
Abstract
The pancreas is an organ with a central role in nutrient breakdown, nutrient sensing and release of hormones regulating whole body nutrient homeostasis. In diabetes mellitus, the balance is broken—cells can be starving in the midst of plenty. There are indications that the incidence of diabetes type 1 and 2, and possibly pancreatogenic diabetes, is rising globally. Events leading to insulin secretion and action are complex, but there is emerging evidence that intracellular nucleotides and nucleotides are not only important as intracellular energy molecules but also as extracellular signalling molecules in purinergic signalling cascades. This signalling takes place at the level of the pancreas, where the close apposition of various cells—endocrine, exocrine, stromal and immune cells—contributes to the integrated function. Following an introduction to diabetes, the pancreas and purinergic signalling, we will focus on the role of purinergic signalling and its changes associated with diabetes in the pancreas and selected tissues/organ systems affected by hyperglycaemia and other stress molecules of diabetes. Since this is the first review of this kind, a comprehensive historical angle is taken, and common and divergent roles of receptors for nucleotides and nucleosides in different organ systems will be given. This integrated picture will aid our understanding of the challenges of the potential and currently used drugs targeted to specific organ/cells or disorders associated with diabetes.
Similar content being viewed by others
Introduction
Diabetes
The incidence of diabetes mellitus (types 1 and 2) is globally rising, and its appearance is shifting towards a younger age group. Type 1 diabetes (T1D), or insulin-dependent diabetes mellitus, is an autoimmune disease that precipitates in genetically predisposed individuals by environmental factors, predominantly viral infections. As a result, pancreatic β-cell mass and function deteriorate and patients become dependent on exogenous insulin [1]. In type 2 diabetes (T2D), insulin secretion may be close to normal, at least in the beginning, but target tissues may be resistant to insulin. With progression of the disease, metabolic stress factors and cytokines, such as interleukin IL-1β, contribute to decrease in β-cell mass and function [2, 3]. T2D is usually a later onset disease; it is often associated with obesity and a low-grade inflammation of adipose tissue and auto-inflammation in islets, and subsequently altered adipokines profiles may in part contribute to an induction of hepatic and skeletal muscle insulin resistance [4, 5].
Another, less well recognised, form of diabetes mellitus is due to exocrine pancreatic dysfunction; the disease accounts for about 10% of diabetic patients, but prevalence may depend on region/population. This pancreatogenic diabetes, referred to as type 3c diabetes, occurs due to inherited or acquired pancreatic disease and has a unique pattern of hormonal and metabolic characteristics and diagnosis may be difficult [6–9]. Links between concurrent exocrine and endocrine pancreatic disease and contributing factors are poorly characterised. Usually, the exocrine pathology is explained as a result of local insulin deficiency or neuropathological changes in pancreas as a result of diabetes or that autoimmune disease could involve both endocrine and exocrine parts of the gland. The close morphological association between exocrine cells, especially ducts, and islets and release of various cytokines from exocrine cells suggests that the exocrine–endocrine axis is important [10–12]. Recent studies also show that genetic mutation coding for the acinar digestive enzyme, carboxyl ester lipase, leads to diabetes [6]. It is proposed that pancreatogenic diabetes may be more common and frequently associated with inflammation and sub-clinical chronic pancreatitis [13, 14].
Generally, diabetic mellitus diseases have in common inadequate insulin-regulated glucose transport and metabolism in major target tissues—liver, skeletal muscle and adipose tissue. This results in high circulating levels of glucose, free fatty acids and pro-inflammatory cytokines that cause serious problems in many organs, such as cardiovascular diseases, neuropathy and pain, renal disease, disturbances in the urogenital and gastrointestinal system, skin healing problems and skeletal muscle weakness. Events leading to insulin production and action are complex, but there is emerging evidence that various components of purinergic signalling may be important regulatory factors, both at the level of the pancreas and at the level of the organs affected. Following the introduction to the pancreas and purinergic signalling, we will focus on purinergic signalling associated with diabetes in selected organ systems.
In order to study the basic mechanism of diabetes, many cell and animal models have been developed (see for reviews [15, 16]). Here, we mention a few common animal models, which have been of particular use in studying the complexity of organ defects in purinergic signalling. Streptozotocin (STZ)-induced diabetes in rats has been widely used [17], but has been questioned as a valuable model for some aspects of diabetes in man. Other animal models include alloxan-induced diabetes [18, 19], Bio Breeder diabetic rats (BBD) [20], non-obese diabetic (NOD) mice [21] and the murine model of T1D, the RIP-I/hIFNβ transgenic mouse treated with very low doses of STZ [22]. For T2D diabetes, leptin-deficient or leptin-resistant mice (ob/ob and db/db) and Zucker diabetic fatty rats (ZDF) [23] are the most common models.
In diabetes mellitus, the basic cellular defects in metabolism lead to altered intracellular nucleotide levels. The next section will show that nucleotides have also important regulatory roles on the outside of the cell.
Purinergic signalling
The purinergic system is a signalling system, where the purine nucleotides, ATP and ADP, and the nucleoside, adenosine, act as extracellular messengers. This concept, which was first proposed 40 years ago [24], met with considerable resistance for many years, largely because ATP had been established as an intracellular energy source involved in various metabolic cycles, and it was thought that such a ubiquitous molecule was unlikely to be involved in selective extracellular signalling. However, ATP was one of the first molecules to appear in biological evolution so that it is not really surprising that it should have been utilised early for extracellular, as well as intracellular, purposes. The existence of potent extracellular enzymes that regulate the amount of ATP and adenosine available for signalling also provides support that ATP has extracellular actions [25]. Implicit in purinergic signalling is the presence of receptors for ATP. A basis for distinguishing adenosine receptors (P1), from ATP/ADP receptors (P2), was proposed in 1978 [26]. The turning point in acceptance of the concept of purinergic signalling was in the early 1990 s, when receptor subtypes were cloned and characterised. Four subtypes of P1 receptors have been cloned, namely, A1, A2A, A2B and A3. A1 and A3 receptors preferentially couple to Gi proteins and inhibit adenylate cyclase, while A2A and A2B couple to Gs and Go and stimulate production of cyclic AMP (cAMP). P2 receptors belong to two families based on molecular structure and second messenger systems, namely P2X ionotropic ligand-gated ion channel receptors and P2Y metabotropic G protein-coupled receptors [27, 28]. There are currently seven subtypes of P2X receptors and eight subtypes of P2Y receptors identified and characterised in mammals [29]. Most P2Y receptors couple to Gq/G11 proteins and thus activate PLC-β, except for P2Y12, P2Y13 and P2Y14 that couple to Gi proteins and inhibit adenylate cyclase, and P2Y11 couples to Gs and Gq [30]. Functional purinoceptors are expressed by neurons and most non-neuronal cells (see [31]), including various cells in the pancreas. Two of the agonist-bound receptors, A2A and P2X4, have been crystallised [32, 33]. Extracellular nucleotide and nucleoside concentrations are regulated by a large number of ecto-nucleotidases, and some of those have also been crystallised [34].
Purinergic signalling in healthy pancreas
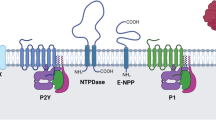
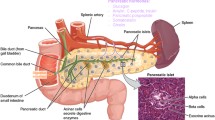
The pancreas is an organ with a central role in nutrient breakdown, nutrient sensing and release of hormones regulating whole body nutrient homeostasis. The close apposition of various cells types, and indications that there is an interrelation between endocrine and exocrine diseases, warrants analysis of the integrated picture and the possibility that purinergic signalling may play a coordinating role (Fig. 1). There have been a number of reviews on the role of purinergic signalling in both endocrine and exocrine pancreas [35–39]. Here, we include only a brief overview, with the latest updates.

Integrated function of pancreas in nutrient breakdown, nutrient sensing and release of pancreatic hormones. Purinergic signalling plays significant roles in physiological responses as well as in diabetes. Distribution of key receptors in pancreatic cells is shown and also locally produced and blood-born factors that could affect insulin release and/or β-cell viability. Pancreatic acini secrete digestive enzymes and ATP. Pancreatic ducts express receptors that are involved in regulation of bicarbonate-rich fluid secretion. Both exocrine cells can contribute to the interstitial milieu in the form of nucleotides/nucleosides or secreted cytokines. β Cells secrete insulin and ATP and purinergic receptors stimulate or inhibit insulin secretion, while others regulate cell viability. In addition, α cells express receptors that regulate glucagon secretion. Figure also shows GLP-1 and GIP that regulate both insulin secretion and β-cell mass. For details, see the text
There are numerous sources of nucleotides/side within the pancreas. ATP/ADP is most likely a cotransmitter in both sympathetic and parasympathetic nerves supplying the pancreas and nucleotides are stored and released from hormone-containing vesicles from endocrine cells [40]. In addition, ATP is released from enzyme-containing zymogen granules from acini, where it is accumulated by the vesicular nucleotide transporter VNUT [41]. Apart from vesicle/granule exocytosis, there may be other mechanisms for ATP release, and this is highly debated and currently an active research field [42]. A number of early biochemical studies have shown that the pancreas expresses several types of ecto-nucleotidases enabling conversion of ATP/ADP to adenosine. In pancreatic islets, ATP pyrophosphatase, alkaline phosphatase, ecto-5′-nucleotidase as well as NTDPase-3 were found [43]. In exocrine pancreas, NTPDase-1 and ecto-5′-nucleotidase were found in acini, in particular in zymogen granules, and these were secreted into pancreatic juice in a particular form (microvesicles). In addition, there are also ATP-generating enzymes, adenylate kinase and nucleoside diphosphate kinase, found in pancreatic juice [44]. NTPDase-1 and NTPDase-2 are also expressed in duct cells, as well as on blood vessels where the ATP/ADPase activity was strongest. For the latest review on the cellular and molecular action of ecto-nucleotidases, see Zimmermann et al. [34].
