
Abstract
Only a percentage of COVID-19 patients develop thrombotic complications. We hypothesized that genetic profiles may explain part of the inter-individual differences. Our goal was to evaluate the genotypic distribution of targeted DNA polymorphisms in COVID-19 patients complicated (PE+) or not (PE−) by pulmonary embolism. We designed a retrospective observational study enrolling N = 94 consecutive patients suffering severe COVID-19 with pulmonary embolism (PE+, N = 47) or not (PE−, N = 47) during hospitalization. A panel of N = 13 prothrombotic DNA polymorphisms (FV R506Q and H1299R, FII G20210A, MTHFR C677T and A1298C, CBS 844ins68, PAI-1 4G/5G, GPIIIa HPA-1 a/b, ACE I/D, AGT T9543C, ATR-1 A1166C, FGB − 455G > A, FXIII103G > T) and N = 2 lipid metabolism-related DNA polymorphisms (APOE T 112C and T158C) were investigated using Reverse Dot Blot technique. Then, we investigated possible associations between genotypic subclasses and demographic, clinical, and laboratory parameters including age, obesity, smoking, pro-inflammatory cytokines, drug therapy, and biomarkers of thrombotic risk such as D-dimer (DD). We found that 58.7% of PE+ had homozygous mutant D/D genotype at ACE I/D locus vs. PE− (40.4%) and 87% of PE+ had homozygous mutant C/C genotype at APOE T158C locus vs. PE− (68.1%). In PE+ group, DD levels were significantly higher in D/D and I/D genotypes at ACE I/D locus (P = 0.00066 and P = 0.00023, respectively) and in C/C and T/C genotypes at APOE T158C locus (P = 1.6e−06 and P = 0.0012, respectively) than PE− group. For the first time, we showed significant associations between higher DD levels and ACE I/D and APOE T158C polymorphisms in PE+ vs. PE− patients suggesting potential useful biomarkers of poor clinical outcome.
Similar content being viewed by others


Highlights
-
Only a fraction of COVID-19 patients develop thrombotic complications.
-
Genetic background may affect the individual susceptibility to develop pulmonary embolism in COVID-19 patients.
-
ACE I/D and APO T158C polymorphisms associated with higher levels of D-dimer in COVID-19 patients with pulmonary embolism.
-
ACE I/D and APO T158C polymorphisms may explain part of the individual susceptibility to poor clinical outcome.
Introduction
Complex genetic and epigenetic molecular circuits can underlie the strong propensity to thrombotic events in patients with coronavirus disease 2019 (COVID-19), especially in those patients who are affected by the severe form of disease; however, the molecular components of the individual risk predisposing to thrombosis have not been fully established yet [1,2,3,4,5]. COVID-19 has a wide spectrum of possible clinical features ranging from asymptomatic disease to severe interstitial pneumoniae and acute respiratory distress syndrome (ARDS) [2, 3].
Retrospective studies showed that increased levels of D-dimer (DD), procalcitonin, and C Reactive Protein (CRP) may be prognostic biomarkers useful to predict the onset of thrombotic complications [6]. Moreover, the incidence of thrombotic events (about 31%) [7] was associated with increased morbidity and mortality in critically ill patients [8]. At molecular level, the thrombotic manifestations of severe COVID-19 patients seem to be consequences of direct SARS-CoV-2 cytotoxic effects, endothelialitis, dysregulated immune response, platelet aggregation, and complement and coagulation cascade activation [1, 8]. But none of these molecular pathways is sufficient to explain why some COVID-19 patients develop thrombotic manifestations and other do not.
Genetic risk factors related to a pre-existing prothrombotic state seem to play a key role in determining the individual susceptibility both to SARS-CoV-2 infection and clinical course of disease [9, 10]. We conducted a retrospective, single-centre, observational study to evaluate the genotypic distribution of N = 15 polymorphisms involved in thrombotic risk and lipid metabolism in COVID-19 patients who developed acute pulmonary embolism (PE+) as compared to those who did not develop acute pulmonary embolism (PE−).
