100°C) was faster still, reached a maximal yield of approximately 9% within one day, and afforded a yield >5% over the course of one week (Fig. 5b). The robustness of the reaction path augurs well for its viability under prebiotic conditions, at least at elevated temperatures.

Reaction timecourse as a function of temperature. Each reaction employed 5 mM ALA and 40 mM 1 in 100 mM sodium acetate (pH 5) under anaerobic conditions. a Reactions over 1.5–2 days at 62°C (open squares), 73°C (closed squares), 80°C (open circles) and 90°C (closed circles). b Reaction over 1 week at 100°C
Although the early Earth is often considered to have been quite hot, recent studies have suggested more modest temperatures (Kasting and Howard 2006). To gain deeper insight into the requirement for high temperature for the reaction in Fig. 3, the pyrrole-forming reaction was examined at lower temperatures. The few reports available concerning Knorr pyrrole formation at room temperature each employed quite high concentrations (Haley and Maitland 1951; Tamura et al. 1971; Fabiano and Golding 1991) compared with the dilute-solution reaction of interest here. The anaerobic reaction of ALA (5 mM) and 1 (40 mM) in water (pH 5, 100 mM sodium acetate buffer) after 24 h at 50°C showed a peak in the ESI-MS spectrum due to the α-methoxymethyl-substituted pyrrole 2, while at 21°C the peak was very weak.
To quantitate the yield, we examined the reaction of ALA and methyl acetoacetate, which affords 4-(2-carboxyethyl)-3-carbomethoxy-2-methylpyrrole (5), a pyrrole that cannot self-condense (Fig. 6). The reaction of such β-dicarbonyl compounds is known to proceed rapidly (15–30 min) at 100°C to give the pyrrole; indeed, this generic reaction forms part of a standard assay for ALA in biological samples (Mauzerall and Granick 1956). To monitor the reaction, we developed an assay wherein the crude reaction sample was treated with an internal standard, the entire reaction mixture was lyophilized, and the resulting residue was treated with dimethylsulfoxide-d 6 (DMSO-d 6 ) for 1H NMR analysis. For the internal standard, we chose 4-methylpyrrole-3-carboxylic acid (6), because (i) the pyrrolic α-proton resonance(s) would be readily detected, (ii) the pyrrole-carboxylate salt is water-soluble and (iii) the compound is relatively non-volatile. The 1H NMR spectrum (DMSO-d 6 ) of the internal standard shows two readily discernible multiplets (δ 7.23–7.26; 6.52–6.57 ppm) corresponding to the two α-protons. Pyrrole 5 has been described in the literature but without any characterization data (Lüönd and Neier 1991, Lüönd et al. 1992). In each case, the pyrrole was prepared (following the procedure for which the homologous ethyl ester was prepared) by condensation of methyl acetoacetate with ALA (Fig. 6). Pyrrole 6 was prepared in 71% yield by saponification of the known ethyl 4-methylpyrrole-3-carboxylate (Cheng et al. 1976) with ethanolic KOH.

Synthesis of pyrrole 5, and the pyrrole (6) used as the internal standard in the pyrrole assay
After 24 h at 21°C, the reaction of ALA and methyl acetoacetate at pH 5.0 (100 mM sodium acetate) was ~10% complete whereas that at pH 7.0 (100 mM sodium phosphate) was ~80% complete. The rapid formation of the pyrrole in comparison with that of the porphyrinogen suggests that the presence of a better leaving group at the α-methyl position would facilitate porphyrinogen formation at lower temperature and at more neutral pH.
Porphyrin Structure and Properties
The reaction of ALA and 1 was carried out at 50-or 100-mL scale for characterization and spectroscopic studies of the porphyrin 4. Treatment of the crude mixture with acidic brine (Rimington and Benson 1967) and 2-butanone caused partitioning of the porphyrin into the organic phase, to which methanol was then added to give the porphyrin in relatively pure form as a crystalline powder. The 1H NMR spectrum of the porphyrin regardless of workup method exhibited predominantly a single peak for the methine protons (δ = 12.56 ppm in TFA-d or 10.15 in DMSO-d 6 ), consistent with the presence of largely if not exclusively the type-I isomer (see ESM for spectral data). We note that the identification of uroporphyrin and coproporphyrin isomers (which bear β-octaalkyl substituents) by 1H NMR spectroscopy has proved exceptionally difficult, given the very slight degree of shielding (e.g., 0.20 to 0.24 ppm) of flanking β-alkyl groups toward meso protons (Scheer and Katz 1975). This problem is quite generic for octaalkylporphyrins (Nguyen and Smith 1996). On the other hand, the deshielding effect of flanking β-carbonyl substituents is strong (up to 1 ppm; see Clezy and Liepa 1971, Clezy et al. 1972), and has enabled facile detection of porphyrin isomers wherein each pyrrole bears one β-alkyl and one β-carbonyl group (Chamberlin and LeGoff 1978). Similar NMR evidence has been used in support of the formation of type-I porphyrin isomers upon cyclotetramerization of β,β’-aryl/ester-substituted pyrroles (Uno et al. 2003) or β,β’-alkyl/acyl-substituted pyrroles (Kobayashi et al. 1978). For a more complete discussion, see the ESM.
