
Abstract
Bovine gammaherpesvirus 4 (BoHV-4) is a common virus detected in bovine with respiratory disease worldwide. In this study, we identified and characterized a novel BoHV-4 strain, referred as HB-ZJK, in vaginal swabs collected from cattle in China, 2022. The long unique region (LUR) of HB-ZJK is 10,9811 bp in length. It shares 99.17% to 99.38% nucleotide identity to five BoHV-4 strains available in GenBank and the highest similarity was seen with BoHV-4V. test (JN133502.1) strain (99.38%). Mutations, insertions or deletions were observed mainly in HB-ZJK gB (ORF8), TK (ORF21), gH (ORF22), MCP (ORF25), PK (ORF36), gM (ORF39), and gL (ORF47) genes compared to its genomic coordinates. Phylogenetic analyses of gB and TK genes showed that HB-ZJK clustered with China 512 (2019), B6010 (2009), and J4034 (2009) strains, demonstrating that the isolated HB-ZJK belongs to genotype 1. This is the first report that has revealed a comprehensive genome profile of BoHV-4 strain in China. This study will provide foundation for epidemiological investigations of BoHV-4 and contribute to the molecular and pathogenic studies of BoHV-4.
Similar content being viewed by others

Introduction
Bovine gammaherpesvirus 4 (BoHV-4) belongs to family Herpesviridae, subfamily Gammaherpesvirinae and genus Rhadinovirus [1]. BoHV-4 was first isolated from sick cattle with respiratory disease in Europe [2]. Then, it has been isolated from cattle with diarrhea, metritis, abortion, vaginitis in cattle [3,4,5]. BoHV-4 has been considered as an important pathogen of bovine respiratory tract and abortion or postpartum metritis [6,7,8,9]. Cattle are the natural hosts of the virus. In addition to infect cattle, BoHV-4 was reported to infect buffaloes, sheep, pig, and goats [10,11,12,13].
BoHV-4 is a double-stranded DNA virus with a genome size of approximately 145 kb. The genome is composed of a long unique coding region (LUR) and tandem repeats flanking at both ends called poly-repetitive DNA (prDNA) [1, 14]. LUR is conserved among BoHV-4 strains, whereas prDNA varies in size depending on the numbers of repetitions [15]. Only the long unique coding region of five BoHV-4 strains has been sequenced so far [1, 16]. BoHV-4 strains showed similar restriction profiles. Based on the restriction patterns, BoHV-4 strains were classified into three genotypes: genotypes 1–3 [17]. Genotype 1 and 2 correspond to or are closely related to the previously reported Movar 33/63-like strains and DN 599-like strains, respectively [17, 18]. BoHV-4 isolates from Argentina (07435 and 09227 strains) were designated as genotype 3 [17].
BoHV-4 glycoprotein B (gB) is a major component of the virion that is essential for viral infectivity, while TK contributes to understanding the regulation of specific genes and their promotor regions [19]. Both gB and TK genes were highly conserved among Herpesviridae family members and have been widely used to analyze the genotypes of BoHV-4 strains [10, 20,21,22].
To date, the transmission and infection of BoHV-4 were found in many countries, such as Belgium, USA, Italy, Germany, and Argentina [16, 22,23,24,25,26]. In China, although the information regarding the prevalence of BoHV-4 is less reported, the outcomes of the diseases caused by BoHV-4 should not be neglected. Therefore, identification and genetically characterization of the wild type strains will be essential for epidemiological investigations of BoHV-4 strains in China. However, there is only one research available regarding the identification of wild type of BoHV-4 strains. Three BoHV-4 strains including 512, BL6010 and J4034 strains were isolated from cattle in China. Phylogenetic analysis of partial sequences of TK and gB genes revealed that these three isolates belong to genotype 1 [10].