Materials and methods
Study population
In this retrospective, single-center, observational study, we enrolled 94 consecutive hospitalized patients (22 F and 72 M) suffering severe COVID-19 who were admitted to the Sub-Intensive Care Unit of A.O.R.N. Ospedali dei Colli, Cotugno Hospital, Naples (Italy) between 13 January 2021 and 2 May 2021. The inclusion criteria were: (1) respiratory rate 30 breaths/min; (2) arterial oxygen saturation 93% at rest; (3) PaO2/FiO2 < 300 mmHg; (4) mechanical ventilation; (5) patients with and without pulmonary embolism. Exclusion criteria were: (1) pediatric age, (2) patients with no genetic testing, and (3) patients missing clinical or laboratory data of interest. Severe COVID-19 patients were stratified in two groups based on the presence of pulmonary embolism (PE+, N = 47) or the absence of pulmonary embolism (PE−, N = 47). Upon admission, we retrieved anamnestic and anthropometric parameters [age, gender, body mass index (BMI), smoking habit], presence of comorbidities [systemic arterial hypertension, type 2 diabetes (T2D), coronary heart disease (CHD), and chronic obstructive pulmonary disease (COPD)], drug therapy, including angiotensin-converting enzyme inhibitors (ACEi) or sartans, and routine laboratory parameters, such as CRP, prothrombin time (PT), activated partial thromboplastin time (aPTT), prothrombin time and international normalized ratio (PT-INR), DD, fibrinogen, interleukin (IL)-2 receptor (IL2R), and IL-6.
Ethics statement
This study was approved by the local ethics committee (AOC-0017432-2020). Each study participant provided written informed consent prior to study enrolment, collection of samples and subsequent genetic analysis. This study was conducted according to the principles and guidelines expressed in the Declaration of Helsinki.
Blood collection and genetic testing
Upon admission, from each study participant we collected about 5 mL of whole blood from a peripheral vein in EDTA tubes. Then, the molecular analysis was performed by our Hospital Central Laboratory by using a Reverse Dot Blot (RDB) kit by Nuclear Laser Medicine (NLM, version 2020.10.19, CVD-14cod. AC084, Milan). We determined the individual genotypes at 15 loci including FV R506Q and FV H1299R, FII G20210A, MTHFR C677T and MTHFR A1298C, CBS 844ins68, PAI-1 4G/5G, GPIIIa HPA-1 a/b, ACE I/D, AGT T9543C, AGTR-1 A1166C, FGB-455G > A, and FXIII103G > T, APOE T112C, and APOE T158C (Table 1).
Statistical analysis
A statistical post-hoc power analysis was performed to compute power values for given sample sizes, effect size, and alpha level. By selecting one-tailed Wilcoxon-Mann–Whitney test with 2 groups (PE+ vs. PE−), with a sample size of 47 for both groups, a medium effect size (f = 0.5) and an alpha level of 0.05, the power resulted around 0.76. Thus, our sample size was more than adequate for the main objective of this study and for possible subgroup analysis. G*Power software version 3.1 was used to compute the power analysis.
Statistical analysis was performed using R Core Team (version 4.0. 0). Continuous variables were expressed as mean and standard deviation (SD). Data distribution was tested for normality through the Shapiro–Wilk test. Unpaired Student’s t-test or Wilcoxon rank-sum test, as required, were performed for comparison between two groups. Categorical variables were expressed as a percentage and were compared using the Chi-Square test or the Fisher’s exact test. A P < 0.05 was considered significant. Bonferroni’s correction was used for multiple hypothesis correction, if necessary. A Spearman’s correlation analysis was run to investigate whether there was an association among variables in two groups separately. A Spearman’s ρ with P ≤ 0 0.05 was set as threshold to identify an agreement between variables. For the multivariate analysis, a logistic regression model was performed to evaluate risk factors associated with pulmonary embolism, considering thrombotic and cardiovascular parameters.
Results
Clinical characteristics of study population
Clinical characteristics of COVID-19 patients are summarized in Table 2. A total of 94 severe COVID-19 patients were included in the study with a mean age of 58.98 ± 12.33 years and 60.94 ± 12.14 years for PE− and PE+ groups, respectively. In both groups there was a prevalence of males (72.3% and 80.9% for PE− and PE+, respectively). Besides, we found a higher percentage of patients falling in the “non-smoking” class both in PE− (57.4%) and PE+ (51.1%) as compared to patients falling in the “smoking” class (PE−, 29.8% and PE+, 17%) and patients falling in the “former-smoking class” (PE−, 12.8% and PE+, 31%). In addition, a higher percentage of patients were not obese (PE−, 74.5% and PE+, 68.1%) or affected by T2D (PE−, 76.6% and PE+, 63.8%) but showed hypertension (PE−, 63.8% and PE+, 59.6%). Only a small percentage of patients was affected by CHD (PE−, 19.1% and PE+, 14.9%) or COPD (PE−, 6.4% and PE+, 6.4%). Regarding pharmacological treatment, we found that a higher percentage of patients did not use ACEi (PE−, 57.4% and PE+, 63.8%) or sartans (PE−, 70.2% and PE+ 61.7%). No statistical differences were detected for all these parameters in PE+ vs. PE− patients. As expected, higher DD levels were found in PE+ vs. PE− groups with a statistical significance (P = 5.4e−09, Wilcoxon rank-sum test after Bonferroni correction) (Table 3 and Supplementary Fig. 1). Otherwise, differences in CRP, PT, aPTT, PT-INR, fibrinogen, IL2R, and IL6 were not statistically significant between the two groups (Table 3). Multivariate logistic regression analysis revealed that D-Dimer was an independent risk factor significantly associated with PE (p-value = 0.015). As reported in Supplementary Table 1, none of the other clinical variables (age, BMI, IL2R, IL6, CRP, PT, aPTT, PT-INR and fibrinogen) reached statistical significance.