It has been shown that human β cells express P2X3, P2X5, P2X7 and P2Y11 and P2Y12 receptors. A large number of studies on rodent pancreas, islet and cell preparations and β-cell lines revealed that these express the following P2 receptor subtypes: P2X1–P2X7, PY1, P2Y2, P2Y4 and P2Y6, P2Y11–P2Y13; although the functions of some are not known and there are likely to be differences between species. In general, some P2 receptors mediate stimulation of insulin release already in non-stimulating glucose concentrations, while others may mediate potentiation of glucose-induced insulin secretion (see [38, 39]). Furthermore, some P2 receptor subtypes (e.g. mP2Y13, rP2X3) have inhibitory effects on insulin secretion, probably by stimulating different signalling pathways. Regarding adenosine receptors, earlier studies established that β cells also express A1 receptors that mediate inhibition of insulin secretion (see [39]). Recent studies indicate that A2B receptors might also be expressed on β cells where they mediate inhibition of insulin secretion directly [45] or via the immune system [46]. Another study also implicates A2A receptors, which mediate increase in β-cell proliferation [47]. Together, activation of P1 and P2 receptors could exert pulsatile and synchronising effects on secretion of insulin and glucagon and thus contribute to balanced blood glucose regulation [35, 37, 48]. Recent studies also show that P2 receptors may mediate regulation of β-cell mass, which would be highly relevant to diabetes and is discussed below.
In contrast to β cells, there is less information about purinergic regulation of other islet cells. α Cells express A2A receptors, which mediate stimulation of glucagon release, while A1 receptors mediate inhibition. The P2Y6 receptor also mediates stimulation of glucagon release, while the P2Y1 receptor mediates inhibition. ADP analogues stimulate somatostatin secretion from δ cells most likely via the P2Y1 receptor. P2X7 receptors have been immunolocalised on α cells [49]. δ Cells express P2Y1 receptors [50].
In exocrine pancreas, acini are a rich site of ATP release, and they also express and release ectonucleotide/side breakdown enzymes (see above). Rodent acini contain transcripts for P2Y2, P2Y4, P2X1 and P2X4 receptors, but their functionality is low, perhaps indicating a protective mechanism curbing release of digestive enzymes. Secreted luminal ATP, and adenosine, and also ATP released from nerve endings and duct cells can regulate pancreatic duct function, which is secretion of HCO3 − rich fluid that together with acinar enzymes contributes to digestive processes within the small intestine. Pancreatic ducts from rodents and human cell lines express P2Y1, P2Y2, P2Y4, P2Y6 and P2Y11–14, and P2X1, P2X2 and P2X4–P2X7 receptors. Functional studies show that luminal P2Y receptors mediate stimulation of mucin secretion and HCO3 −/H+ transport, and in particular, they stimulate fluid secretion by activating Cl− channels (TMEM16A and CFTR), as well as K+ channels (IK, KCa3.1), which potentiate the secretory effect [51]. P2X7 receptors also mediate regulation of pancreatic secretion. On the basolateral membrane some receptors, e.g. P2Y2, mediate down-regulation of secretion, probably a safety mechanism in over-distended duct. A recent study on the human adenocarcinoma cell line, PANC-1, shows that P2Y1 and P2Y6 receptors mediate increase in proliferation [52]. Pancreatic ducts also express A2A and A2B receptors that mediate stimulation of CFTR Cl− channels and therefore secretion.
Pancreatic stellate cells (PSCs) are thought to be important in inflammation and fibrosis, and their role in pancreas cancer and chronic pancreatitis has been well studied, but their role in diabetes is less clear. At the messenger RNA (mRNA) level, PSCs express P2Y1, P2Y2, P2Y6, P2X1, P2X4 and P2X6 receptors. Micromolar concentrations of ATP stimulate nuclear Ca2+ signals, which may stimulate proliferation of this cell type [53]. A recent study shows that the P2X7 receptor on PSC also can stimulate proliferation of these cells but also cell death, depending on the ATP concentrations [54].
Purinergic signalling in diabetes
Pancreas
Let us start with intracellular events in β cells, where the primary defect in intracellular signalling, membrane transporters, metabolic processes or regulation thereof, may lead to a decrease in insulin secretion and, in the worst case, cell death. In normal β cells, glucose entry via GLUT (GLUT2 in rodents, GLUT1/3 in humans) and its metabolism leads to the production of ATP, which closes ATP sensitive K+ channels (KATP), and this in turn leads to depolarisation of the plasma membrane, influx of Ca2+ and a chain of signalling events that culminate in exocytosis of insulin (and ATP). Two incretins, GIP and GLP-1, potentiate insulin secretion and synthesis, as well as preserving β-cell mass [55]. In normal pancreatic β cells, glucose also stimulates polyphosphoinositide (PPI) hydrolysis via activation of a phosphoinositide-specific phospholipase C. In STZ-injected neonatal rats, glucose-induced PPI hydrolysis was severely diminished and was associated with reduced insulin-secreting responses to glucose [56]. A further cytotoxic effect of STZ on β cells may be due to a reduction in the intracellular level of ATP and thus activation of KATP, hyperpolarisation of cell membrane and reduction of insulin release [57]. Nevertheless, KATP channel openers, such as diazoxide, can counteract chronic over-stimulation of β cells and have been proposed to have beneficial effects in a subgroup of T2D [58]. Gain-of-function mutations in KATP channel subunits SUR1 or Kir6.2 can cause transient or permanent neonatal diabetes, for example, as demonstrated in a study of R826W mutation in SUR1 [59]. The intracellular ATP/ADP ratio is a coupling factor between glucose metabolism and insulin release [60]. Glycation end products, which are implicated in diabetic complications, inhibit cytochrome c oxidase and ATP production, resulting in impairment of glucose-stimulated insulin secretion [61].
In addition to intracellular functions, extracellular nucleotides/sides regulate pancreatic islet cells (Fig. 1), as well as a range of organs affected in diabetes. Evidence for the role of purinergic signalling in relation to diabetes was obtained in early experiments in the 1970–1980 s. Studies on a diabetic rat model using alloxan and dithizone [62, 63] showed that ATP was protective by reducing blood sugar levels. Infusion of ATP into the carotid artery increased the sensitivity of alloxan-diabetic rats to glucose, suggesting that a possible cause of diabetes was a defect in purinergic innervation of the islet cells [64].
Two early studies on different animal models of T1D and T2D showed that, although the insulin response to glucose was lost, insulin secretion to P2 receptor agonists was preserved (Fig. 2). In the isolated perfused pancreas of STZ-rats, ADPβS, a potent P2Y1 receptor agonist induced insulin release that was similar to that in control rats, and this was independent of glucose concentration 5–28 mM [65, 66]. In ZDF rats, increases in [Ca2+]i and insulin secretion by pancreatic β cells were preserved and mediated by P2Y receptors, again with ADPβS being most potent [66]. Consequently, a number of studies focussed on developing stable P2Y1 receptor agonists as potential insulin secretagogues (e.g. 2-methylthio ATP-α-β, A isomer and stable dinucleoside polyphosphate analogues); however, a vasodilatory effect was a risk factor [67–70]. Also using a mouse model, another pattern regarding the P2Y1 receptor appears. In islets isolated from P2Y1 −/− mice, insulin secretion was significantly increased at high stimulating glucose concentrations compared to wild type [71].

Perfusion of isolated rat pancreas with ADPβS and glucose. a Pancreata isolated from Zucker lean controls (ZLC) were perfused for 30 min with perfusate containing 5 mM glucose (first 15 min not shown). ADPβS (15 μM) was applied into perfusate as shown by horizontal bar. ADPβS produced a biphasic insulin release. Raising glucose to 10 mM produced a biphasic insulin secretion. b Pancreata isolated from Zucker diabetic fatty (ZDF) rats were perfused using the same protocol as in a. ADPβS (15 μM) caused a much larger biphasic insulin secretion while glucose induced a small and transient insulin release. c Perfusion of isolated rat pancreas from Wistar rats with ADPβS and glucose. (Reproduced from [66], with permission)
In a histochemical study on STZ-diabetic rats, the following picture was revealed. In pancreatic islets, P2Y1 receptors were present in intra-islet capillaries, while P2X4 receptors were present on β and δ cells. Pancreatic duct cells still expressed P2Y1 and P2Y2 receptors, while P2X1, P2X3, P2Y1 and P2Y2 receptors were expressed in small pancreatic blood vessels [72]. α Cells expressed P2X7 receptors in healthy pancreas on the periphery of islets. These P2X7-immunoreactive cells migrated to the centres of islets to replace the lost β cells in both STZ-diabetic rats and NOD mice [72, 73]. In another study, P2X7 receptors were also claimed to be expressed β cells; P2X7 receptors were down-regulated in T2D, but up-regulated in human obesity [74]. In human islets, the receptor seems to be involved in secretion of insulin and the IL-1 receptor antagonist IL-1Ra.
Regarding adenosine receptors, older studies showed that STZ diabetes suppressed the stimulatory action of adenosine on glucagon secretion from pancreatic α cells and reduced vasodilation of the vascular bed [75, 76], via A2 receptors [77]. Several studies showed that a nonspecific adenosine receptor agonist, adenosine-5′-N-ethylcarboxamide (NECA), decreased insulin secretion and increased blood glucose and decreased glucose uptake. Studies on A1 −/− mice showed increased insulin and glucagon secretion [78]. A recent study on A1 receptor knockout mice showed that fasting glucose and insulin secretion were significantly higher, but insulin sensitivity was impaired as reflected by reduced glucose uptake in muscles and adipose tissue [79]. However, the effects could not simply be explained by an A1 receptor effect on insulin secretion of β cells. Several studies with non-selective adenosine receptor agonists showed the following. NECA decreased blood glucose in STZ-diabetic and cyclophosphamide-treated NOD mice; it increased pancreas insulin content and suppressed expression of pancreatic proinflammatory cytokines (TNF-α, MIP-1α, IL-12 and INF-γ) and in immunoreactive cells [80]. This effect was most likely mediated via the A2B receptor, although the authors proposed that this was not at the level of the pancreas. In other studies, infusion of specific A2B receptor blockers increased insulin secretion in INS-1 cells and also in T2D model of Goto-Kakizaki rats, indicating that the effect could be at the pancreas level [45]. Furthermore, a study on a mouse model for T2D suggested that the increased expression of A2B receptors on endothelial cells and macrophages enhances production of IL-6 and that this results in stimulation of an inflammatory response and insulin resistance in skeletal muscle, adipose tissue and liver; effects on pancreas were also considered [46].