In 2022, we found some cattle developed severe respiratory inflammation and abortion symptoms on a cattle farm in Hebei, China. Given the clinical symptoms, we suspected that the causative agent of this disease might be BoHV. In this study, a BoHV-4 strain was isolated from the vaginal swabs of the sick cattle. Phylogenetic analysis based on gB and TK genes revealed that the isolated BoHV-4 strain belong to genotype 1, which is closely related to the strains identified in the mainland China.
Materials and methods
Cells and virus isolation
Madin-Darby bovine kidney cells (MDBK) were purchased from the American Type Culture Collection (ATCC) and maintained in the laboratory. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (BI, China) supplemented with 8% fetal bovine serum (FBS) (PAN Biotech, Germany) and incubated at 37 °C in 5% CO2 incubator. Twenty cattle vaginal swabs were collected from a cattle farm in Hebei Province, China, 2022. The positive samples of BoHV-4 were homogenized with DMEM and centrifuged at 6000×g for 5 min. The supernatant was filtered by 0.22 μm filter unit. Then, the supernatant was inoculated onto MDBK cell monolayers and grew for virus isolation by monitoring cytopathic effects (CPEs).
Polymerase chain reaction (PCR)
DNA was extracted from vaginal swabs and the supernatant of the infected cells using the Ezup column viral DNA kit (Sangon Biotech, China) separately. Specific primers were designed to amplify BoHV-4 gB (329 bp) and TK (216 bp) genes (Table 1). PCR reactions were done using DNA polymerase (Vazyme Biotech, China), 2 × Phanta Max Buffer (Vazyme Biotech, China), 10 mM dNTP Mix (Vazyme Biotech, China), 1 μL template, and 10 μM specific primers. PCR reactions were run under the following conditions: 95 °C for 1 min, 35 cycles at 95 °C for 10 s, 56 °C for 30 s, and 72 °C for 5 min.
Genomic sequencing and sequence analysis
Viral DNA was extracted from the supernatant of the infected cells using Viral DNA kit (Omega, USA) according to the manufactory’s instructions. The genome sequence was obtained using the second-generation sequencing platform in GeneScript company. The obtained genome sequence was compared and aligned with the published BoHV-4 genomes in the NCBI database. The obtained genome was further analyzed using the viral genome with the highest homology as reference. Phylogenetic analyses based on BoHV-4 gB and TK sequences were performed using MEGA software (version 7.0.26). Information of BoHV-4 gB and TK genes retrieved from GenBank for phylogenetic analysis were listed in Table 2. ORF8 (gB) amino acid sequences were aligned using the MegAlign process in DNASTAR Lasergene 11 software. Phylogenetic analysis of BoHV-4 (HB-ZJK) complete genome sequence was done using Neighbor Joining method in Molecular Evolutionary Genetics on NCBI.
Immunofluorescence assay (IFA)
To investigate virus infection and replication, MDBK cell monolayers grew on coverslips were infected with BoHV-4 at 0.1, 0.25, 0.5, 1, and 2.5 MOI. At 72 h post infection (hpi), the cells were fixed with 4% formaldehyde at 37 °C for 40 min. Then, the cells were washed with PBS buffer and blocked with PBS containing 5% non-fat milk at 37 °C for 45 min. After three washes with PBS buffer, the cells were incubated with BoHV-4 polysera (1:100 dilution) as primary antibody for 40 min. The cells were washed with PBS and incubated with anti-mouse IgG antibody as secondary antibody for 40 min. Finally, the cells were washed with PBS and stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were captured under an ECHO fluorescence microscope (RVL-100-g, USA). Notably, BoHV-4 polysera was prepared from mice challenged with BoHV-4.
Results
Isolation and biological characteristics of HB-ZJK
Three samples were identified as BoHV-4 positive from twenty cattle vaginal swabs (Fig S1). To isolate the strains, positive samples were inoculated on MDBK cells. Only one showed a visible CPE with round cells either shrunk or detached at 72 hpi (Fig. 1A). After three times purification, two fragments were amplified from the infected cells by PCR (Fig. 1B). Further sequence alignment revealed that these two amplified fragments showed high similarity to BoHV-4 gB gene (329 bp) and TK gene (216 bp), respectively. These data suggested that isolated virus was BoHV-4 strain. We referred it as HB-ZJK.