Genotyping
We evaluated a panel of 15 polymorphisms already known to be associated with increased thrombotic risk and lipid metabolism dysfunction (Table 1). We found that the genotypic distribution at ACE I/D (P = 0.04), CBS 844ins68 (P = 0.03), and APOE T158C (P = 0.02) loci significantly discriminated PE+ vs. PE− (Table 1). According to genotypic subclass distribution, we found that ACE I/D polymorphism showed: (1) D/D genotype (homozygous mutant) in 58.7% of PE+ vs. 40.4% of PE−; (2) I/D genotype (heterozygous) in 23.9% of PE+ vs. 48.9% of PE−; and (3) I/I genotype (homozygous wild type) in 17.4% PE+ vs. 10.6% of PE−; CBS 844ins68 polymorphism showed: (1) D/D genotype (homozygous wild type) in 95.7% of PE+ vs. 80.9% of PE−, (2) I/D genotype (heterozygous) in 4.3% of PE+ vs. 19.1% of PE−; and APOE T158C polymorphism showed: (1) C/C genotype (homozygous mutant) in 87% of PE+ vs. 68.1% of PE−, (2) T/C genotype (heterozygous) in 10.9% of PE+ vs. 31.9% of PE−, and (3) T/T genotype (homozygous wild-type) in 2.2% of PE+ vs. 0% of PE−. In contrast, FV R506Q and H1299R, FII G20210A, MTHFR C677T and A1298C, PAI-1 4G/5G, GPIIIa HPA-1 a/b, AGT T9543C, ATR-1 A1166C, FGB-455G > A, FXIII103G > T, and APOE T 112C did not show a significant different distribution between PE+ and PE− patients (Table 1).
Associations between ACE I/D polymorphism and clinical parameters
Based on our interest on ACE I/D polymorphism and its documented association with the severity of COVID-19 in Europe [11,12,13,14,15], we investigated potential associations between genotypic subclasses and clinical and laboratory parameters. As showed in Supplementary Fig. 2, in PE+ patients we observed: (1) a moderate positive correlation between IL2-R and fibrinogen (FB) (ρ = 0.44, P = 0.02) and a significant negative correlation between PT and PT-INR (ρ = 0.4, P = 0.045) for D/D genotype; (2) a significant positive correlation between BMI and PT (ρ = 0.67, P = 0.03) for I/D genotype; and (3) a significant positive correlation between CRP and DD levels (ρ = 0.7, P = 0.004) and between IL6 levels and aPTT (ρ = 0.88, P = 0.004), as well as a significant negative correlation between IL6 levels and PT-INR (ρ = − 0.71, P = 0.048) for I/I genotype. In PE− patients (Supplementary Fig. 3), we observed: (1) a significant positive correlation between IL2R and PT-INR (ρ = 0.64, P = 0.003), a moderate positive correlation between PT-INR and FB (ρ = 0.5, P = 0.03) and a significant negative correlation between PT and age (ρ = − 0.63, P = 0.004) for D/D genotype; (2) a moderate positive correlation between CRP and IL6 levels (ρ = 0.46, P = 0.03), age and BMI (ρ = 0.43, P = 0.04), PT-INR and IL2R (ρ = 0. 41, P = 0.049) and PT-INR and DD levels (ρ = 0.45, P = 0.03) as well as a moderate negative correlation between CRP and age (ρ = − 0.43, P = 0.03), PT and aPTT (ρ = − 0.47, P = 0.02) and PT and PT-INR (ρ = − 0.45, P = 0.03) for I/D genotype; and (3) a significant positive correlation between CRP, IL6 and DD levels (ρ = 1, P < < 0.01; ρ = 0.9, P = 0.04) as well as between IL6 and DD levels (ρ = 0.9, P = 0.04) for I/I genotype.