A key factor in the pathogenesis of diabetes is the pancreatic β-cell mass (especially T1D). In order to understand β-cell function and survival at the integrative level, exploration of the mechanisms of purinergic signalling together with incretins and inflammatory signals will be necessary. Here, are some trends in these directions. Pro- and anti-inflammatory cytokines, originating from various local or invading cells or from other organs, can influence proliferation and apoptosis of β cells [81]. A number of purinergic receptors have similar abilities to mediate cell proliferation and apoptosis and activities via the P2X7 receptor may be able to support both functions [82, 83]. The P2X7 receptor and cytokine systems can be related. For example, in macrophages, the P2X7 receptor is involved IL-1β secretion, and in T cells, it caused MHC-I shedding and extravasation [82]. The two incretins, GLP-1 and GIP, augment insulin secretion, but also have proliferative and anti-apoptotic effects on β-cell mass [55].
Recently, there have been studies addressing the question of purinergic signalling and β-cell survival, and the summary of data is depicted in Fig. 3. P2Y6 receptor agonists not only increase insulin secretion in MIN6 mouse β cells, but they also prevent β-cell death induced by tumour necrosis factor-α [84]. In contrast, activation of the P2Y13 receptor of the mouse pancreatic insulinoma cell line, MIN6C4, has pro-apoptotic effects [85]. Furthermore, high glucose and free fatty acids induce β-cell apoptosis via autocrine ADP action on the P2Y13 receptor [86]. Extracellular ATP (1 μM) increased insulin secretion in mouse β-cell lines, but at higher ATP concentrations, cell viability decreased and P2Y1 and P2X4 receptors were implicated [87]. The P2X7 receptor knockout mice had lower β-cell mass, impaired glucose tolerance and defective insulin and interleukin secretion [74]. Recent studies address the question of adenosine in β-cell mass using screening assays. Using a zebra fish model of diabetes, it was found that the non-specific adenosine agonist, NECA, increased proliferation of β cells but not other endocrine cells, and the data suggest that this was via A2Aa receptors. In STZ-diabetic mice, NECA also increased the number of β cells and improved glucose control [47]. In another screening study, it was found that adenosine kinase inhibitors increased rodent and porcine β-cell replication. It was proposed that the nuclear enzyme regulates adenosine levels and the mTOR cell proliferation pathway [88].

Purinergic receptors have effects on β-cell mass. Receptors marked in green increase β-cell mass (proliferation/replication), while those marked in red mediate β-cell death (apoptosis). Some purinergic receptors exert cytoprotective actions when cells are exposed to other factors, e.g. cytokines. The effects of P1 and P2 receptor stimulation on cell viability and/or insulin release may be dependent on concentrations of nucleotides/sides. For details, see the text
Earlier reviews describing the roles of purinergic signalling in insulin secretion and diabetes in relation to the pancreas are available [89–91]. Below, we will review evidence for the role of purinergic signalling in various organs affected by hyperglycaemia in diabetes and indicate whether any of those can be potential targets for organ-specific treatments in diabetes.
Cardiovascular system
Problems associated with diabetes and the cardiovascular system are many and include hypertension, atherosclerosis, cardiac disease, microvascular pathology in several organs and disturbances in blood cells. In particular adenosine receptors, but also P2 receptors, nucleotide/side converting enzymes and transporters, are affected in the diabetic vascular system; effects vary depending on the organ and local regulatory system. In general, in a healthy vessel, there is P2X receptor-mediated vasoconstriction and P2Y receptor-mediated vasodilation via stimulation of nitric oxide (NO) synthase and NO release from endothelial cells. On endothelial cells, A1 receptors also mediate stimulation of NO release in some vessels. In the heart, adenosine is cytoprotective, and it slows sinoatrial and atrioventricular conduction, resulting in decreased heart rate, coronary vasodilatation, and it attenuates the functional and metabolic effects of β-adrenergic receptor stimulation, and in particular it has significant effects on glucose and fatty acid metabolism [92, 93]. Thus, adenosine helps to restore the balance in myocardial O2 supply–demand, and there is evidence that all four adenosine receptor subtypes expressed in various cells in the heart exert cardioprotective effects [92]. In the following paragraphs, we will review the original studies that support the notion that purinergic signalling is involved in the diabetic cardiovascular system.
Both microvascular pathology and sympathetic denervation are present in alloxan-induced diabetes in rats [94]. Twelve weeks after induction of STZ diabetes, there was prejunctional impairment of sympathetic transmission via P1 receptors and impaired endothelium-mediated vasodilation by ATP of the rat mesenteric arterial bed [95]. In contrast, at 8 weeks STZ diabetes, the functions were unimpaired, although sensory-motor nerve-mediated vasodilation was attenuated [96]. Enhanced ATP-induced contraction of mesenteric arteries from diabetic Goto-Kakizaki rats at the chronic stage of diabetes was shown to be due to increased cPLA2/COX pathway activity in smooth muscle [97]. It was shown further that the angiotensin II type 1 antagonist, losartan, normalises the P2Y receptor-mediated contraction. P2Y receptor-mediated insulin stimulating responses of β cells and of the pancreas vascular bed were preserved in STZ-diabetic rat pancreas [65]. In the tail artery of STZ-diabetic rats, there is an increased neurotransmitter role for ATP compared to its cotransmitter noradrenaline (NA) in sympathetic nerves and an increased potency of ATP via P2X receptors [98].
The sensitivity of platelet aggregation by ADP is increased in diabetic patients, and this may contribute to microangiopathy [99]. Platelets of T2D patients were characterised by high ATP content [100]. The activity of both NTPDase and 5′-nucleotidase of platelets (and synaptosomes) showed increased activity in alloxan-induced diabetes [101, 102]. Adenosine deaminase and 5′-nucleotidase activities were higher in platelets in diabetic patients than control subjects [103]. In erythrocytes, ATP concentration is influenced by insulin levels in plasma [104], but there is impairment of ATP release from human erythrocytes in T2D, which may contribute to the vascular disease [105]. Interestingly, in blood serum of STZ-treated rats, nucleotide hydrolysis rates were increased, but these could return to control in rats subjected to physical training [106].
In human subjects with T2D, the vasodilator actions of ATP, UTP and adenosine in skeletal muscle were diminished by 50% compared to controls, and this effect was most likely due to altered receptor sensitivity [107]. A similar conclusion was reached in a study of another vascular bed; soon after the onset of alloxan-induced diabetes in rabbits, retinal blood flow velocity decreased following ATP or 2′(3′)-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate infusion and P2X7 receptors were implicated [108]. However, vasodilator effects of adenosine on retinal arterioles were preserved in STZ-diabetic rats [109].
Adenosine receptors are major regulators of vascular beds in many organs. Adenosine and AMP enhanced the NO synthase response to inflammatory cytokines in diabetic vascular smooth muscle cells from rat aorta [110]. Relaxation of the rat aorta by adenosine via A2 receptors and endothelial release of endothelium-derived relaxing factor (EDRF) was attenuated in the STZ-diabetic rat [111], although an earlier study found similar release of EDRF in response to cholinergic stimulation, but enhanced release of oxygen free radicals in diabetic preparations [112]. It has been proposed that foetal endothelial dysfunction in gestational diabetes mellitus involves a functional link between adenosine and insulin signalling [113]. Adenosine inhibited the enhanced growth of aortic smooth muscle cells in STZ-diabetic rats [114].
In the heart, adenosine protects the myocardium against ischaemic and reperfusion injury; it has negative inotropic and chronotropic effects and attenuates proliferation of fibroblasts. Diabetic patients have ventricular hypertrophy and reduced tolerance to stress. Atria from 6 week STZ-diabetic rats exhibited supersensitivity to the negative inotropic and chronotropic effect of adenosine, postulated to be due to impairment of the adenosine uptake mechanism on plasma membranes [115]. Also in diabetic rat cardiac fibroblasts, altered expression of nucleoside transporters was detected, and this was proposed to lead to increased uptake but decreased release of adenosine [116].
Myocardial over-expression of adenine nucleotide translocase 1 on inner mitochondrial membranes accelerates mitochondrial ATP/ADP exchange and ameliorates diabetic cardiomyopathy in mice, a promising target for diabetic cardiomyopathy [117]. Diabetic cardiomyocytes from rats treated with losartan maintained the capacity to respond to ATP depletion leading to contractile failure [118]. STZ diabetes in rats resulted in an increase in A1 and A3 receptor protein levels in cardiac myocytes, while A2A receptor protein expression remained unchanged [119]. Heart rate response to adenosine infusion (increase due to A2 receptors) is diminished in patients with diabetes mellitus, probably due to cardiovascular autonomic neuropathy [120, 121]. In accordance, a preliminary study showed that ATP and ADP hydrolysis was decreased in cardiac synaptosomes of STZ-rats, and this effect was reversed with insulin treatment [122].
One of the receptors linked to many diseases is the P2X7 receptor. It was hypothesised that P2X7 receptors participate in the pathogenesis of vascular complications in diabetes, based on experiments showing that high glucose concentrations triggered the assembly of P2X7 receptors and apoptosis in skin fibroblasts that share some of the features with smooth muscle cells [123]. Accordingly, human fibroblasts from diabetic patients had enhanced P2X7 receptor activity [124]. Nevertheless, another paper from this group also showed that P2Y receptor-dependent GLUT1 activation was defective in fibroblasts from the T2D patients [125].
Gestational diabetes is first recognised in pregnancy and is associated with abnormal foetal development and perinatal complications. There is a low capacity of adenosine transport by the foetal endothelium of umbilical vein in gestational diabetes leading to accumulation of extracellular adenosine and its action on endothelial A2A receptors [126–128]) and insulin reversed these effects [129].