Identification and biological characterization of isolated HB-ZJK strain. A Isolation of virus in MDBK cells. CPE was observed on HB-ZJK-infected cells at 72 hpi. B Amplification of conserved gene in HB-ZJK-infected cells. Fragments was amplified from genomic DNA extracted from supernatant of infected cells using specific primers for BoHV-4 gB (lane 1) and TK genes (lane 2). C Viral infection was detected by IFA. The cells were infected with the virus at MOI = 0.1, 0.25, 0.5, 1, and 2.5 individually and incubated for 72 h. HB-ZJK virus was recognized using BoHV-4 polysera
To investigate the infection characteristics of the BoHV-4 (HB-ZJK) in MDBK cells, cells was infected with the purified BoHV-4 (MOI = 0.1, 0.25, 0.5, 1, and 2.5) for 72 h and subsequently analyzed by IFA. The results showed that the isolated BoHV-4 could be detected in MDBK cells infected with the virus (0.5, 1, and 2.5 MOI) at 72 hpi.
Characterization of HB-ZJK genome
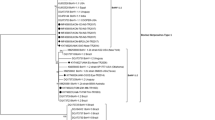
The genome of HB-ZJK was sequenced by the next-generation sequencing. After assembly, a 10,9811 bp genome of HB-ZJK was obtained. Although BoHV-4 has been reported in China, there is no genome map of BoHV-4 strain available to date. Here, we analyzed and organized the genome map of HB-ZJK. The results showed that HB-ZJK genome was collinear with USA SD16-49 (MN551084.1). The genome of HB-ZJK contains long unique sequence region with about 42% G-C content. Sequence analysis showed that HB-ZJK genome encoded 77 open reading frames (ORFs) in long unique region of the genome (Fig. 2). The gB, TK, gH, MCP, PK, gM, and gL genes were encoded by ORF8, ORF21, ORF22, ORF25, ORF36, ORF39, and ORF47, respectively (Fig. 2). These genes were marked in different colors in the organized genome map.

Organization of HB-ZJK genome map. The genome of HB-ZJK contains long unique sequence region (LUR) and 77 open reading frames (ORFs) in the LUR were illustrated
Analysis of HB-ZJK genome
We found that HB-ZJK genome shares 99.17% to 99.38% nucleotide identity to five BoHV-4 strains available in GenBank and the highest nucleotide identity (99.38%) was seen with V. test (JN133502.1). Genome sequence alignment showed that insertions, mutations, or deletions were observed in HB-ZJK gB (ORF8), TK (ORF21), gH (ORF22), MCP (ORF25), PK (ORF36), gM (ORF39), and gL (ORF47) genes compared to five previously reported BoHV-4 strains (Fig. 3A). Amino acid alignment of gB sequences showed that P (Pro) was deleted at position 23 compared with USA-SD16-49 and Canada-FMV09-1180503 strain (Fig. 3B); At position 45 of HB-ZJK gB, K (Lys) was mutated to Q (Gln). The position of 456 showed that S (Ser) was deleted compared with compared with USA-SD16-49 and Canada-FMV09-1180503 strain. Another mutation site was found in 468 L (Leu) when aligned with USA-SD16-49 and Canada-FMV09-1180503 stains. These analyses suggested that the genome of HB-ZJK is different compared with previous reported BoHV-4 strains.