The presence of the type-I isomer, rather than the statistical mixture of isomers, indicates that under these conditions the porphyrinogen-forming reaction proceeds via kinetic rather than thermodynamic control. The occurrence of kinetic control does not imply that the product is not thermodynamically stable (Eschenmoser 2007b). In this case, the porphyrinogen 3 is thermodynamically stable relative to the acyclic precursor; however, thermodynamic equilibrium is not attained given that the full set of porphyrinogen isomers (types I-IV) is not formed. The absence of the full set of porphyrinogen isomers indicates the reaction is largely irreversible. The preferred formation of the type-I isomer in this case, versus the statistical formation of uroporphyrinogens (Mauzerall 1960a, b), can be understood in the context of the differences in molecular structure of pyrrole 2 versus porphobilinogen, respectively, and the mechanisms of statistical randomization. One mechanism for statistical randomization entails cleavage at the site between a protonated α-carbon and the adjacent meso-methylene group of a pyrromethane or porphyrinogen unit (Chevalier et al 2002). Randomization also would result from repetition of a biosynthetic-like process (Battersby 2000) that entails (i) closure of the bilane to give the spiropyrrolenine-containing macrocycle, (ii) ring-opening to give a bilane or equivalent, and (iii) closure to form the rearranged porphyrinogen. Subsequent ring opening of the porphyrinogen to initiate repetition of steps i-iii requires pyrrole protonation and is facilitated in an acidic medium (Mauzerall 1960a). The requirement for protonation of the pyrrole in both mechanisms provides a rationale for the absence of statistical randomization herein, given that electronically deactivated pyrroles (owing to the presence of a β-ester substituent) are little susceptible to protonation (Chevalier et al 2002). The poor leaving group (CH3O-) of 2 is expected to make only a minor contribution to the observed irreversibility. By contrast, porphobilinogen is activated by two β-alkyl units, contains a superb leaving group (+H3N-), and readily condenses to give the statistical mixture of uroporphyrinogens.
It deserves emphasis that porphyrin 4 is similar to, yet distinct from, uroporphyrins and coproporphyrins (Fig. 7). Uroporphyrins bear four propionic acid and four acetic acid moieties, whereas coproporphyrins bear four propionic acid and four methyl groups. Porphyrin 4 bears four propionic acid and four carbomethoxy groups, and thus is expected to have polarity intermediate between that of the analogous, naturally derived uroporphyrins and coproporphyrins.

Structure of uroporphyrin (type III) and coproporphyrin (type III)
The absorption spectrum of porphyrin 4 is shown in Fig. 8. In aqueous buffer solutions at neutral or alkaline pH, the Soret (B) band is broad with maximum at 410 nm, and the visible (Q) bands are broadened and indistinct. By contrast, in N,N-dimethylformamide (DMF) or DMF/H2O (9:1), the B band appears at 427 nm and the Q bands are relatively sharp; the spectrum is nearly identical to that of the similarly substituted synthetic compound 2,7,12,17-tetracarboethoxy-3,8,13,18-tetramethylporphyrin in CHCl3 (Kleinspehn et al. 1966). Similar behavior in aqueous versus organic media is exhibited by coproporphyrin and other porphyrins that bear four or fewer alkyl carboxy groups (Mauzerall 1965; Brown et al. 1976; Morales-Rojas and Yatsimirsky 1999). The broadened, blue-shifted spectrum in aqueous solution is a consequence of aggregation, most likely via hydrophobic stacking to form multimeric assemblies (Brown et al. 1976). By contrast, examination of 4 in the presence of cationic micelles (10 mM cetyltrimethylammonium bromide, 50 mM sodium phosphate, pH 7) resulted in a sharp absorption spectrum (λmax = 429 nm) nearly identical to that in DMF. No such change was observed upon exposure to anionic (10 mM sodium dodecylsulfate) or neutral (2% Triton X-100) micelles.