Nervous system
Diabetic neuropathy, characterised by nerve fibre atrophy and loss, was recognised early (for reviews, see [130–133]). In T2D animal models, there was early evidence for increased sympathetic activity in STZ-diabetic rats. Mice with spontaneous diabetes show changes of sympathetic function similar to those found in diabetic patients with autonomic nerve pathology [134]. In addition, sympathetic nerves in pancreatic islets are impaired in BBD rats [135]. STZ-diabetic mice at 7 weeks showed reduced cutaneous sensory innervation and reduced expression of P2X3 receptors in footpad skin [136].
Painful diabetic neuropathy is a complication of diabetes; it causes hyperalgesia and allodynia [137]. Modulation of cutaneous polymodal receptors in diabetic rats by sympathetic nerves (which release ATP and NA as cotransmitters) has been reported [138]. Adenosine seems to be protective. An adenosine kinase inhibitor, which increases extracellular levels of adenosine, attenuates tactile allodynia in a rat model of diabetic neuropathic pain [139]. Allosteric enhancers for A1 receptors are targets for neuropathic drug development [140]. Peripheral neuropathy, vascular disease and oedema are some of the factors responsible for impaired healing after trauma and infection. Adenosine receptor agonists have been proposed for promotion of dermal wound healing, particularly for diabetic foot ulcers [141]. In some neuropathic pain models, there is activation of dorsal horn microglia and P2 receptors (P2X4 and P2Y12) [142]. In STZ-diabetic mice, the levels of P2X2 and P2X3 receptor mRNA were significantly increased in dorsal root ganglion, suggesting that the up-regulation of these receptors is associated with mechanical allodynia [143]. A substantial enhancement of P2X3 receptor activity and an increase in expression of P2X3 receptors was reported recently and claimed to contribute to the development of chronic pain in STZ-induced diabetic rats [144]. As another approach, the protective actions of adenosine were investigated in STZ-diabetic rat models of neuropathic pain. Using antagonists, it was concluded that analgesic actions of adenosine were exerted via A1 receptors [145].
Regarding the effect on the brain, in alloxan-diabetic rats, there are abnormalities in activity but not expression of G proteins in the striatum [146]. Diabetic encephalopathy results in cognitive impairment and modification of hippocampal function. In STZ-diabetic rats, there was a decrease in ATP concentrations in cerebrospinal fluid, decrease in density of P2X3,5,7 and P2Y2,6,11 receptors in hippocampal nerve terminals, but an increase in P2X1,2.5,6,7 and P2Y6 (but not P2Y2) receptors in membranes of astrocytes/neurones, indicating changes neuro- and gliotransmission [147]. There is decreased adenosine uptake in hippocampus of STZ-diabetic rats. This can increase adenosine sensitivity of synaptic potentials [148] and accelerate ischaemic block of population spikes in hippocampal slices [149]. The balance between inhibitory A1 and facilitatory A2A receptor activation was modified in the hippocampus of STZ-diabetic rats; A1 receptors were down-regulated, while A2A receptors were up-regulated [150]. In patients with diabetic neuropathy, there are abnormalities of Ca2+/Mg2+ ATPase activity in erythrocytes, and the results were interpreted in favour of altered Ca2+ homeostasis and microangiopathy playing a role in the pathogenesis of diabetic neuropathy [151].
Retina
Diabetic retinopathy, involving capillary abnormalities, is often seen in the early stages of diabetes (see [152]). The involvement of damaged sympathetic nerves (that release the cotransmitters NA and ATP) in the deterioration of capillaries and loss of ganglion cells was proposed [153]. High glucose alters the purinergic signalling system in the retina. Firstly, it increases the exocytotic release of ATP from cultured retinal cells and also decreases its extracellular degradation, both of which result in high levels of ATP [154]. Second, retinal neurons and microglia cultured in high glucose media augmented Ca2+ responses to P2 receptor stimulation, which may increase release of neurotransmitters and inflammatory mediators and thus lead to the inflammation involved in the pathogenesis of diabetic retinopathy [155].
ATP and ADP but not adenosine stimulate phosphoinositide metabolism in endothelial cells from bovine retinal microvessels, and it was proposed that this may be involved in the pathophysiology of diabetic retinopathy [156]. In addition, the formation of P2X7 receptor pores is enhanced in retinal microvessels early in the course of experimental diabetes [157]. Extracellular ATP induces cell death of retinal microvessels via P2X7 receptors and voltage-activated Ca2+ channels, and it was proposed that activation of P2Y4 receptors triggers a series of events that prevents P2X7 receptor-mediated pores and toxicity in retinal microvessels [108, 158]. Diabetic retinopathy is associated with macula oedema, which may be due to breakdown of blood–brain barrier and other effects, such as dysfunction of glial cell volume regulation. Activation of A1 receptors restores cell volume regulation of glial cells in the diabetic rat retina [159].
Kidney
Glomerular hyperfiltration, hypertrophy and microvascular dysfunction are the leading hallmarks of early diabetes leading to progressive nephropathy and hypertension. Diabetic nephropathy leads to end-stage renal disease, morbidity and mortality. The kidney is an interface between vascular and epithelial systems, and both are regulated by P1 and P2 receptors. Regarding the vasculature, adenosine signalling has a prominent but complex role. In STZ-diabetic rats, adenosine enhanced vasoconstriction of the kidney vascular bed via A1 receptors [160]. It was suggested that the increased vasoconstrictor effect of adenosine on the diabetic renal vasculature was caused by defective NO-dependent renal vasodilation of the afferent arterioles [161]. A1 receptor knockout mice developed diabetic-induced glomerular hyperfiltration, which suggested that the tubuloglomerular feedback (TGF) mechanism is not involved in the development of hyperfiltration [162]. However, in a later paper, it was shown that A1 receptor knockout blunts glomerular hyperfiltration and the salt paradox in early STZ diabetes [163]. Furthermore, a study on an A1 receptor over-expression mouse model shows that it is a crucial receptor for regulation of afferent arteriole tone [164]. The general scenario is that in response to salt load, macula densa cells release ATP, which can be hydrolyzed to adenosine, and A1 (and P2X1) receptors on the afferent arteriole mediate TGF responses (see [165, 166]). The tubulo-centric hypothesis, which requires further support, states that increased glucose load and absorption in proximal tubule (as in diabetes mellitus) decreases electrolyte to macula densa and thus down-regulated TGF causes increased GFR and hyperfiltration. Vasodilatory effects are mediated via A2 receptors, A2A on endothelial cells increase NO production; A2B receptors are expressed in podocytes. In addition, A2A receptors are also general anti-inflammatory receptors (see [167]).
There is evidence for altered adenosine level and adenosine receptor expression in diabetes. Reasoning that glomerular hyperfiltration in diabetes may be due to decreased vasoconstriction by adenosine, it was shown that inhibiting adenosine uptake by dipyridamole prevented early alterations in kidney function associated with diabetes [168]. In addition, administration of an adenosine analogue decreased diuresis and glycosuria in STZ-diabetic rats, although the non-specific adenosine receptor antagonist seemed to have different effects [169]. Agonists to adenosine receptors attenuated glucose and protein excretion in diabetic Wistar rat kidneys [170]. However, if adenosine levels are too high and low sensitivity A2 receptors are activated, the effects may become different. There is a significant increase in levels of adenosine (and purine metabolites) in plasma of patients with diabetic nephropathy compared to T2D patients without nephropathy [171]. Glomeruli of diabetic rats accumulate six times more adenosine than control tissues, and this is due to decreased nucleoside uptake activity and increased AMP hydrolysis; a possible consequence is activation of A2B receptors that then cause release of transforming growth factor β1 (TFG-1β) that may contribute to glomerulopathy [172]. In contrast, A2A receptor activation attenuates inflammation, injury and diabetic nephropathy [173].
In STZ-diabetic rats, A1 and A3 receptor mRNA and protein increased in both kidney cortex and medulla; A2A receptor expression increased in the cortex, but not medulla; A2B receptor expression was unchanged; and immunohistochemistry showed receptor localisation mainly on renal tubules [174]. cAMP-mediated inhibition of distal phosphate transport may explain the observation that adenosine enhances the antiphosphaturic effect in STZ diabetes [175]. In general, purinergic receptors on renal tubules exert negative regulator effects on electrolyte transport [176].
Adenosine receptors are being suggested as a therapeutic option for diabetic nephropathy [167, 172]. The vascular ectonucleotidase ENTPD-1 is a novel factor considered, as it prevents chronic microvascular injury, inflammation and thromboregulation, in STZ-mice [177]. For both approaches, differential effects of adenosine via A1 vs A2B receptors should be considered.
Urinogenital system
A high incidence of bladder dysfunction has been reported in patients with diabetes mellitus; the symptoms may progress with time and range from an overactive bladder and hypercontractive detrusor to voiding problems with urinary retention and acontractile detrusor. It is not certain whether bladder dysfunction is secondary to neuropathology or bladder overdistension with smooth muscle and urothelial dysfunction. Again, animal studies have been useful. In STZ-induced diabetic rats, there is bladder hypertrophy and distension [178]. After 3 months, STZ-treated rats showed reduced contractile responses to nerve stimulation, but no change in sensitivity to acetylcholine and ATP [179]. Later, it was claimed that there was a reduction in the non-cholinergic contractile component of parasympathetic nerve stimulation in 12 weeks STZ-rats, probably caused by a reduction in release of the non-cholinergic transmitter [180]. It was proposed that cholinergic and purinergic parasympathetic nerve components of contraction were minimally affected by STZ treatment, but in M2-muscarinic knockout mouse bladder, STZ treatment reduced both the cholinergic and purinergic components [181]. There are conflicting reports about the changes in ATP-mediated neural responses in STZ-diabetic bladder. This appears to depend largely on the time course; it seems likely that there is an increase in the purinergic component in the early stages (1–8 weeks), but decreased responses after 8 weeks. Up-regulation of P2X1 receptors was claimed in the early stages of STZ diabetes and down-regulation of P2X2 receptors in the later stages [182, 183]. Similarly, there was increased expression of P2Y2, P2Y4 and P2X4 receptors in STZ-rats in 2–4 months, but not at 8 months [184]. Furthermore, 6 months after alloxan-induced diabetes in rabbits, there was enhancement of purinergic, but reduction of cholinergic neurotransmission to the detrusor muscle of the bladder [185]. The P2X3 receptor is important for afferent pathways controlling urinary bladder volume reflexes [173], and this may be a candidate regulator. Impairment of the initiation of voiding reflexes via sensory nerve pathways activated by ATP released from urothelial cells in the bladder in diabetes has also been implicated [186–188]. In addition, there is impaired ATP-induced release of prostaglandins from urothelial cells [189], and urothelium itself releases ATP [190]. Both ATP and NO are released from the urothelium in the bladder. In early diabetic bladders from STZ-treated rats showing overactivity and a ‘diuretic’ underactivity model, the release ratio of ATP and NO was correlated with bladder contraction frequency, being enhanced in overactive and diminished in underactive bladders [191]. It was suggested that the ATP/NO ratio could be used to monitor changes in bladder activity during drug therapy.