Phylogenetic analysis of complete genome and alignment of ORF8 (gB) sequence. A Analysis of complete genome sequence for HB-ZJK. B Alignment of ORF8 (gB) amino acid sequence using the MegAlign process in DNASTAR Lasergene 11 software. C Phylogenetic analysis of HB-ZJK complete genome sequence using Neighbor Joining method in Molecular Evolutionary Genetics on NCBI
To explore the evolutionary status of HB-ZJK, phylogenetic tree was conducted and analyzed based on genome of BoHV-4. Phylogenetic analysis showed that the tree was divided into two branches (Fig. 3C). The HB-ZJK (China, 2022) was clustered with V. test (Belgium, 1981), SD16-38 (USA, 2019), and BoHV-4 (Germany, 2001) on one branch. FMV09-1180503 (Canada, 2009) and SD16-49 (USA, 2019) were clustered on another branch (Fig. 3C). These results suggested that the identified HB-ZJK might be a new outbreak strain in China.
Phylogenetic analysis of gB gene
To explore the genotype and evolutionary status of HB-ZJK. Phylogenetic tree of HB-ZJK gB was constructed based on 33 reference nucleotide sequences available in NCBI database. Phylogenetic analysis of gB showed that the tree was clustered in three major clades, including clade 1, 2, and 3 (Fig. 4). Clade 1 was mainly composed of HB-ZJK (China, 2022), 512 (China, 2019), B6010 and J4034 (China, 2009), TR/BHV4/2018/Bozdogan1 (Turkey, 2018), Uruguay (2018), and Italy (2014). Clade 2 was further divided into three Lineages. Lineage 1 included strains isolated in Argentina (2013, 2015, 2018), USA (2016), Turkey (2017). Lineages 2 consisted of strains isolated in Turkey (2007, 2009, 2010, 2014) and Argentina (2016). Lineages 3 was composed of T-6 strain isolated in Turkey (2009). Clade 3 consisted of strains isolated in Brazil (2011) and Turkey (2008, 2018). Based on these analysis, HB-ZJK was closely related to China 512 (2019), B6010 (2009), and J4034 (2009) strains, which belongs to genotype 1.

Maximum-likelihood analysis of HB-ZJK gB gene sequence. Thirty-three BoHV-4 gB nucleotide sequences available in GenBank were compared using MEGA7 software. Phylogenetic analysis of gB showed that the tree was clustered into clades 1, 2 and 3
Phylogenetic analysis of TK gene
Next, we constructed phylogenetic tree of HB-ZJK TK gene based on 30 reference nucleotide sequences available in GenBank. Phylogenetic analysis of TK gene showed that the tree was clustered into three clades, including clade 1, 2 and 3 (Fig. 5). Clade 1 was further divided into two lineages. Lineage 1 consists of HB-ZJK and other strains isolated in China (2009, 2019) and Italy (2018), which belong to genotype 1. Lineage 2 includes strains isolated in Argentina (2012, 2013, 2018), which are far away from the isolated strains in China (Fig. 5A). Clade 2 mainly includes strains isolated in Turkey (2017, 2018) and Argentina (2018). The strains isolated in Turkey (2009, 2017, 2018), Italy (2014), and Argentina (2018) were clustered on another branch in the clade 2. Clade 3 mainly clustered with strains isolated in Argentina (2015, 2016), USA (2016), Brazil (2007), and Turkey (2014).

Maximum-likelihood analysis of HB-ZJK TK gene sequence. Thirty BoHV-4 TK nucleotide sequences available in GenBank were compared using MEGA7 software. Phylogenetic analysis of TK gene showed that the tree was clustered into clades 1, 2 and 3
These data suggested that HB-ZJK was closely related to strains isolated in China (2009, 2019), Italy IZSM (2018), which belongs to genotype 1. Combing with the phylogenetic analysis based on gB gene, we can conclude that the isolated strain HB-ZJK belongs to genotype 1 and is closely related to the prevalent BoHV-4 strains in China.