Absorption spectra of equal concentrations of porphyrin 4 at room temperature in DMF/H2O (9:1; dotted line, λmax = 427 nm), aqueous sodium phosphate (50 mM, pH 7.0; dashed line, λmax = 410 nm), and cationic micellar solution (10 mM cetyltrimethylammonium bromide and 50 mM sodium phosphate at pH 7.0; solid line, λmax = 429 nm). The Q-band region of each spectrum is displayed at 7-fold the intensity for clarity. The inset shows the fluorescence emission spectrum (λem 657, 728 nm) observed upon 429-nm excitation of porphyrin 4 in the cationic micellar solution
Fluorescence spectroscopy of 4 in neutral aqueous solution indicates the species that absorbs at 410 nm is non-fluorescent, with a Φf at least 350-fold less than that of uroporphyrin (Φf ~0.055) (Feitelson and Mauzerall 1996) under identical conditions. Moreover, excitation spectroscopy indicates that the residual fluorescence stems from a trace of unassociated, monomeric porphyrin 4 (λmax 426 nm). In DMF, porphyrin 4 exhibited fluorescence similar in spectrum (λem 656, 725 nm) and 0.46 times the integrated intensity of that of the benchmark meso-tetraphenylporphyrin. The fluorescence spectrum obtained upon excitation at 429 nm of the cationic micellar solution of 4 (Fig. 8, inset) was nearly identical to that in DMF (λem 656, 725 nm), consistent with a monomolecular dispersion. The fluorescence quantum yield of 4 in the cationic micellar solution was 0.52 times that of coproporphyrin under identical conditions (which is known to assemble into cationic micelles) (Reddi et al. 1983), and 0.59 times that of uroporphyrin in aqueous sodium phosphate solution. Thus, porphyrin 4 upon assembly into cationic micelles exhibits photophysical features typical of monomeric porphyrins. Porphyrins are known to participate in efficient photoinduced electron-transfer reactions upon assembly into diverse lipid assemblies (Ilani et al. 1989). In this regard, porphyrin-containing micelles have been proposed as photocatalytic systems in the emergence of primitive photosynthesis (Sivash et al. 1991).
Prebiotic Implications
The facile assembly of four molecules each of ALA and β-ketoester 1 to yield the porphyrinogen via the α-methoxymethyl-substituted pyrrole illustrates the Eschenmoser concept (Eschenmoser 2007a) that “constitutionally complex biomolecules can be generationally intrinsically simple.” While the environment of the early Earth remains a subject of vigorous discussion (Kasting and Howard 2006), the simple conditions employed here (few mM concentration of reactants in slightly acidic-to-neutral water at 70–100°C for ≤ 1 day) would seem to be compatible with a prebiotic milieu. If so, the results have a number of implications.
First, a family of β-ketoesters is expected to react similarly with ALA to give porphyrinogens. Derivatization at the carboxylic acid unit with other alkyl esters or amides would give diverse substituted macrocycles, while 4-substituents that constitute leaving groups superior to methoxy (e.g., cyano, thiocyanato, acyloxy, N-acylamino, N-sulfonylamino, N-phosphorylamino, halo, O-phosphonooxy, S-phosphonothio) may enable porphyrinogen formation under milder conditions. Such a 4-substituent also may better balance the competing pH requirements of Knorr pyrrole formation (neutral pH; Haley and Maitland 1951) and the condensation to give the porphyrinogen.
Second, the tetrapyrrole macrocycles are expected to undergo molecular processing over time in the prebiotic milieu that results in hydro/dehydrogenation, metalation, side-chain alteration, and selective binding with other molecules. Together with the diverse carboxylic acid-substituted β-ketoesters, a very rich collection of (photo)redox-active catalytic molecules should readily form.