Adenosine, a direct vasodilator of corpus cavernosum, was recommended for the treatment of diabetic erectile impotence [192]. Adenosine-induced inhibition of sympathetic nerve-mediated contractile responses of mouse corpus cavernosum is impaired in T2D db/db mice [193]. It was suggested that the relaxant response of the corpus cavernosum to adenosine and ATP in both men and rats was largely endothelium-dependent via release of NO [194]. A functional study of purinergic signalling in the alloxan-diabetic rabbit corpus cavernosum led to the conclusion that relaxations mediated by both P2Y1 receptors (via ADP and ATP) on endothelial cells and P2Y4 receptors (via UTP and ATP) on cavernosal smooth muscle, were impaired [195]. Another paper showed that ATP can contract corpus cavernosum smooth muscle via P2X1 receptors and relax via P2Y1 and P2Y2 receptors and that the P2Y receptor relaxant effects were significantly decreased in STZ-diabetic rats [196].
Gastrointestinal system
Many gastrointestinal complications of diabetes seem to be related to dysfunction of the neurons supplying the enteric nervous system. Thus, delayed gastric emptying, abnormal motility, secretion or absorption could lead to some of the symptoms of pain, constipation, diarrhoea, irritable bowel, ulcers, etc. Non-adrenergic, non-cholinergic (NANC) relaxant responses of the gastric fundus were impaired in 8 and 12 week STZ-induced diabetic rats [197, 198]. However, in 8 week STZ-diabetic rats, there appeared to be an increase in the purinergic component of the vagal NANC responses of the gastric fundus to vagal nerve stimulation [199]. Another study described impairment of the nitrergic component of NANC nerve-mediated relaxations of the rat gastric fundus [200]. Moreover, impairment of the nitrergic-mediated relaxation of rat duodenum was described in 3–4 week STZ-rats; responses to the NO donor sodium nitroprusside were unchanged, but the response to ATP was enhanced [201]. It was suggested that reduction in NO synthase activity was associated with the impairment of NANC relaxation. Impairment of both purinergic and nitrergic components of NANC inhibitory neurotransmission was claimed in the gastrointestinal tract of T1D diabetic RIP-I/hIFNβ transgenic mice [22]. Electrophysiological responses to NA, but not to acetylcholine or ATP, were potentiated in the caecum of 8 week STZ-diabetic rats, perhaps resulting from supersensitivity of α-adrenoceptors after sympathetic nerve damage [202].
Fatty infiltrations of the liver are common in T2D patients and liver tests are abnormal, but it is not clear whether this is due to obesity. ATP administrated to alloxan-diabetic rats lowered blood glucose, decreased liver fat, increased serum albumin and decreased β-globulin [203]. Development of STZ diabetes in rats resulted in a significant increase in expression of A2A and A3 (but not A1) receptor mRNA levels and protein content in the rat liver, while expression of A2B was markedly decreased [204]. Adenosine induced hepatic glucose production, and this was inhibited by an A2B receptor antagonist that also reduced blood glucose in KK-Ay T2D diabetic mice [205]. In another study on the liver of STZ-induced diabetic rats, an increase in adenosine A1 receptor expression was detected in hepatocytes, as well as increased glycogen synthesis with the adenosine analogue, cyclopentyladenosine [206].
ATP reduced binding to insulin receptor degradation in rat adipocytes [207]. A similar process may happen in hepatocytes that are the main site of insulin degradation. However, in later studies, it was shown that it was cellular ATP that had a direct allosteric effect on insulin-degrading enzyme [208, 209].
Adipocytes
One of the manifestations of diabetes is an enhancement of the lipolytic process, release of free fatty acids, adipokines and cytokines. Adenosine is known to have anti-lipolytic effects; it stimulates lipogenesis, modulates insulin sensitivity, and mediates leptin secretion in isolated adipocytes. Activation of A1 receptors on adipocytes for the treatment of non-insulin-dependent diabetes mellitus was proposed early [210]. Accordingly, over-expression of A1 receptors in adipose tissue protects mice from obesity-related insulin resistance [211]. Knockdown of A1 receptors impaired insulin sensitivity of glucose uptake by adipocytes [79]. Although A1 agonists, by inhibiting adenylate cyclase, inhibited lipolysis in vitro and in vivo in STZ-diabetic animals, oral administration also produced significant bradycardia [212]. Recent reviews summarise adenosine receptor physiology and challenges in therapeutic approach for treatment of diabetes and obesity [213, 214]. In addition, A1 receptors stimulate adipocyte differentiation, while A2B receptors inhibit adipogenesis, and targeting these may be useful in management of obesity and diabetes [215].
ATP originates from sympathetic nerves and seems to have dual effect on white adipocyte: stimulating lipolysis and inhibiting insulin-induced leptin secretion [216]. Although adipocytes express several P2 receptors (P2Y1,2,4,6,11), the leptin effect may be due to P2Y1 receptors, since specific antagonist reduced leptin release in isolated adipocytes and circulating levels of leptin was lower in P2Y1 knockout mice [217]. High ATP dosages stimulated inflammatory responses and insulin resistance in rat adipocytes [218]. Human adipocytes express P2X7 receptors that modulate the release of inflammatory cytokines, and interestingly, patients with metabolic syndrome showed enhanced expression of P2X7 receptors [219]. Brown adipocytes that have a capacity to convert metabolic energy to heat also express a number of P2 receptors (P2Y2, P2Y6, P2Y12, P2X1–7), that can increase Ca2+ signalling and membrane trafficking [220], and their role in metabolic syndromes needs to be evaluated. In patients with T2D, insulin resistance is related to lowered ATP synthesis in liver, which is at least partly accounted for by fat deposits assessed from waist circumference [221].
Skeletal muscle
Skeletal muscles are the major site of insulin-sensitive glucose uptake, and therefore, insulin resistance has a profound effect on hyperglycaemia and glucose intolerance in T2D. Diabetes also leads to muscle weakness that is linked most strongly to impairment of glucose uptake via the insulin-sensitive GLUT4 transporter and subsequent metabolic disturbances. For example, ATP synthesis is impaired in isolated mitochondria from myotubes isolated from T2D patients [222]. There are a number of studies indicating that purinergic receptors also regulate glucose transport into muscles and vascular perfusion, which can alter muscle performance. There are studies that show that A1 receptors are involved in insulin sensitivity of glucose uptake and utilisation in isolated muscle fibres [223, 224]. In addition, extracellular ATP stimulates translocation of GLUT4 to plasma membranes of muscle fibres [225]. Exercise increases interstitial concentrations of ATP [226], and a recent study showed immunohistochemical localisation of several P2 receptors on skeletal muscle plasma membrane, and on blood vessels, although the distribution was similar in samples from normal and T2D individuals [227].
On the issue related to the vascular supply of skeletal muscles, there are also a number studies implicating purinergic signalling. Pre-diabetic ZDF rats have high insulin levels, which impair the ability of red blood cells to release ATP in response to low pO2. It was suggested that this O2-dependent release of ATP may contribute to the failure in the regulation of O2 supply to meet the demand in skeletal muscle in pre-diabetes [228]. ATP release was also shown to be impaired in erythrocytes of humans with T2D, and it was suggested that this could contribute to peripheral vascular disease in skeletal muscle in T2D [229]. In human experiments, it was shown that the vasodilator action of ATP, UTP and adenosine in skeletal muscle was diminished by 50% in T2D patients. This does not seem to be due to altered receptor expression, but rather to their altered sensitivity, and it may underlie reduced vascular function to limit exercise capacity [107]. A recent study showed that 7-day bed rest induced insulin resistance and lowered content/activity of proteins responsible for glucose transport, phosphorylation and storage in muscles [230].
Skin
Impaired wound healing is one of the major problems in patients with diabetes, and it can lead to ulcers, pain and eventually amputation. The exact mechanisms are not understood, but eventually, alterations in fibroblast proliferation lead to formation of granulation tissue that delays wound healing. It has been suggested that adenosine A2A receptor activation by polydeoxyribonucleotide might represent a therapeutic strategy to overcome the diabetes-impaired cell-cycle machinery during impaired skin wound healing in genetically diabetic mice [231]. Improved healing of skin wounds in diabetic rabbits has been reported during direct delivery of intracellular ATP via lipid vesicles [232].
Therapeutic approaches
Purinergic signalling offers potential for the development of novel therapeutic approaches to treat the primary disorder in diabetes, i.e. the pancreas, as well as diabetes-related problems in other organs/tissues. Strategies for the pancreas could include drugs that would increase insulin secretion, protect and support β-cell regeneration, curb inflammation within the pancreas, and for T1D, pretreatment of islets before transplantation and stem cell therapy (the latter two are beyond this review). There are a number of promising studies on model animals and systems that could be translated to human applications.