Discussion
BoHV-4, a common pathogen that causes respiratory diseases, is prevalent on cattle farms worldwide. In 1963, it was first isolated from sick bovine with respiratory disease [2]. From 1989 to 2005, the global seroprevalence of BoHV-4 ranged from 4.2 to 30% on cattle farms [23, 27, 28]. In an investigation of Turkey, the seroprevalence of BoHV-4 reached 42% and 60% for cattle before 2-year-old and cattle after 2-year-old, respectively [29]. In China, transmission and infection of BoHV-4 were rarely reported in recent years [10]. In 2022, we isolated a BoHV-4 strain from sick cattle in Hebei, China. Transmission and infection of BoHV-4 might be occurring in the cattle farm in Hebei, China, 2022.
The genome of BoHV-4 was approximately 145 kb in length. It contains LUR with a size of about 110 kb and displayed about 41.4% G-C content [17, 30]. The LUR of BoHV-4 was reported to encode about 79 open reading frames (ORFs) [21]. Here, we identified that the LUR of the BoHV-4 HB-ZJK genome was 10, 9811 bp in length and displayed about 42% G-C content. This was consistent with previous reported. Unfortunately, we failed to identify the prDNA sequences flanking on both ends of the genome after several sequencing trials. Subsequently, we organized the LUR of the BoHV-4 genome using SD16-49 (MN551084.1) as reference. 77 ORFs were annotated as shown in Fig. 2. The comparison of the LUR with that of other BoHV-4 strains revealed insertions, mutations, or deletions occurred in HB-ZJK gB, TK, gH, MCP, PK, gM, and gL genes (Fig. 3). These data suggested that HB-ZJK strain had some antigenic changes.
The gB and TK genes have been used to construct phylogenetic tree for clustering BoHV-4 genotype. Based on gB and TK genes, phylogenetic analyses revealed that the isolated BoHV-4 (HB-ZJK) belongs to genotype 1 and was closely related to previous Chinese BoHV-4 isolates (Figs. 4 and 5). Due to BoHV-4 was capable of antigenic drift [3], we suspect that HB-ZJK might be originated from the same ancestor with 512 (2019), B6010 (2009), and J4034 (2009) strains. In addition, the sequence changes (insertions, mutations, or deletions of gene bases) of the LUR of the genome could be one reason that the transmission and infection of BoHV-4 were difficult to prevent and control on cattle farms.
BoHV-4 has been used as virus vector to deliver antigens. For instance, antigens from several viruses, such as Ebola virus, BHV-1, and PPRV were successfully expressed on BoHV-4 vector and immunization of these BoHV-4-antigens induced well neutralizing antibody responses [31,32,33,34,35]. Immunization of a recombinant vector (BoHV-4-A-PPRV-H-ΔTK) that delivers PPRV-H protein showed effective protection against PPRV infection [12]. Hence, the novel BoHV-4 strain might be used as virus vector for vaccine development.
Taken together, we have successfully isolated and characterized a BoHV-4 strain in China. The genome of BoHV-4 HB-ZJK was sequenced and a comprehensive map of the conserved long unique coding region was drawn. Phylogenetic analyses suggested that the BoHV-4 HB-ZJK belongs to genotype 1. This study will favor the epidemiological investigations of the prevalence of BoHV-4 strains on cattle farms in China.
Data availability
The data presented in the study are deposited in the GenBank, accession number OP631674.