Third, although the yields observed herein are not high, in a prebiotic setting the amount of porphyrins (such as 4) formed would depend on the flux of precursors and the competitive dynamics of condensation and oxidation processes. The structural simplicity of β-dicarbonyl compounds and their ease of formation (e.g., from an activated carboxylic acid and a malonate species) suggests their likely availability under prebiotic conditions. Although prebiotic routes to β-dicarbonyl compounds apparently have not yet been demonstrated, it should be noted that β-dicarbonyl compounds occur across biological systems in fatty acid biosynthesis and degradation, and in microbial polyketide synthesis. The β-ketothioester acetoacetyl-acyl carrier protein (ACP) is a common intermediate in such metabolic processes (Smith and Tsai 2007). In addition, methoxymalonyl-ACP (derived from 1,3-bisphoshoglycerate) is employed as an extender unit via decarboxylative Claisen condensation to introduce the methoxyacetyl unit in polyketide synthesis (Chan et al. 2009). The occurrence of a thioester analogue of 4-methoxyacetoacetate would likely require methoxyacetyl-coenzyme A (CoA) or analogous thioester as a starter unit. In this regard, an early acetyl-CoA based metabolism via metal-sulfide chemistry has been proposed at volcanic sites (Wächtershäuser 2006) or hydrothermal vents (Martin and Russell 2007), although the available quantity of such products has recently been questioned (Ross 2008). In practice, the availability of β-ketoesters would depend in part on the competition with reactions typical of β-ketoesters such as the formation of dihydropyridines (Hantzsch reaction), dihydropyrimidines (Biginelli reaction), α,β-unsaturated carbonyl compounds (Knoevenagel reaction), and fatty acids, as well as diverse alkylation processes (Hamby and Hodges 1993). A prebiotic route to ALA also has not yet been developed, although ALA is available by transamination of 4,5-dioxovaleric acid with glycine (Beale et al. 1979). The finding herein of a 1% yield of porphyrin even in the presence of oxidants (in this case, O2) augurs well for successful procession through the required oligopyrromethane intermediates, the oxidation of which would be deleterious (Lindsey et al. 1994), prior to reaching the porphyrinogen.
Fourth, a mixed condensation similar to that of ALA and 1 may lead to plausible prebiotic routes to uroporphyrinogens, where ALA condenses with an ALA analogue that itself is incapable of self-condensation yet contains a 5-substituent (other than amino) that (1) directs enamine formation to the 3-carbon for subsequent aldol condensation at this site, and (2) provides a viable leaving group on the resulting pyrrole for porphyrinogen formation. Prior studies have shown that neither the 5-chloro (Jaffe and Rajagopalan 1990) nor 5-dimethylamino-substituted levulinic acid (Butler and George 1992) was suitable in this regard. The 5-methoxy group is an obvious choice given the results herein as well as the faster exchange measured at the α-methyl versus α-methylene of methoxyacetone (Bothner-By and Sun 1967). The key ring-closure step in pyrrole formation is an aldol condensation (Granick and Mauzerall 1958; Nandi and Shemin 1968). On the basis of consideration of substrates and mechanisms of aldolase enzymes (Periana et al. 1980), 5-(O-phosphonooxy) derivatives of levulinic acid also may provide appropriate reactivity. Studies along these lines are underway.
Finally, tetrapyrrole macrocycles persist for hundreds of millions of years in anaerobic environments (Callot and Ocampo 2000), and hence would be expected to accumulate during the Hadean and Archaean eras. Minute quantities of porphyrins are readily detectable. Porphyrin 4 is not found in extant metabolism, yet its facile formation raises the intriguing question as to whether diagenesis of 4 or other abiotic analogues would yield detectable fossil relics of prebiotic metabolism. In this regard, the identification of tetrapyrrole macrocycles alone in exobiological settings (Suo et al. 2007) may not be an unambiguous indication of current or previous life.
Much remains to be done to establish a comprehensive model for the prebiogenesis of tetrapyrrole macrocycles, where plausible prebiotically available precursors to 1 and ALA yield hydroporphyrins and porphyrins such as 4 in lipid assemblies. The results described herein address a key part of this longstanding puzzle, namely how selected acyclic compounds can react under simple conditions to give a porphyrinogen. The development of an integrated route beginning with simple precursors of ALA and β-ketoesters would provide strong argument for the self-constitution of tetrapyrrole macrocycles in a pre-RNA, pre-protein, prebiotic world, thus providing the cofactors and pigments essential for the spontaneous origin of redox-rich metabolic and proto-photosynthetic processes.