In order to increase insulin secretion for treatment of T2D, stable and tissue specific analogues for the P2Y1 receptor have been developed, although no drug is in clinical trials (see above). Other P2Y receptors that could be considered are P2Y6 receptors that mediate increase in insulin secretion and prevent β-cell death [84]. Inhibition of the pro-apoptotic P2Y13 receptor may also be considered [85]. The P2X7 receptor may be an interesting target for diabetes and obesity, but due to its wide expression in several pancreatic cells, inflammatory cells and CNS, more basic knowledge is needed.
Adenosine is a potent endogenous autocrine anti-inflammatory and immunosuppressive molecule, it is released or formed after breakdown of at the site of injury and therefore adenosine receptors are also potential targets. The methylxanthine, caffeine, is a nonspecific adenosine receptor blocker, and it is no surprise that a role of coffee consumption in lowering the risk of T2D has been widely debated. Well-controlled studies in diabetic mice models provide support for human studies [233]. Nevertheless, more specific adenosine receptor drugs would provide a better control. A2A receptor agonists may be exploited for β-cell regeneration [47]. Antagonists of A2B receptors would improve insulin secretion as well as decrease the inflammatory response and improve insulin resistance [45, 46]. In fact, A2B blockers are being developed for reversal of insulin resistance in T2D: ATL 844 as a joint venture by Clinical Data Inc. and Novartis; and GS 6201 (CVT-6883) by Gilead Sciences [234, 235].
Regarding other diabetic associated maladies, Sandoz/Novartis is developing an A1 receptor agonist SDZ WAG94; Gilead has GS 9667 in clinical I trials for the treatment of hypertriglyceridemia associated with diabetes. The specific A2A agonist BVT.115959 by Biovitrum is in clinical II trials for diabetic neuropathic pain, and Sonedenoson by King is in clinical trials for diabetic foot ulcers and wound healing [235].
In addition to purinergic signalling, the energy/nucleotide status of pancreatic and other cells could be considered for therapeutic approaches. Enhancement of ATP synthesis in pancreatic islets, e.g. by biotin, reinforces glucose-induced insulin secretion [236]. Direct delivery of intracellular ATP via lipid vesicles appears to be possible, at least in the skin [232].
Conclusions and perspectives
The pancreas is a central organ in nutrient and energy homeostasis with endocrine, exocrine, stromal and immunoreactive cells, which participate in complex processes that have consequences for whole body physiology (Fig. 1). This review has focussed on the role of purinergic signalling in the regulation of insulin secretion and β-cell viability, and in the regulation of various tissues/organs that are affected by diabetes. The enormous flexibility and diversity of the purinergic system can be exploited in drug design for the treatment of primary and secondary sites of diabetes, although integrated understanding is needed. Some purinergic drugs are already in clinical trials, and it is hoped that finer regulation of diabetes 2 and 1 and their complications will be possible in the near future.
References
van Belle TL, Coppieters KT, von Herrath MG (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91:79–118
Stumvoll M, Goldstein BJ, van Haeften TW (2005) Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365:1333–1346
Dinarello CA, Donath MY, Mandrup-Poulsen T (2010) Role of IL-1 β in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 17:314–321
Kalupahana NS, Moustaid-Moussa N, Claycombe KJ (2012) Immunity as a link between obesity and insulin resistance. Mol Aspects Med 33:26–34
Osborn O, Olefsky JM (2012) The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 18:363–374
Johansson BB, Torsvik J, Bjorkhaug L, Vesterhus M, Ragvin A, Tjora E, Fjeld K, Hoem D, Johansson S, Raeder H, Lindquist S, Hernell O, Cnop M, Saraste J, Flatmark T, Molven A, Njolstad PR (2011) Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene-maturity onset diabetes of the young (CEL-MODY): a protein misfolding disease. J Biol Chem 286:34593–34605
Leeds JS, Oppong K, Sanders DS (2011) The role of fecal elastase-1 in detecting exocrine pancreatic disease. Nat Rev Gastroenterol Hepatol 8:405–415
Andersen DK (2012) The practical importance of recognizing pancreatogenic or type 3c diabetes. Diabetes Metab Res Rev 28:326–328
Cui Y, Andersen DK (2011) Pancreatogenic diabetes: special considerations for management. Pancreatology 11:279–294
Bertelli E, Bendayan M (2005) Association between endocrine pancreas and ductal system. More than an epiphenomenon of endocrine differentiation and development? J Histochem Cytochem 53:1071–1086
Movahedi B, Van de Casteele M, Caluwe N, Stange G, Breckpot K, Thielemans K, Vreugdenhil G, Mathieu C, Pipeleers D (2004) Human pancreatic duct cells can produce tumour necrosis factor-α that damages neighbouring beta cells and activates dendritic cells. Diabetologia 47:998–1008
Movahedi B, Gysemans C, Jacobs-Tulleneers-Thevissen D, Mathieu C, Pipeleers D (2008) Pancreatic duct cells in human islet cell preparations are a source of angiogenic cytokines interleukin-8 and vascular endothelial growth factor. Diabetes 57:2128–2136
Ewald N, Raspe A, Kaufmann C, Bretzel RG, Kloer HU, Hardt PD (2009) Determinants of exocrine pancreatic function as measured by fecal elastase-1 concentrations (FEC) in patients with diabetes mellitus. Eur J Med Res 14:118–122
Ewald N, Kaufmann C, Raspe A, Kloer HU, Bretzel RG, Hardt PD (2012) Prevalence of diabetes mellitus secondary to pancreatic diseases (type 3c). Diabetes Metab Res Rev 28:338–342
Rees DA, Alcolado JC (2005) Animal models of diabetes mellitus. Diabet Med 22:359–370
Chatzigeorgiou A, Halapas A, Kalafatakis K, Kamper E (2009) The use of animal models in the study of diabetes mellitus. In Vivo 23:245–258
Rakieten N, Rakieten ML, Nadkarni MV (1963) Studies on the diabetogenic action of streptozotocin (Nsc-37917). Cancer Chemother Reports 29:91–98
Jacobs HR (1937) Hyperglycemic actions of alloxan. Proc Soc Exp Biol Med 37:404–409
Rerup CC (1970) Drugs producing diabetes through damage of the insulin secreting cells. Pharmacol Rev 22:485–518
Nakhooda AF, Like AA, Chappel CI, Murray FT, Marliss EB (1977) The spontaneously diabetic Wistar rat. Metabolic and morphologic studies. Diabetes 26:100–112
Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y (1980) Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 29:1–13
Domènech A, Pasquinelli G, De Giorgio R, Gori A, Bosch F, Pumarola M, Jiménez M (2011) Morphofunctional changes underlying intestinal dysmotility in diabetic RIP-I/hIFNβ transgenic mice. Int J Exp Pathol 92:400–412
Clark JB, Palmer CJ, Shaw WN (1983) The diabetic Zucker fatty rat. Proc Soc Exp Biol Med 173:68–75
Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24:509–581
Yegutkin GG (2008) Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta 1783:673–694
Burnstock G (1978) A basis for distinguishing two types of purinergic receptor. In: Straub RW, Bolis L (eds) Cell membrane receptors for drugs and hormones: a multidisciplinary approach. Raven Press, New York, pp 107–118
Burnstock G, Kennedy C (1985) Is there a basis for distinguishing two types of P2-purinoceptor? Gen Pharmacol 16:433–440
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–492
Burnstock G (2007) Purine and pyrimidine receptors. Cell Mol Life Sci 64:1471–1483
Abbracchio MP, Burnstock G, Boeynaems J-M, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA (2006) International Union of Pharmacology. Update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341
Burnstock G, Knight GE (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240:31–304
Hattori M, Gouaux E (2012) Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485:207–212
Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332:322–327
Zimmermann H, Zebisch M, Strater N (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8:437–502
Novak I (2008) Purinergic receptors in the endocrine and exocrine pancreas. Purinergic Signal 4:237–253
Novak I (2011) Purinergic signalling in epithelial ion transport—regulation of secretion and absorption. Acta Physiologica 202:501–522
Hellman B (2009) Pulsatility of insulin release—a clinically important phenomenon. Ups J Med Sci 114:193–205
Petit P, Lajoix AD, Gross R (2009) P2 purinergic signalling in the pancreatic beta-cell: control of insulin secretion and pharmacology. Eur J Pharm Sci 37:67–75
Burnstock G, Novak I (2012) Purinergic signalling in the pancreas in health and disease. J Endocrinol 213:123–141
Karanauskaite J, Hoppa MB, Braun M, Galvanovskis J, Rorsman P (2009) Quantal ATP release in rat beta-cells by exocytosis of insulin-containing LDCVs. Pflugers Arch 458:389–401
Haanes KA, Novak I (2010) ATP storage and uptake by isolated pancreatic zymogen granules. Biochem J 429:303–311
Lazarowski ER (2012) Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal 8:359–373