Association of DD levels with ACE I/D and APOE T158C polymorphisms
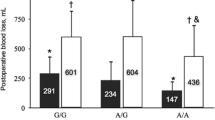
We investigated the distribution of DD levels according to the ACE I/D and APOE T158C (Fig. 1, on the left panel) genotypic subclasses. First, in PE+ patients DD levels were significantly higher in carriers of D/D and I/D genotypes at ACE I/D locus (respectively, P = 0.00066 and P = 0.00023) (Fig. 1, on the right panel) and in carriers of C/C and T/C genotypes at APOE T158C locus (respectively P = 1.6e−06 and P = 0.0012) as compared to PE− patients. No significant difference was observed in DD levels between the three genotypic subclasses at ACE I/D and APOE T158C loci both in PE+ and PE− patients (Supplementary Fig. 4).

Association of DD plasma levels with ACE I/D and APOE T158C genotypic subclasses in severe COVID-19 patients. In PE+ patients, DD levels were significantly higher in carriers of D/D and I/D genotypes at ACE I/D locus (P = 0.00066 and P = 0.00023, respectively) (on the right panel) and in carriers of C/C and T/C genotypes at APOE T158C locus (P = 1.6e−06 and P = 0.0012, respectively) (on the left panel)
Discussion
Our major findings were as follow: (1) genotypic differences at ACE I/D (P = 0.04), CBS 844ins68 (P = 0.03), and APOE T158C (P = 0.02) loci discriminated significantly PE+ vs. PE−; (2) a higher percentage of homozygous mutant ACE D/D and APOE C/C genotypes was found in PE+ vs. PE− patients; (3) in PE+ patients, DD levels were significantly higher in carriers of D/D and I/D genotypes at ACE I/D locus and in carriers of C/C and T/C genotypes at APOE T158C locus.
Previous data showed a higher prevalence of ACE D/D genotype in severe COVID-19 patients as compared to those with less severe disease [12, 13, 16]. At genetic level, the ACE I/D polymorphism is distinguished by either an insertion (I) or deletion (D) of 287 base pairs (Alu repeat segment) in the intron 16. In the general population, the carriers of the mutant D allele had higher ACE protein plasma and tissue levels (ACE levels in D/D carriers were approximately twice that in I/I carriers) as well as elevated levels of the vasopressor angiotensin II and reduced half-life of the vasodilator bradykinin as compared to carriers of the I allele [17, 18]. Beyond the systemic hypertension, ACE D/D genotype was significantly associated to cardiometabolic diseases, such as obesity and T2D, which are known risk factors for COVID-19 [19]. Therefore, it is plausible that ACE I/D polymorphism may play a key role in COVID-19 patients who are susceptible to develop severe lung injury or ARDS. Also, the racial difference of ACE I/D polymorphism is well established. In European populations (Italy, Spain, and France), there is a higher frequency of D allele up to 82% to 87% [20] than Eastern Asian populations (Chinese, Korean, Taiwanese, and Japanese) which have a higher frequency of ACE I allele (33% to 51%) [21]. Globally, it seems that the racial variance of ACE I/D genotype coincides with the differences of outcome; in fact, populations with higher D allele frequency (e.g., Italian) experienced higher fatality (https://www.worldometers.info/coronavirus/#countries). ACE gene shows potent vasoconstrictive effects, attenuation of fibrinolysis, and platelet activation and aggregation, and ACE I/D polymorphism represents a susceptibility risk biomarker for thrombosis. In particular, ACE D/D genotype was associated to thromboembolic manifestations in patients affected by other diseases with no pre-existing risk factors and traditional thrombophilia-related polymorphisms [22], increased venous thromboembolism risk in patients with a thrombogenic condition [23, 24], and hypercoagulability and endothelial damage in hypertensive patients [25]. At global level, our data corroborated previous evidence about the increased DD plasma levels in severe COVID-19 patients with thrombotic complications [6, 26]. Screening the three genotypic subclasses, we found for the first time a statistically significant association between increased plasma DD levels and carriers both of D/D and I/D genotypes in PE+ vs. PE− patients. Despite there is need to validate molecular mechanisms underlying this association, we hypothesized that the unbalance of ACE/ACE2 levels characterizing the D/D and I/D genotypes might induce apoptotic processes which target the endothelial cells of the vascular structure leading to coagulopathy and thus increased DD levels in COVID-19 patients.
The APOE locus has been associated with increased vulnerability to severe COVID-19 and mortality, especially for the APOE4 homozygous genotype (ε4/ε4) [27], which is the strongest genetic risk factor for sporadic Alzheimer’s disease. The APOE ε4 allele is characterized by the presence of a C at the APOE T112C locus and a C at the APOE T158C [28]. Our analysis showed that PE+ patients had a higher percentage of homozygous mutant C/C genotype at APOE T158C locus vs. PE− patients, even if we did not find statistically significant difference for the APOE T 112C locus. Also, DD levels were significantly higher in carriers of C/C and T/C genotypes at APOE T158C locus.