References
Abe K, Konaka R (1989) Quantitation of urinary porphyrins by liquid chromatography after oxidation of porphyrinogens. Clin Chem 35:1619–1622
Aylward N, Bofinger N (2005) Possible origin for porphin derivatives in prebiotic chemistry–a computational study. Orig Life Evol Biosph 35:345–368
Bates RG (1962) Revised standard values for pH measurements from 0 to 95°C. J Res Natl Bur Stand 66A:179–184
Battersby AR (2000) Tetrapyrroles: the pigments of life. Nat Prod Rep 17:507–526
Battersby AR, Fookes CJR, Matcham GWJ, McDonald E (1980) Biosynthesis of the pigments of life: formation of the macrocycle. Nature 285:17–21
Beale SI, Gold MH, Granick S (1979) Chemical synthesis of 4,5-dioxovaleric acid and its nonenzymatic transamination to 5-aminolevulinic acid. Phytochem 18:441–444
Bothner-By AA, Sun C (1967) Acid- and base-catalyzed hydrogen-deuterium exchange between deuterium oxide and simple ketones. J Org Chem 32:492–493
Brown SB, Shillcock M, Jones P (1976) Equilibrium and kinetic studies of the aggregation of porphyrins in aqueous solution. Biochem J 153:279–285
Bunke A, Zerbe O, Schmid H, Burmeister G, Merkle HP, Gander B (2000) Degradation mechanism and stability of 5-aminolevulinic acid. J Pharm Sci 89:1335–1341
Butler AR, George SD (1992) The nonenzymatic cyclic dimerisation of 5-aminolevulinic acid. Tetrahedron 48:7879–7886
Butler AR, George SD (1993) Reaction of some diketones with 5-aminolevulinic acid in acid solution. Tetrahedron 49:7017–7026
Butler AR, George SD (1994) Mechanism of acid catalysis in the cyclisation of 5-aminolevulinic acid and acetylacetone to 3-acetyl-4-(2-carboxyethyl)-2-methylpyrrole. J Chem Soc Perkin Trans 2:315–318
Callot HJ, Ocampo R (2000) In The Porphyrin Handbook. Kadish KM, Smith KM, Guilard, R (eds) Academic, San Diego, Vol 1, pp 349–398
Canfield DE, Rosing MT, Bjerrum C (2006) Early anaerobic metabolisms. Phil Trans R Soc B 361:1819–1836
Chamberlin KS, LeGoff E (1978) Synthesis of octasubstituted porphyrins. Synth Commun 8:579–585
Chan YA, Podevels AM, Kevany BM, Thomas MG (2009) Biosynthesis of polyketide synthase extender units. Nat Prod Rep 26:90–114
Chaperon AR, Bertschy H, Franz-Schrumpf A-L, Hugelet B, Neels A, Stoeckli-Evans H, Neier R (2003) The synthesis of a pyrazol analogon of porphobilinogen with the help of the Mukaiyama aldol reaction. Chimia 57:601–606
Cheng DO, Bowman TL, LeGoff E (1976) Synthesis and Michael reaction of 3,4-dimethylpyrrole. J Heterocycl Chem 13:1145–1147
Chevalier F, Geier GR III, Lindsey JS (2002) Acidolysis of intermediates used in the preparation of core-modified porphyrinic macrocycles. J Porphyrins Phthalocyanines 6:186–197
Clezy PS, Liepa AJ (1971) The chemistry of pyrrolic compounds. XV. Porphyrins from electronegatively substituted bilenes and biladienes. Aust J Chem 24:1027–1040
Clezy PS, Liepa AJ, Webb NW (1972) The chemistry of pyrrolic compounds. XX. Bilenes-b as intermediates in the synthesis of porphyrins related to protoporphyrin IX. Meso-pyrrolylporphyrins. Aust J Chem 25:1991–2001
Comer E, Murphy WS (2003) The bromoquinone annulation reaction: a formal total synthesis of EO9. Arkivoc 286–296
Corwin AH (1950) In Heterocyclic Compounds. Elderfield RC (ed) Wiley & Sons, Inc., New York, Vol 1, pp 286–289
Danton M, Lim CK (2006) Porphyrin profiles in blood, urine and faeces by HPLC/electrospray ionization tandem mass spectrometry. Biomed Chromatogr 20:612–621
De Blois AW, Grouls RJE, Ackerman EW, Wijdeven WJA (2002) Development of a stable solution of 5-aminolaevulinic acid for intracutaneous injection in photodynamic therapy. Lasers Med Sci 17:208–215
Donnelly RF, McCarron PA, Woolfson AD (2007) Derivatives of 5-aminolevulinic acid for photodynamic therapy. Pers Med Chem 1:49–63
Eisner U, Gore PH (1958) The light absorption of pyrroles. Part I. Ultraviolet spectra. J Chem Soc 922–927