Lavoie EG, Fausther M, Kauffenstein G, Kukulski F, Kunzli BM, Friess H, Sevigny J (2010) Identification of the ectonucleotidases expressed in mouse, rat, and human Langerhans islets: potential role of NTPDase3 in insulin secretion. Am J Physiol Endocrinol Metab 299:E647–E656
Yegutkin GG, Samburski SS, Jalkalen S, Novak I (2006) ATP-consuming and ATP-generating enzymes secreted by pancreas. J Biol Chem 281:29441–29447
Rusing D, Muller CE, Verspohl EJ (2006) The impact of adenosine and A2B receptors on glucose homoeostasis. J Pharm Pharmacol 58:1639–1645
Figler RA, Wang G, Srinivasan S, Jung DY, Zhang Z, Pankow JS, Ravid K, Fredholm B, Hedrick CC, Rich SS, Kim JK, LaNoue KF, Linden J (2011) Links between insulin resistance, adenosine A2B receptors, and inflammatory markers in mice and humans. Diabetes 60:669–679
Andersson O, Adams BA, Yoo D, Ellis GC, Gut P, Anderson RM, German MS, Stainier DY (2012) Adenosine signaling promotes regeneration of pancreatic β cells in vivo. Cell Metab 15:885–894
Salehi A, Qader SS, Grapengiesser E, Hellman B (2005) Inhibition of purinoceptors amplifies glucose-stimulated insulin release with removal of its pulsatility. Diabetes 54:2126–2131
Coutinho-Silva R, Parsons M, Robson T, Burnstock G (2001) Changes in expression of P2 receptors in rat and mouse pancreas during development and aging. Cell Tissue Res 306:373–383
Salehi A, Qader SS, Grapengiesser E, Hellman B (2007) Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regul Pept 144:43–49
Wang J, Haanes KA, Novak I (2013) Purinergic regulation of CFTR and Ca2+-activated Cl- channels and K+ channels in human pancreatic duct epithelium. Am J Physiol Cell Physiol. doi:10.1152/ajpcell.00196.2012
Ko T, An HJ, Ji YG, Kim OJ, Lee DH (2012) P2Y receptors regulate proliferation of human pancreatic duct epithelial cells. Pancreas 41:797–803
Won JH, Zhang Y, Ji B, Logsdon CD, Yule DI (2011) Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol Biol Cell 22:421–436
Haanes KA, Schwab A, Novak I (2012) The P2X7 receptor supports both life and death in the fibrogenic pancreatic stellate cells. PLoS One 7(12):e51164
Yabe D, Seino Y (2011) Two incretin hormones GLP-1 and GIP: comparison of their actions in insulin secretion and beta cell preservation. Prog Biophys Mol Biol 107:248–256
Morin L, Giroix MH, Portha B (1996) Decreased ATP-induced synthesis and Ca2+-stimulated degradation of polyphosphoinositides in pancreatic islets from neonatally streptozotocin-diabetic rats. Biochem Biophys Res Commun 228:573–578
Nukatsuka M, Yoshimura Y, Nishida M, Kawada J (1990) Importance of the concentration of ATP in rat pancreatic beta cells in the mechanism of streptozotocin-induced cytotoxicity. J Endocrinol 127:161–165
Grill V, Radtke M, Qvigstad E, Kollind M, Björklund A (2009) Beneficial effects of K-ATP channel openers in diabetes: an update on mechanisms and clinical experiences. Diabetes Obes Metab 11:143–148
de Wet H, Proks P, Lafond M, Aittoniemi J, Sansom MS, Flanagan SE, Pearson ER, Hattersley AT, Ashcroft FM (2008) A mutation (R826W) in nucleotide-binding domain 1 of ABCC8 reduces ATPase activity and causes transient neonatal diabetes. EMBO Rep 9:648–654
Detimary P, Jonas J-C, Henquin J-C (1995) Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. J Clin Invest 96:1738–1745
Zhao Z, Zhao C, Zhang XH, Zheng F, Cai W, Vlassara H, Ma ZA (2009) Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 150:2569–2576
Mikhail TH, Awadallah R (1977) The effect of ATP and certain trace elements on the induction of experimental diabetes. Z Ernährungswiss 16:176–183
Awadallah R, Tahani HM, El-Dessoukey EA (1979) Serum mineral changes due to exogenous ATP and certain trace elements in experimental diabetes. Z Ernährungswiss 18:1–7
Tahani HM (1979) The purinergic nerve hypothesis and insulin secretion. Z Ernährungswiss 18:128–138
Hillaire-Buys D, Gross R, Chapal J, Ribes G, Loubatieres-Mariani MM (1992) P2y purinoceptor responses of β cells and vascular bed are preserved in diabetic rat pancreas. Br J Pharmacol 106:610–615
Tang J, Pugh W, Polonsky KS, Zhang H (1996) Preservation of insulin secretory responses to P2 purinoceptor agonists in Zucker diabetic fatty rats. Am J Physiol 270:E504–E512
Fischer B, Chulkin A, Boyer JL, Harden KT, Gendron FP, Beaudoin AR, Chapal J, Hillaire-Buys D, Petit P (1999) 2-thioether 5′-O-(1-thiotriphosphate)adenosine derivatives as new insulin secretagogues acting through P2Y-Receptors. J Med Chem 42:3636–3646
Fischer B et al (2000) 2-Thioether-5'-O-(1-thiotriphosphate)-adenosine derivatives: new insulin secretagogues acting through P2Y-receptors. Isr Med Assoc J 2(92–8):92–98
Farret A, Vignaud M, Dietz S, Vignon J, Petit P, Gross R (2004) P2Y purinergic potentiation of glucose-induced insulin secretion and pancreatic beta-cell metabolism. Diabetes 53(Suppl 3):S63–S66, S63-S66
Eliahu S, Barr HM, Camden J, Weisman GA, Fischer B (2010) A novel insulin secretagogue based on a dinucleoside polyphosphate scaffold. J Med Chem 53:2472–2481
Léon C, Freund M, Latchoumanin O, Farret A, Petit P, Cazenave JP, Gachet C (2005) The P2Y1 receptor is involved in the maintenance of glucose homeostasis and in insulin secretion in mice. Purinergic Signal 1:145–151
Coutinho-Silva R, Parsons M, Robson T, Lincoln J, Burnstock G (2003) P2X and P2Y purinoceptor expression in pancreas from streptozotocin-diabetic rats. Mol Cell Endocrinol 204:141–154
Coutinho-Silva R, Robson T, Beales PE, Burnstock G (2007) Changes in expression of P2X7 receptors in NOD mouse pancreas during the development of diabetes. Autoimmunity 40:108–116
Glas R, Sauter NS, Schulthess FT, Shu L, Oberholzer J, Maedler K (2009) Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia 52:1579–1588
Gross R, Hillaire-Buys D, Bertrand G, Ribes G, Loubatières-Mariani MM (1989) Diabetes and impaired response of glucagon cells and vascular bed to adenosine in rat pancreas. Diabetes 38:1291–1295
Laurent F, Hillaire-Buys D, Chapal J, Dietz S, Portet K, Cros G, Petit P, Michel A (1999) Contrasting effects of streptozotocin-induced diabetes on the in vitro relaxant properties of adenosine in rat pancreatic vascular bed and thoracic aorta. Naunyn Schmiedebergs Arch Pharmacol 360:309–316
Gross R, Hillaire-Buys D, Ribes G, Loubatières-Mariani MM (1991) Diabetes alters the responses of glucagon secreting cells and vascular bed to isoprenaline and forskolin in vitro in rat pancreas. Life Sci 48:2349–2358
Johansson SM, Salehi A, Sandstrom ME, Westerblad H, Lundquist I, Carlsson PO, Fredholm BB, Katz A (2007) A1 receptor deficiency causes increased insulin and glucagon secretion in mice. Biochem Pharmacol 74:1628–1635
Faulhaber-Walter R, Jou W, Mizel D, Li L, Zhang J, Kim SM, Huang Y, Chen M, Briggs JP, Gavrilova O, Schnermann JB (2011) Impaired glucose tolerance in the absence of adenosine A1 receptor signaling. Diabetes 60:2578–2587
Németh ZH, Bleich D, Csóka B, Pacher P, Mabley JG, Himer L, Vizi ES, Deitch EA, Szabo C, Cronstein BN, Hasko G (2007) Adenosine receptor activation ameliorates type 1 diabetes. FASEB J 21:2379–2388
Maedler K, Dharmadhikari G, Schumann DM, Storling J (2009) Interleukin-1β targeted therapy for type 2 diabetes. Expert Opin Biol Ther 9:1177–1188
Elliott JI, Higgins CF (2004) Major histocompatibility complex class I shedding and programmed cell death stimulated through the proinflammatory P2X7 receptor: a candidate susceptibility gene for NOD diabetes. Diabetes 53:2012–2017
Lenertz LY, Gavala ML, Zhu Y, Bertics PJ (2011) Transcriptional control mechanisms associated with the nucleotide receptor P2X7, a critical regulator of immunologic, osteogenic, and neurologic functions. Immunol Res 50:22–38
Balasubramanian R, Ruiz de Azua I, Wess J, Jacobson KA (2010) Activation of distinct P2Y receptor subtypes stimulates insulin secretion in MIN6 mouse pancreatic beta cells. Biochem Pharmacol 79:1317–1326
Tan C, Salehi A, Svensson S, Olde B, Erlinge D (2010) ADP receptor P2Y13 induce apoptosis in pancreatic beta-cells. Cell Mol Life Sci 67:445–453
Tan C, Voss U, Svensson S, Erlinge D, Olde B (2012) High glucose and free fatty acids induces beta-cell apoptosis via autocrine effects of ADP acting on the P2Y13 receptor. Purinergic Signal 2012 [Epub ahead of print 1/9/12]
Ohtani M, Ohura K, Oka T (2011) Involvement of P2X receptors in the regulation of insulin secretion, proliferation and survival in mouse pancreatic β-cells. Cell Physiol Biochem 28:355–366
Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA (2012) Adenosine kinase inhibition selectively promotes rodent and porcine islet beta-cell replication. Proc Natl Acad Sci U S A 109:3915–3920
Loubatières-Mariani MM, Hillaire-Buys D, Chapal J, Bertrand G, Petit P (1997) P2 purinoceptor agonists: new insulin secretagogues potentially useful in the treatment of non-insulin-dependent diabetes mellitus. In: Jacobson KA, Jarvis MF (eds) Purinergic approaches in experimental therapeutics. Wiley-Liss, New York, pp 253–260