During pandemic, inherited thrombophilia has been associated with severe COVID-19 [29,30,31]. However, there is not a rigorous consensus regarding the genes which are truly involved in inherited thrombophilia. For example, the MTHFR polymorphism is considered as “benign” without a validated link to thrombosis thus questioning its inclusion in the current thrombophilia laboratory test panels [32]. In our study, we did not find statistical differences for MTHFR C677T polymorphism in PE+ vs. PE−, as well as for the other polymorphisms included in the panel. Considering the MTHFR C677T polymorphism, the prevalence of the homozygous TT genotype was 25.5% and 19.6% in PE− vs. PE+ patients (Table 1). These distributions are quite similar to known global levels of the MTHFR C677T polymorphism, for which the prevalence of the homozygous TT genotype was about 26% and 20% in Campania and Sicily (South Italy), respectively [33].
Nevertheless, our small sample size did not allow a secure finding but limited our observation to a hypothesis generating study.
The unique exception was the CBS 844ins68 polymorphism. Surprisingly, D/D homozygous wild-type genotype was most represented in PE+ (95.7%) vs. PE− (80.9%) patients. This might be due to the relatively small number of patients recruited in the study. As in oncology and cardiovascular diseases, the study of genetic regulation should be coupled with the investigation of epigenetic regulation of prothrombotic genes in severe COVID-19 patients and their possible involvement in immune reactivity [5, 34,35,36,37,38,39,40,41,42].
Our study has some limitations. First, this is a retrospective study. Second, we focused on COVID-19 patients with and without PE, with no control group (non-COVID-19 patients and/or healthy subjects) limiting the attribution of results to a specific association with COVID-19. Second, PE+ patients showed a higher prevalence of homozygous mutant ACE D/D genotype and APOE C/C genotype vs. PE− patients; but homozygous mutant ACE D/D genotype and APOE C/C genotype were also found in 40.4% and 68.1% of PE− patients (Table 1). This suggests a quite high absolute prevalence of the homozygous mutant polymorphisms in these genes. This somehow limits the predictivity of the mutant genotype for PE+.
Conclusions
Our data documented a differential distribution of homozygous mutant ACE D/D and APOE C/C genotypes between PE+ and PE− patients. Higher DD plasma levels significantly associated with carriers of D/D and I/D genotypes at ACE I/D locus as well as with carriers of C/C and T/C genotypes at APOE T158C locus in PE+ vs. PE− patients. These associations and the potential pathogenic role of these polymorphisms needs to be further evaluated in larger prospective studies.
Data availability
The data that support the findings of this study are available from the corresponding author, [GB], upon reasonable request.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- ACEi:
-
Angiotensin-converting enzyme inhibitors
- AGT:
-
Angiotensinogen
- AGTR-1:
-
Angiotensin II type 1 receptor
- APOE:
-
Apolipoprotein
- aPTT:
-
Activated partial thromboplastin time
- ARDS:
-
Acute respiratory distress syndrome
- BMI:
-
Body mass index
- CBS:
-
Cystathionine β-synthase
- CHD:
-
Coronary artery disease
- COPD:
-
Chronic obstructive pulmonary disease
- COVID-19:
-
Coronavirus disease 2019
- CRP:
-
C reactive protein
- DD:
-
D-Dimer
- FGB:
-
Fibrinogen β
- FII:
-
Factor II
- FiO2:
-
Fractional inspired oxygen
- FV:
-
Factor V
- FXIII:
-
Factor XIII
- GPIIIa:
-
Platelet glyciprotein IIIA
- IL:
-
Interleukin
- MTHFR:
-
Methylenetetrahydrofolate reductase
- PAI-1:
-
Plasminogen activator inhibitor-1
- PaO2:
-
Arterial oxygen partial pressure
- CRP:
-
C reactive protein
- PE:
-
Pulmonary embolism
- PT:
-
Prothrombin time PT-INR
- PT-INR:
-
Protrombin time-international normalized ratio
- RDB:
-
Reverse dot blot
- T2D:
-
Type 2 diabetes
References