Elfsson B, Wallin I, Eksborg S, Rudaeus K, Ros AM, Ehrsson H (1998) Stability of 5-aminolevulinic acid in aqueous solution. Eur J Pharm Sci 7:87–91
Eschenmoser A (1986) Chemistry of corphinoids. Ann NY Acad Sci 471:108–129
Eschenmoser A (1988) Vitamin B12: experiments concerning the origin of its molecular structure. Angew Chem Int Ed Engl 27:5–39
Eschenmoser A (2007a) The search for the chemistry of life’s origin. Tetrahedron 63:12821–12844
Eschenmoser A (2007b) Question 1: commentary referring to the statement “the origin of life can be traced back to the origin of kinetic control” and the question “do you agree with this statement; and how would you envisage the prebiotic evolutionary bridge between thermodynamic and kinetic control?” stated in section 1.1”. Orig Life Evol Biosph 37:309–314
Fabiano E, Golding BT (1991) On the mechanism of pyrrole formation in the Knorr pyrrole synthesis and by porphobilinogen synthase. J Chem Soc Perkin Trans 1 3371–3375
Feitelson J, Mauzerall D (1996) Photoacoustic evaluation of volume and entropy changes in energy and electron transfer. Triplet state porphyrin with oxygen and naphthoquinone-2-sulfonate. J Phys Chem 100:7698–7703
Franck B, Stratmann H (1981) Condensation products of the porphyrin precursor 5-aminolevulinic acid. Heterocycles 15:919–923
Goldberg RN, Kishore N, Lennen RM (2002) Thermodynamic quantities for the ionization reactions of buffers. J Phys Chem Ref Data 31:231–370
Granick S (1965) In Evolving Genes and Proteins. Bryson V, Vogel HJ (ed) Academic, New York pp 67–88
Granick S, Mauzerall D (1958) Porphyrin biosynthesis in erythrocytes II. Enzymes converting δ-aminolevulinic acid to coproporphyrinogen. J Biol Chem 232:1119–1140
Greenhill JV (1977) Enaminones. Chem Soc Rev 6:277–294
Haley CAC, Maitland P (1951) Organic reactions in aqueous solution at room temperature. Part I. The influence of pH on condensations involving the linking of carbon to nitrogen and of carbon to carbon. J Chem Soc 3155–3174
Hamby JM, Hodges JC (1993) α-Amino ketones from amino acids as precursors for the Knorr pyrrole synthesis. Heterocycles 35:843–850
Hartley H, Campbell NP (1908) The solubility of iodine in water. J Chem Soc Trans 741–745
Hayes A, Kenner GW, Williams NR (1958) Pyrroles and related compounds. Part I. Syntheses of some unsymmetrical pyrrolylmethylpyrroles (pyrromethanes). J Chem Soc 3779–3788
Hildebrand JH, Benesi HA, Mower LM (1950) Solubility of iodine in ethyl alcohol, ethyl ether, mesitylene, p-xylene, 2,2-dimethylbutane, cyclohexane and perfluoro-n-heptane. J Am Chem Soc 72:1017–1020
Hunter GA, Rivera E, Ferreira GC (2005) Supraphysiological concentrations of 5-aminolevulinic acid dimerize in solution to produce superoxide radical anions via a protonated dihydropyrazine intermediate. Arch Biochem Biophys 437:128–137
Ilani A, Woodle M, Mauzerall D (1989) Photoinduced electron transfer across lipid bilayers containing magnesium octaethylporphyrin. Photochem Photobiol 49:673–679
Jaffe EK, Rajagopalan JS (1990) Nuclear magnetic resonance studies of 5-aminolevulinate demonstrate multiple forms in aqueous solution. Bioorg Chem 18:381–394
Jeandon C, Callot HJ (1993) Cyclotetramerization of modified Knorr pyrroles into porphyrins. A reinvestigation. Bull Soc Chim Fr 130:625–629
Kasting JF, Howard MT (2006) Atmospheric composition and climate on the early Earth. Phil Trans R Soc B 361:1733–1742
Kay IT (1962) The formation of coproporphyrins by the polymerization of monopyrroles in acidic solution. Proc Natl Acad Sci USA 48:901–905
Kinoshita H, Tanaka S, Nishimori N, Dejima H, Inomata K (1992) Synthesis of 2-(substituted methyl)-3,4-disubstituted pyrroles and their conversion into the corresponding porphyrins. Bull Chem Soc Jpn 65:2660–2667
Kleinspehn GG, Briod AE, Caughey WS (1966) The synthesis of four isomeric tetramethyltetracarbethoxyporphyrins. J Org Chem 31:1613–1617
Kobayashi H, Archibald JL, MacDonald SF (1978) Porphyrin syntheses requiring negative substituents. Can J Chem 56:2430–2436