References
Zimmermann W, Broll H, Ehlers B, Buhk HJ, Rosenthal A, Goltz M (2001) Genome sequence of bovine herpesvirus 4, a bovine Rhadinovirus, and identification of an origin of DNA replication. J Virol 75:1186–1194. https://doi.org/10.1128/jvi.75.3.1186-1194.2001
Bartha A, Juhász M, Liebermann H (1966) Isolation of a bovine herpesvirus from calves with respiratory disease and keratoconjunctivitis. A preliminary report. Acta Vet Acad Sci Hung 16:357–358
Frazier KS, Baldwin CA, Pence M, West J, Bernard J, Liggett A, Miller D, Hines ME 2nd (2002) Seroprevalence and comparison of isolates of endometriotropic bovine herpesvirus-4. J Vet Diagn Invest 14:457–462. https://doi.org/10.1177/104063870201400602
Izumi Y, Tsuduku S, Murakami K, Tsuboi T, Konishi M, Haritani M, Kamiyoshi T, Kimura K, Sentsui H (2006) Characterization of Bovine herpesvirus type 4 isolated from cattle with mastitis and subclinical infection by the virus among cattle. J Vet Med Sci 68:189–193. https://doi.org/10.1292/jvms.68.189
Miyano H, Haritani M, Sentsui H, Tsuboi T, Tanimura N, Kimura KM, Kobayashi M, Obara N, Akimoto Y (2004) Mammary lesions associated with bovine herpesvirus type 4 in a cow with clinical mastitis. J Vet Med Sci 66:457–460. https://doi.org/10.1292/jvms.66.457
Chastant-Maillard S (2015) Impact of Bovine Herpesvirus 4 (BoHV-4) on reproduction. Transbound Emerg Dis 62:245–251. https://doi.org/10.1111/tbed.12155
Egyed L, Sassi G, Tibold J, Mádl I, Szenci O (2011) Symptomless intrauterine transmission of bovine herpesvirus 4 to bovine fetuses. Microb Pathog 50:322–325. https://doi.org/10.1016/j.micpath.2010.10.006
Nikolin VM, Donofrio G, Milosevic B, Taddei S, Radosavljevic V, Milicevic V (2007) First Serbian isolates of bovine herpesvirus 4 (BoHV-4) from a herd with a history of postpartum metritis. New Microbiol 30:53–57
Florencia R, Julieta M, Sandra P, Enrique LU, Maia M, German C, Leunda MR, Erika GA, Susana P, Maximiliano S, Anselmo O, Leandro J, Verna AE (2020) Characterization of the first bovine gammaherpesvirus 4 strain isolated from an aborted bovine fetus in Argentina. Arch Virol 165:719–723. https://doi.org/10.1007/s00705-019-04507-3
Lin J, Chen RH, Yang MJ, Zhu YM, Xue F (2021) Isolation and molecular characterization of bovine herpesvirus 4 from cattle in mainland China. Arch Virol 166:619–626. https://doi.org/10.1007/s00705-020-04896-w
Dewals B, Thirion M, Markine-Goriaynoff N, Gillet L, de Fays K, Minner F, Daix V, Sharp PM, Vanderplasschen A (2006) Evolution of Bovine herpesvirus 4: recombination and transmission between African buffalo and cattle. J Gen Virol 87:1509–1519. https://doi.org/10.1099/vir.0.81757-0
Rodríguez-Martín D, Rojas JM, Macchi F, Franceschi V, Russo L, Sevilla N, Donofrío G, Martín V (2021) Immunization with bovine herpesvirus-4-based vector delivering PPRV-H protein protects sheep from PPRV challenge. Front Immunol 12:705539. https://doi.org/10.3389/fimmu.2021.705539
Pedrera M, Macchi F, McLean RK, Franceschi V, Thakur N, Russo L, Medfai L, Todd S, Tchilian EZ, Audonnet JC, Chappell K, Isaacs A, Watterson D, Young PR, Marsh GA, Bailey D, Graham SP, Donofrio G (2020) Bovine herpesvirus-4-vectored delivery of Nipah virus glycoproteins enhances T cell immunogenicity in pigs. Vaccines. https://doi.org/10.3390/vaccines8010115
Thiry E, Bublot M, Dubuisson J, Van Bressem MF, Lequarre AS, Lomonte P, Vanderplasschen A, Pastoret PP (1992) Molecular biology of bovine herpesvirus type 4. Vet Microbiol 33:79–92. https://doi.org/10.1016/0378-1135(92)90037-t