Napoli C, Tritto I, Benincasa G, Mansueto G, Ambrosio G (2020) Cardiovascular involvement during COVID-19 and clinical implications in elderly patients. A review. Ann Med Surg (Lond) 57:236–243. https://doi.org/10.1016/j.amsu.2020.07.054
Becker RC (2020) Toward understanding the 2019 Coronavirus and its impact on the heart. J Thromb Thrombolysis 50:33–42. https://doi.org/10.1007/s11239-020-02107-6
Grimaldi V, Benincasa G, Moccia G, Sansone A, Signoriello G, Napoli C (2022) Evaluation of circulating leucocyte populations both in subjects with previous SARS-COV-2 infection and in healthy subjects after vaccination. J Immunol Methods 502:113230. https://doi.org/10.1016/j.jim.2022.113230
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3
Crimi E, Benincasa G, Figueroa-Marrero N, Galdiero M, Napoli C (2020) Epigenetic susceptibility to severe respiratory viral infections and its therapeutic implications: a narrative review. Br J Anaesth 125:1002–1017. https://doi.org/10.1016/j.bja.2020.06.060
Gorog DA, Storey RF, Gurbel PA, Tantry US, Berger JS, Chan MY, Duerschmied D, Smyth SS, Parker WAE, Ajjan RA, Vilahur G, Badimon L, Berg JMT, Cate HT, Peyvandi F, Wang TT, Becker RC (2022) Current and novel biomarkers of thrombotic risk in COVID-19: a Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat Rev Cardiol 19:475–495. https://doi.org/10.1038/s41569-021-00665-7
Rico-Mesa JS, Rosas D, Ahmadian-Tehrani A, White A, Anderson AS, Chilton R (2020) The role of anticoagulation in COVID-19-induced hypercoagulability. Curr Cardiol Rep 22:53. https://doi.org/10.1007/s11886-020-01328-8
McFadyen JD, Stevens H, Peter K (2020) The emerging threat of (Micro) thrombosis in COVID-19 and its therapeutic implications. Circ Res 127:571–587. https://doi.org/10.1161/CIRCRESAHA.120.317447
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, the Northwell COVID-19 Research Consortium, Barnaby DP, Becker LB, Chelico JD, Cohen SL, Cookingham J, Coppa K, Diefenbach MA, Dominello AJ, Duer-Hefele J, Falzon L, Gitlin J, Hajizadeh N, Harvin TG, Hirschwerk DA, Kim EJ, Kozel ZM, Marrast LM, Mogavero JN, Osorio GA, Qiu M, Zanos TP (2020) Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City Area. JAMA 323:2052–2059. https://doi.org/10.1001/jama.2020.6775
Scully EP, Haverfield J, Ursin RL, Tannenbaum C, Klein SL (2020) Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat Rev Immunol 20:442–447. https://doi.org/10.1038/s41577-020-0348-8
Oscanoa TJ, Vidal X, Coto E, Romero-Ortuno R (2021) ACE gene I/D polymorphism and severity of SARS-CoV-2 infection in hospitalized patients: a meta-analysis. Arterial Hypertens 25:112–118. https://doi.org/10.5603/AH.a2021.0018
Annunziata A, Coppola A, Di Spirito V, Cauteruccio R, Marotta A, Micco PD, Fiorentino G (2021) The angiotensin converting enzyme deletion/deletion genotype is a risk factor for severe COVID-19: implication and utility for patients admitted to emergency department. Medicina (Kaunas) 57:844. https://doi.org/10.3390/medicina57080844
Calabrese C, Annunziata A, Coppola A, Pafundi PC, Guarino S, Di Spirito V, Maddaloni V, Pepe N, Fiorentino G (2021) ACE gene I/D polymorphism and acute pulmonary embolism in COVID19 pneumonia: a potential predisposing role. Front Med (Lausanne) 7:631148. https://doi.org/10.3389/fmed.2020.631148
Delanghe JR, Speeckaert MM, De Buyzere ML (2020) The host’s angiotensin-converting enzyme polymorphism may explain epidemiological findings in COVID-19 infections. Clin Chim Acta 505:192–193. https://doi.org/10.1016/j.cca.2020.03.031
Yamamoto N, Ariumi Y, Nishida N, Yamamoto R, Bauer G, Gojobori T, Shimotohno K, Mizokami M (2020) SARS-CoV-2 infections and COVID-19 mortalities strongly correlate with ACE1 I/D genotype. Gene 758:144944. https://doi.org/10.1016/j.gene.2020.144944
Gómez J, Albaiceta GM, García-Clemente M, López-Larrea C, Amado-Rodríguez L, Lopez-Alonso I, Hermida T, Enriquez AI, Herrero P, Melón S, Alvarez-Argüelles ME, Boga JA, Rojo-Alba S, Cuesta-Llavona E, Alvarez V, Lorca R, Coto E (2020) Angiotensin-converting enzymes (ACE, ACE2) gene variants and COVID-19 outcome. Gene 762:145102. https://doi.org/10.1016/j.gene.2020.145102