Krasnovsky AA (1976) Chemical evolution of photosynthesis. Orig Life 7:133–143
Ksander G, Bold G, Lattmann R, Lehmann C, Fruh T, Xiang Y-B, Inomata K, Buser H-P, Schreiber J, Zass E, Eschenmoser A (1987) Chemie der α-aminonitrile. Einleitung und wege zu uroporphyrinogen-octanitrilen (Chemistry of α-aminonitriles I: Introduction and pathways to uroporphyrinogen-octanitriles). Helv Chim Acta 70:1115–1172
Lazcano A, Miller SL (1999) On the origin of metabolic pathways. J Mol Evol 49:424–431
Lin W, Timkovich R (1994) Oxygenated tetrapyyroles produced from porphyrinogens. Bioorg Chem 22:72–94
Lindsey JS, MacCrum KA, Tyhonas JS, Chuang YY (1994) Investigation of a synthesis of meso-porphyrins employing high concentration conditions and an electron transport chain for aerobic oxidation. J Org Chem 59:579–587
Lindsey JS, Schreiman IC, Hsu HC, Kearney PC, Marguerettaz AM (1987) Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. J Org Chem 52:827–836
Lüönd RM, Neier R (1991) Michael reactions of α-unsubstituted trisubstituted 1H-pyrroles. Helv Chim Acta 74:91–102
Lüönd RM, Walker J, Neier RW (1992) Assessment of the active-site requirements of 5-aminolaevulinic acid dehydratase: evaluation of substrate and product analogues as competitive inhibitors. J Org Chem 57:5005–5013
Martin W, Russell MJ (2007) On the origin of biochemistry at an alkaline hydrothermal vent. Phil Trans R Soc B 362:1887–1925
Mauzerall D (1960a) The thermodynamic stability of porphyrinogens. J Am Chem Soc 82:2601–2605
Mauzerall D (1960b) The condensation of porphobilinogen to uroporphyrinogen. J Am Chem Soc 82:2605–2609
Mauzerall D (1965) Spectra of molecular complexes of porphyrins in aqueous solution. Biochemistry 4:1801–1810
Mauzerall D (1976) Chlorophyll and photosynthesis. Phil Trans R Soc Lond B 273:287–294
Mauzerall D (1978) In Bioorganic Chemistry. van Tamelen EE. (ed) Academic, New York, Vol IV, pp 303–314
Mauzerall D (1992) Light, iron, Sam Granick and the origin of life. Photosyn Res 33:163–170
Mauzerall D, Granick S (1956) The occurrence and determination of δ-aminolevulinic acid and porphobilinogen in urine. J Biol Chem 219:435–446
Mauzerall D, Granick S (1958) Porphyrin biosynthesis in erythocytes III. Uroporphyrinogen and its decarboxylase. J Biol Chem 232:1141–1162
Mauzerall DC (1990) The photochemical origins of life and photoreaction of ferrous ion in the Archaean oceans. Orig Life Evol Biosph 20:293–302
Mauzerall DC (1998) Evolution of porphyrins. Clin Dermatol 16:195–201
Meierhenrich UJ, Munoz Caro GM, Schutte WA, Thiemann WH-P, Barbier B, Brack A (2005) Precursors of biological cofactors from ultraviolet irradiation of circumstellar/interstellar ice analogues. Chem Eur J 11:4895–4900
Mercer-Smith JA, Mauzerall DC (1985) A model for the origin of photosynthesis-III. The ultraviolet photochemistry of uroporphyrinogen. Photochem Photobiol 42:239–244
Mercer-Smith JA, Raudino A, Mauzerall DC (1984) Photochemistry of porphyrins: a model for the origin of photosynthesis. Photochem Photobiol 39:397–405
Miller SL (1986) Current status of the prebiotic synthesis of small molecules. Chemica Scripta 26B:5–11
Miller SL (1987) Which organic compounds could have occurred on the prebiotic Earth? Cold Spr Harbor Symp Quant Biol 52:17–27
Miller SL (1998) In The Molecular Origins of Life. Brack A (ed) Cambridge University Press, Cambridge pp 59–85
Morales-Rojas H, Yatsimirsky AK (1999) Medium effects on the dimerization of coproporphyrin-I free base. J Phys Org Chem 12:377–387
Nandi DL, Shemin D (1968) δ-Aminolevulinic acid dehydratase of Rhodopseudomonas spheroides III. Mechanism of porphobilinogen synthesis. J Biol Chem 243:1236–1242
Nguyen LT, Smith KM (1996) Syntheses of type-I porphyrins via monopyrrole tetramerization. Tetrahedron Lett 37:7177–7180