Ehlers B, Buhk HJ, Ludwig H (1985) Analysis of bovine cytomegalovirus genome structure: cloning and mapping of the monomeric polyrepetitive DNA unit, and comparison of European and American strains. J Gen Virol 66(Pt 1):55–68. https://doi.org/10.1099/0022-1317-66-1-55
Palmeira L, Machiels B, Lete C, Vanderplasschen A, Gillet L (2011) Sequencing of bovine herpesvirus 4 v.test strain reveals important genome features. Virol J 8:406. https://doi.org/10.1186/1743-422X-8-406
Verna AE, Manrique JM, Perez SE, Leunda MR, Pereyra SB, Jones LR, Odeon AC (2012) Genomic analysis of bovine herpesvirus type 4 (BoHV-4) from Argentina: high genetic variability and novel phylogenetic groups. Vet Microbiol 160:1–8. https://doi.org/10.1016/j.vetmic.2012.04.039
Thiry E, Dubuisson J, Bublot M, Van Bressem MF, Pastoret PP (1990) The biology of bovine herpesvirus-4 infection of cattle. Dtsch Tierarztl Wochenschr 97:72–77
Lomonte P, Bublot M, Pastoret PP, Thiry E (1992) Location and characterization of the bovine herpesvirus type 4 thymidine kinase gene; comparison with thymidine kinase genes of other herpesviruses. Arch Virol 127:327–337. https://doi.org/10.1007/BF01309595
Pérez S, Manrique J, Morán P, Romeo F, Angelini H, Leunda MR, Pereyra S, Spetter M, González Altamiranda E, Odeón A, Jones L, Verna A (2020) Genetic characterization of bovine herpesvirus 4 (BoHV-4) isolates from Argentine cattle suggests a complex evolutionary scenario. Mol Biol Rep 47:4905–4909. https://doi.org/10.1007/s11033-020-05449-9
Gagnon CA, Traesel CK, Music N, Laroche J, Tison N, Auger JP, Music S, Provost C, Bellehumeur C, Abrahamyan L, Carman S, DesCôteaux L, Charette SJ (2017) Whole genome sequencing of a Canadian bovine gammaherpesvirus 4 strain and the possible link between the viral infection and respiratory and reproductive clinical manifestations in dairy cattle. Front Vet Sci 4:92. https://doi.org/10.3389/fvets.2017.00092
Areda D, Chigerwe M, Crossley B (2018) Bovine herpes virus type-4 infection among postpartum dairy cows in California: risk factors and phylogenetic analysis. Epidemiol Infect 146:904–912. https://doi.org/10.1017/s0950268818000791
Graham DA, McNeill GJ, Calvert V, Mawhinney K, Curran W, Ball NW, Todd D (2005) Virological and serological evidence of bovine herpesvirus type 4 in cattle in Northern Ireland. Vet Rec 157:539–543. https://doi.org/10.1136/vr.157.18.539
Bellino C, Iussich S, Biasato I, Peletto S, Caruso C, Gianella P, Cagnasso A, D’Angelo A (2015) Potential pathogenetic role of bovine herpesvirus 4 in two dairy cows with dermatitis-pyrexia-hemorrhagic syndrome. J Clin Microbiol 53:2763–2767. https://doi.org/10.1128/jcm.00717-15
Bilge Dağalp S, Dogan F, Babaoglu AR, Aligholipour Farzani T, Alkan F (2021) Genetic variability of bovine herpesvirus type 4 (BoHV-4) field strains from Turkish cattle herds. Vet Ital 57:49–59. https://doi.org/10.12834/VetIt.2095.11150.1
González Altamiranda E, Manrique JM, Pérez SE, Ríos GL, Odeón AC, Leunda MR, Jones LR, Verna A (2015) Molecular characterization of the first bovine herpesvirus 4 (BoHV-4) strains isolated from in vitro bovine embryos production in Argentina. PLoS One 10:e0132212. https://doi.org/10.1371/journal.pone.0132212
Naeem K, Goyal SM, Werdin RE (1989) Prevalence of bovid herpesvirus-4 and its antibody in cattle in Minnesota. Am J Vet Res 50:1931–1935