Rice T, Rankinen T, Province MA, Chagnon YC, Perusse L, Borecki IB, Bouchard C, Rao DC (2000) Genome-wide linkage analysis of systolic and diastolic blood pressure: the Quebec Family Study. Circulation 102:1956–1963. https://doi.org/10.1161/01.cir.102.16.1956
Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F (1990) An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 86:1343–1346. https://doi.org/10.1172/JCI114844
Sarangarajan R, Winn R, Kiebish MA, Bountra C, Granger E, Narain NR (2020) Ethnic prevalence of angiotensin-converting enzyme deletion (D) polymorphism and COVID-19 risk: rationale for use of angiotensinconverting enzyme inhibitors/angiotensin receptor blockers. J Racial Ethn Health Dispar 8:1–8. https://doi.org/10.1007/s40615-020-00853-0
Lee YJ, Tsai JC (2002) ACE gene insertion/deletion polymorphism associated with 1998 World Health Organization definition of metabolic syndrome in Chinese type 2 diabetic patients. Diabetes Care 25:1002–1008. https://doi.org/10.2337/diacare.25.6.1002
Saab YB, Gard PR, Overall AD (2007) The geographic distribution of the ACE II genotype: a novel finding. Genet Res 89:259–267. https://doi.org/10.1017/S0016672307009019
Güngör Y, Kayatas M, Yildiz G, Özdemir Ö, Candan F (2011) The presence of PAI-1 4G/5G and ACE DD genotypes increases the risk of earlystage AVF thrombosis in hemodialysis patients. Ren Fail 33:169–175. https://doi.org/10.3109/0886022X.2011.552151
Di Tano G, Dede M, Pellicelli I, Martinelli E, Moschini L, Calvaruso E, Danzi GB (2022) Pulmonary embolism in patients with COVID-19 pneumonia on adequate oral anticoagulation. J Thromb Thrombolysis 53:576–580. https://doi.org/10.1007/s11239-021-02589-y
Lubbe L, Cozier GE, Oosthuizen D, Acharya KR, Sturrock ED (2020) ACE2 and ACE: structure-based insights into mechanism, regulation and receptor recognition by SARS-CoV. Clin Sci 134:2851–2871. https://doi.org/10.1042/CS20200899
Makris TK, Stavroulakis GA, Dafni UG, Gialeraki AE, Krespi PG, Hatzizacharias AN, Tsoukala CG, Vythoulkas JS, Kyriakidis MK (2000) ACE/DD genotype is associated with hemostasis balance disturbances reflecting hypercoagulability and endothelial dysfunction in patients with untreated hypertension. Am Heart J 140:760–765. https://doi.org/10.1067/mhj.2000.110764
Ibañez C, Perdomo J, Calvo A, Ferrando C, Reverter JC, Tassies D, Blasi A (2021) High D dimers and low global fibrinolysis coexist in COVID19 patients: what is going on in there? J Thromb Thrombolysis 51:308–312. https://doi.org/10.1007/s11239-020-02226-0
Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, Melzer D (2020) APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. J Gerontol A Biol Sci Med Sci 75:2231–2232. https://doi.org/10.1093/gerona/glaa131
Chaudhary S, Kaushik M, Ahmed S, Kukreti R, Kukreti S (2018) Structural Switch from Hairpin to Duplex/Antiparallel G-Quadruplex at Single-Nucleotide Polymorphism (SNP) Site of human Apolipoprotein E (APOE) gene coding region. ACS Omega 3:3173–3182. https://doi.org/10.1021/acsomega.7b01654
Stevens H, Canovas R, Tran H, Peter K, McFadyen JD (2022) Inherited thrombophilias are associated with a higher risk of COVID-19-associated venous thromboembolism: a prospective population-based cohort study. Circulation 2022(145):940–942. https://doi.org/10.1161/CIRCULATIONAHA.121.057394
Gardner AJ, Kirkin DJ, Rodriguez-Villar S, Leoz Abellanas G, Tee A, Valentin A (2021) Antithrombin III deficiency-induced coagulopathy in the context of COVID-19: a case series. Br J Haematol 194:1007–1009. https://doi.org/10.1111/bjh.17575
Lu H, Chen M, Tang S, Yu W (2021) Association of coagulation disturbances with severity of COVID-19: a longitudinal study. Hematology 26:656–662. https://doi.org/10.1080/16078454.2021.1968648
Deloughery TG, Hunt BJ, Barnes GD, Connors JM, Steering Committee WTD (2022) A call to action: MTHFR polymorphisms should not be a part of inherited thrombophilia testing. Res Pract Thromb Haemost 2022(6):e12739. https://doi.org/10.1002/rth2.12739