Novo M, Hüttmann G, Diddens H (1996) Chemical instability of 5-aminolevulinic acid used in the fluorescence diagnosis of bladder tumours. J Photochem Photobiol B Biol 34:143–148
Olofsson G (1984) Temperature dependence of the heat-capacity change for the dissociation of acetic acid and of propionic acid in water. J Chem Thermodyn 16:39–44
Olson JM, Pierson BK (1987) Origin and evolution of photosynthetic reaction centers. Orig Life 17:419–430
Periana RA, Motiu-DeGrood R, Chiang Y, Hupe DJ (1980) Does substrate rather than protein provide the catalyst for α-proton abstraction in aldolase? J Am Chem Soc 102:3923–3927
Rapoport H, Bordner J (1964) Synthesis of substituted 2, 2’-bipyrroles. J Org Chem 29:2727–2731
Reddi E, Jori G, Rodgers MAJ, Spikes JD (1983) Flash photolysis studies of hemato- and copro-porphyrins in homogeneous and microheterogeneous aqueous dispersions. Photochem Photobiol 38:639–645
Rimington C, Benson A (1967) Partition of porphyrins between cyclohexanone and aqueous sodium acetate as a function of pH. Biochem J 105:1085–1090
Ross DS (2008) A quantitative evaluation of the iron-sulfur world and its relevance to life’s origins. Astrobiol 8:267–272
Scheer H, Katz JJ (1975) In Porphyrins and Metalloporphyrins. Smith KM (ed) Elsevier Scientific, Amsterdam pp 399–524
Scott AI (1997) How were porphyrins and lipids synthesized in the RNA world? Tetrahedron Lett 38:4961–4964
Sivash AA, Masinovsky Z, Lozovaya GI (1991) Surfactant micelles containing protoporphyrin IX as models of primitive photocatalytic systems: a spectral study. Biosystems 25:131–140
Smith S, Tsai S-C (2007) The type I fatty acid and polyketide synthases: a tale of two megasynthases. Nat Prod Rep 24:1041–1072
Stauffer F, Zizzari E, Soldermann-Pissot C, Faurite J-P, Neier R (2001) Porphobilinogen synthase: a challenge for the chemist? Chimia 55:314–319
Suo Z, Avci R, Schweitzer MH, Deliorman M (2007) Porphyrin as an ideal biomarker in the search for extraterrestrial life. Astrobiol 7:605–615
Tamura Y, Kato S, Ikeda M (1971) One-step Knorr pyrrole synthesis with hydroxylamine O-sulphonic acid. Chem Ind 767.
Thyrann T, Lightner DA (1996) Oxidation of pyrrole α-methyl to methoxymethyl with ceric triflate. Tetrahedron Lett 37:315–318
Uno H, Inoue K, Inoue T, Ono N (2003) Oligocyclization of 2-(hydroxymethyl) pyrroles with electron-withdrawing groups at β-positions: formation and structural elucidation of porphyrinogens and hexaphyrinogens. Org Biomol Chem 1:3857–3865
Wächtershäuser G (2006) From volcanic origins of chemoautotrophic life to Bacteria, Archaea and Eukarya. Phil Trans R Soc B 361:1787–1808
Wnuk M (1983) The probable ways of the synthesis of porphyrin compounds during chemical evolution. Roczniki Filozoficzne 31:185–195
Acknowledgments
J.S.L. wishes to thank Dr. David C. Mauzerall for stimulating conversations over a 30-year period concerning tetrapyrrole biosynthesis, evolution, and photochemistry. J.S.L. also wishes to acknowledge Maya Mohan, Qingliu Lee, and Eric Bortz, who worked on predecessors of this project in the early 1990s. This work was supported by North Carolina State University including a leave of absence to J.S.L. Mass spectra were obtained at the NCSU Department of Chemistry Mass Spectrometry Facility. Funding for the facility was obtained from the North Carolina Biotechnology Center, and the NCSU Department of Chemistry.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
ESI-MS spectra; 1H NMR spectra; discussion concerning expected spectra for distinct porphyrin regioisomers; and full experimental procedures for anaerobic reactions as well as the preparation and isolation of porphyrin 4. (PDF 751 kb)
Rights and permissions
About this article
Cite this article
Lindsey, J.S., Ptaszek, M. & Taniguchi, M. Simple Formation of an Abiotic Porphyrinogen in Aqueous Solution. Orig Life Evol Biosph 39, 495–515 (2009). https://doi.org/10.1007/s11084-009-9168-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-009-9168-3