Wellenberg GJ, van Rooij EM, Maissan J, Van Oirschot JT (1999) Evaluation of newly developed immunoperoxidase monolayer assays for detection of antibodies against bovine herpesvirus 4. Clin Diagn Lab Immunol 6:447–451. https://doi.org/10.1128/cdli.6.4.447-451.1999
BilgeDagalp S, Demir AB, Gungor E, Alkan F (2007) The seroprevalence of bovine herpesvirus type 4 (BHV4) infection in dairy herds in Turkey and possible interaction with reproductive disorders. Rev Med Vet 158:201–205
Yamamoto Y, Murakami K, Inoshima Y, Nakane T, Saika K, Sentsui H (2000) Characterization of a bovine herpesvirus type 4 isolated from the spinal cord of a cow with astasia. Arch Virol 145:2363–2370. https://doi.org/10.1007/s007050070026
Rosamilia A, Jacca S, Tebaldi G, Tiberti S, Franceschi V, Macchi F, Cavirani S, Kobinger G, Knowles DP, Donofrio G (2016) BoHV-4-based vector delivering Ebola virus surface glycoprotein. J Transl Med 14:325. https://doi.org/10.1186/s12967-016-1084-5
Bilge-Dagalp S, Farzani TA, Dogan F, Akkutay Yoldar Z, Ozkul A, Alkan F, Donofrio G (2021) Development of a BoHV-4 viral vector expressing tgD of BoHV-1 and evaluation of its immunogenicity in mouse model. Braz J Microbiol 52:1119–1133. https://doi.org/10.1007/s42770-021-00525-z
Donofrio G, Cavirani S, Vanderplasschen A, Gillet L, Flammini CF (2006) Recombinant bovine herpesvirus 4 (BoHV-4) expressing glycoprotein D of BoHV-1 is immunogenic and elicits serum-neutralizing antibodies against BoHV-1 in a rabbit model. Clin Vaccine Immunol 13:1246–1254. https://doi.org/10.1128/cvi.00200-06
Shringi S, O’Toole D, Cole E, Baker KN, White SN, Donofrio G, Li H, Cunha CW (2021) OvHV-2 glycoprotein B delivered by a recombinant BoHV-4 is immunogenic and induces partial protection against sheep-associated malignant catarrhal fever in a rabbit model. Vaccines (Basel). https://doi.org/10.3390/vaccines9020090
Macchi F, Rojas JM, Verna AE, Sevilla N, Franceschi V, Tebaldi G, Cavirani S, Martín V, Donofrio G (2018) Bovine herpesvirus-4-based vector delivering Peste des Petits ruminants virus hemagglutinin ORF induces both neutralizing antibodies and cytotoxic T cell responses. Front Immunol 9:421. https://doi.org/10.3389/fimmu.2018.00421
Acknowledgements
This research work was funded by the National Key Research and Development Program of China and the National Nature Science Foundation. This research work is conducted in Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences. The authors would like to thank these.
Funding
This experiment was supported by the National Key Research and Development Program of China (No. 2021YFD1801401-4), National Nature Science Foundation of China (grant numbers 32170161 and U19A2039).
Author information
Authors and Affiliations
Contributions
WG, and HC designed the experiments. WG, TS, XD, CT et al. performed these experiments. WG, HC, and JL organized and analyzed the data. Reagents, materials and analytical tool were helped with HC and JL. The manuscript was written by WG together with HC, and JL. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Edited by: Zhen Fu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guo, W., Sun, T., Liu, Y. et al. Characterization and phylogenetic analysis of bovine gammaherpesvirus 4 isolated in China, 2022. Virus Genes 59, 417–426 (2023). https://doi.org/10.1007/s11262-023-01981-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-023-01981-5