Ponti G, Pastorino L, Manfredini M, Ozben T, Oliva G, Kaleci S, Iannella R, Tomasi A (2021) COVID-19 spreading across world correlates with C677T allele of the methylenetetrahydrofolate reductase (MTHFR) gene prevalence. J Clin Lab Anal 35(7):e23798. https://doi.org/10.1002/jcla.23798
Benincasa G, Costa D, Infante T, Lucchese R, Donatelli F, Napoli C (2019) Interplay between genetics and epigenetics in modulating the risk of venous thromboembolism: a new challenge for personalized therapy. Thromb Res 177:145–153. https://doi.org/10.1016/j.thromres.2019.03.008
Napoli C, Benincasa G, Criscuolo C, Faenza M, Liberato C, Rusciano M (2021) Immune reactivity during COVID-19: implications for treatment. Immunol Lett 231:28–34. https://doi.org/10.1016/j.imlet.2021.01.001
Vietri MT, Albanese L, Passariello L, D’Elia G, Caliendo G, Molinari AM, Angelillo IF (2022) Evaluation of neutralizing antibodies after vaccine BNT162b2: preliminary data. J Clin Virol 146:105057. https://doi.org/10.1016/j.jcv.2021.105057
Napoli C, Tritto I, Mansueto G, Coscioni E, Ambrosio G (2020) Immunosenescence exacerbates the COVID-19. Arch Gerontol Geriatr 90:104174. https://doi.org/10.1016/j.archger.2020.104174
Sarno F, Benincasa G, List M, Barabasi AL, Baumbach J, Ciardiello F, Filetti S, Glass K, Loscalzo J, Marchese C, Maron BA, Paci P, Parini P, Petrillo E, Silverman EK, Verrienti A, Altucci L, Napoli C; International Network Medicine Consortium (2021) Clinical epigenetics settings for cancer and cardiovascular diseases: real-life applications of network medicine at the bedside. Clin Epigenetics 13(1):66. https://doi.org/10.1186/s13148-021-01047-z
Vietri MT, Caliendo G, Schiano C, Casamassimi A, Molinari AM, Napoli C, Cioffi M (2015) Analysis of PALB2 in a cohort of Italian breast cancer patients: identification of a novel PALB2 truncating mutation. Fam Cancer 14(3):341–348. https://doi.org/10.1007/s10689-015-9786-z
Businaro R, Scaccia E, Bordin A, Pagano F, Corsi M, Siciliano C, Capoano R, Procaccini E, Salvati B, Petrozza V, Totta P, Vietri MT, Frati G, De Falco E (2018) Platelet lysate-derived neuropeptide y influences migration and angiogenesis of human adipose tissue-derived stromal cells. Sci Rep 8(1):14365. https://doi.org/10.1038/s41598-018-32623-8
Vietri MT, Caliendo G, Casamassimi A, Cioffi M, De Paola ML, Napoli C, Molinari AM (2015) A novel PALB2 truncating mutation in an Italian family with male breast cancer. Oncol Rep 33(3):1243–1247. https://doi.org/10.3892/or.2014.3685
Benincasa G, Maron BA, Affinito O, D'Alto M, Franzese M, Argiento P, Schiano C, Romeo E, Bontempo P, Golino P, Berrino L, Loscalzo J, Napoli C (2022) Association between circulating CD4+ T cell methylation signatures of network-oriented SOCS3 gene and hemodynamics in patients suffering pulmonary arterial hypertension. J Cardiovasc Transl Res. https://doi.org/10.1007/s12265-022-10294-1
Funding
Open access funding provided by Università degli Studi della Campania Luigi Vanvitelli within the CRUI-CARE Agreement. There are no funding supporting this study.
Author information
Authors and Affiliations
Contributions
GF, GB, and CN designed the study. GB, AC, AA, MF, and OF wrote the manuscript. MF and OA performed all statistical analysis. MV contributed to literature search and draft of tables and figures. GF and CN revised the final manuscript and provided their intellectual content.
Corresponding author
Ethics declarations
Conflict of interest
The Authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fiorentino, G., Benincasa, G., Coppola, A. et al. Targeted genetic analysis unveils novel associations between ACE I/D and APO T158C polymorphisms with D-dimer levels in severe COVID-19 patients with pulmonary embolism. J Thromb Thrombolysis 55, 51–59 (2023). https://doi.org/10.1007/s11239-022-02728-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-022-02728-z