Petit P, Hillaire-Buys D, Loubatières-Mariani MM, Chapal J (2001) Purinergic receptors and the pharmacology of type 2 diabetes. In: Abbracchio MP, Williams M (eds) Handbook of experimental pharmacology. Purinergic and pyrimidinergic signalling II—cardiovascular, respiratory, immune, metabolic and gastrointestinal tract function. Springer, Berlin, pp 337–391
Farret A, Lugo-Garcia L, Galtier F, Gross R, Petit P (2005) Pharmacological interventions that directly stimulate or modulate insulin secretion from pancreatic beta-cell: implications for the treatment of type 2 diabetes. Fund Clin Pharmacol 19:647–656
McIntosh VJ, Lasley RD (2012) Adenosine receptor-mediated cardioprotection: are all 4 subtypes required or redundant? J Cardiovasc Pharmacol Ther 17:21–33
Headrick JP, Peart JN, Reichelt ME, Haseler LJ (2011) Adenosine and its receptors in the heart: regulation, retaliation and adaptation. Biochim Biophy Acta Biomembranes 1808:1413–1428
Mueller SM, Mueller TM, Ertel PJ (1982) Sympathetic and vascular dysfunction in early experimental juvenile diabetes mellitus. Am J Physiol 243:H139–H144
Ralevic V, Belai A, Burnstock G (1995) Effects of streptozotocin-diabetes on sympathetic nerve, endothelial and smooth muscle function in the rat mesenteric arterial bed. Eur J Pharmacol 286:193–199
Ralevic V, Belai A, Burnstock G (1993) Impaired sensory-motor nerve function in the isolated mesenteric arterial bed of streptozotocin-diabetic and ganglioside-treated streptozotocin-diabetic rats. Br J Pharmacol 110:1105–1111
Ishida K, Matsumoto T, Taguchi K, Kamata K, Kobayashi T (2011) Mechanisms underlying altered extracellular nucleotide-induced contractions in mesenteric arteries from rats in later-stage type 2 diabetes: effect of ANG II type 1 receptor antagonism. Am J Physiol Heart Circ Physiol 301:H1850–H1861
Speier S, Rupnik M (2003) A novel approach to in situ characterization of pancreatic beta-cells. Pflugers Arch 446:553–558
Onodera H, Hirata T, Sugawara H, Sugai K, Yoda B, Toyota T, Goto Y (1982) Platelet sensitivity to adenosine diphosphate and to prostacyclin in diabetic patients. Tohoku J Exp Med 137:423–428
Guo X, Wu J, Du J, Ran J, Xu J (2009) Platelets of type 2 diabetic patients are characterized by high ATP content and low mitochondrial membrane potential. Platelets 20:588–593
Lunkes GI, Lunkes DS, Morsch VM, Mazzanti CM, Morsch AL, Miron VR, Schetinger MR (2004) NTPDase and 5′-nucleotidase activities in rats with alloxan-induced diabetes. Diabetes Res Clin Pract 65:1–6
Miron VR, Bauermann L, Morsch AL, Zanin RF, Correa M, da Silva AC, Mazzanti C, Morsch VM, Lunkes GI, Schetinger MR (2007) Enhanced NTPDase and 5′-nucleotidase activities in diabetes mellitus and iron-overload model. Mol Cell Biochem 298:101–107
De Bona KS, Belle LP, Sari MH, Thome G, Schetinger MR, Morsch VM, Boligon A, Athayde ML, Pigatto AS, Moretto MB (2010) Syzygium cumini extract decrease adenosine deaminase, 5′nucleotidase activities and oxidative damage in platelets of diabetic patients. Cell Physiol Biochem 26:729–738
Aursnes I, Dahl-Jørgensen K, Hanssen KF (1986) ATP-concentrations in erythrocytes influenced by insulin levels in plasma. Clin Hemorheol 6:429–433
Sprague R, Stephenson A, Bowles E, Stumpf M, Ricketts G, Lonigro A (2012) Expression of the heterotrimeric G protein Gi and ATP release are impaired in erythrocytes of humans with diabetes mellitus. Adv Exp Med Biol 588:207–216
Moritz CE, Abreu-Vieira G, Piroli C, De Senna PN, Cardoso VV, Wink MR, Harthmann AD, Rucker B, Casali EA (2012) Physical training normalizes nucleotide hydrolysis and biochemical parameters in blood serum from streptozotocin-diabetic rats. Arch Physiol Biochem 118:253–259
Thaning P, Bune LT, Hellsten Y, Pilegaard H, Saltin B, Rosenmeier JB (2010) Attenuated purinergic receptor function in patients with type 2 diabetes. Diabetes 59:182–189
Sugiyama T, Oku H, Komori A, Ikeda T (2006) Effect of P2X7 receptor activation on the retinal blood velocity of diabetic rabbits. Arch Ophthalmol 124:1143–1149
Nakazawa T, Mori A, Saito M, Sakamoto K, Nakahara T, Ishii K (2008) Vasodilator effects of adenosine on retinal arterioles in streptozotocin-induced diabetic rats. Naunyn Schmiedebergs Arch Pharmacol 376:423–430
Malorgio F, Cignarella A, Pelosi V, Bolego C, Gaion RM (2010) Alterations of adenosine-related inflammatory pathways in vascular smooth muscle cells from diabetic rats. Purinergic Signal 6:96
Fahim M, Hussain T, Mustafa SJ (2001) Relaxation of rat aorta by adenosine in diabetes with and without hypertension: role of endothelium. Eur J Pharmacol 412:51–59
Pieper GM, Mei DA, Langenstroer P, O’Rourke ST (1992) Bioassay of endothelium-derived relaxing factor in diabetic rat aorta. Am J Physiol 263:H676–H680
Guzman-Gutierrez E, Abarzua F, Belmar C, Nien JK, Ramirez MA, Arroyo P, Salomon C, Westermeier F, Puebla C, Leiva A, Casanello P, Sobrevia L (2011) Functional link between adenosine and insulin: a hypothesis for fetoplacental vascular endothelial dysfunction in gestational diabetes. Curr Vasc Pharmacol 9:750–762
Parés-Herbuté N, Hillaire-Buys D, Etienne P, Gross R, Loubatières-Mariani MM, Monnier L (1996) Adenosine inhibitory effect on enhanced growth of aortic smooth muscle cells from streptozotocin-induced diabetic rats. Br J Pharmacol 118:783–789
Usta CK, Adan G, Özdem SS (2001) The effects of adenosine on isolated right atrial preparations from streptozotocin-diabetic rats. J Auton Pharmacol 21:191–195
Podgorska M, Kocbuch K, Grden M, Szutowicz A, Pawelczyk T (2006) Reduced ability to release adenosine by diabetic rat cardiac fibroblasts due to altered expression of nucleoside transporters. J Physiol 576:179–189
Wang Y, Ebermann L, Sterner-Kock A, Wika S, Schultheiss HP, Dörner A, Walther T (2009) Myocardial overexpression of adenine nucleotide translocase 1 ameliorates diabetic cardiomyopathy in mice. Exp Physiol 94:220–227
Alfarano C, Suffredini S, Fantappie O, Mugelli A, Cerbai E, Manni ME, Raimondi L (2011) The effect of losartan treatment on the response of diabetic cardiomyocytes to ATP depletion. Pharmacol Res 63:225–232
Grden M, Podgórska M, Szutowicz A, Pawelczyk T (2005) Altered expression of adenosine receptors in heart of diabetic rat. J Physiol Pharmacol 56:587–597
Bravo PE, Hage FG, Woodham RM, Heo J, Editor IA (2008) Heart rate response to adenosine in patients with diabetes mellitus and normal myocardial perfusion imaging. Am J Cardiol 102:1103–1106
Hage FG, Heo J, Franks B, Belardinelli L, Blackburn B, Wang W, Iskandrian AE (2009) Differences in heart rate response to adenosine and regadenoson in patients with and without diabetes mellitus. Am Heart J 157:771–776
Fürstenau CR, Rücker B, Pochmann D, Vieira G, Bischoff LB, Sarkis JJ, Zerbini LF, Casali EA, Wink MR (2010) Streptozotocin-induced diabetes alters ATP and ADP hydrolysid in rat heart left ventricle. Purinergic Signalling 6:S114
Solini A, Chiozzi P, Falzoni S, Morelli A, Fellin R, Di Virgilio F (2000) High glucose modulates P2X7 receptor-mediated function in human primary fibroblasts. Diabetologia 43:1248–1256
Solini A, Chiozzi P, Morelli A, Adinolfi E, Rizzo R, Baricordi OR, Di Virgilio F (2004) Enhanced P2X7 activity in human fibroblasts from diabetic patients: a possible pathogenetic mechanism for vascular damage in diabetes. Arterioscler Thromb Vasc Biol 24:1240–1245
Solini A, Chiozzi P, Morelli A, Passaro A, Fellin R, Di Virgilio F (2003) Defective P2Y purinergic receptor function: A possible novel mechanism for impaired glucose transport. J Cell Physiol 197:435–444
Sobrevia L, Jarvis SM, Yudilevich DL (1994) Adenosine transport in cultured human umbilical vein endothelial cells is reduced in diabetes. Am J Physiol 267:C39–C47
San Martín R, Sobrevia L (2006) Gestational diabetes and the adenosine/L-arginine/nitric oxide (ALANO) pathway in human umbilical vein endothelium. Placenta 27:1–10
Farías M, Puebla C, Westermeier F, Jo MJ, Pastor-Anglada M, Casanello P, Sobrevia L (2010) Nitric oxide reduces SLC29A1 promoter activity and adenosine transport involving transcription factor complex hCHOP-C/EBPα in human umbilical vein endothelial cells from gestational diabetes. Cardiovasc Res 86:45–54
Westermeier F, Salomon C, Gonzalez M, Puebla C, Guzman-Gutierrez E, Cifuentes F, Leiva A, Casanello P, Sobrevia L (2011) Insulin restores gestational diabetes mellitus-reduced adenosine transport involving differential expression of insulin receptor isoforms in human umbilical vein endothelium. Diabetes 60:1677–1687
Thomas PK, Tomlinson DR (1993) Diabetic and hypoglycaemic neuropathy. In: Dyck PI, Thomas PK, Griffin JW, Low PA, Poduslo JF (eds) Peripheral neuropathy. W.B. Saunders, Philadelphia, pp 1219–1250
Yasuda H, Terada M, Maeda K, Kogawa S, Sanada M, Haneda M, Kashiwagi A, Kikkawa R (2003) Diabetic neuropathy and nerve regeneration. Prog Neurobiol 69:229–285
Anand P (1996) Neurotrophins and peripheral neuropathy. Philos Trans R Soc Lond B Biol Sci 351:449–454
Low PA (2009) Experimental diabetic autonomic neuropathy. In: Veves A, Malik R (eds) Contemporary diabetes: diabetic neuropathy. Clinical management. Humana, Totowa, pp 153–164
Giachetti A (1978) The functional state of sympathetic nerves in spontaneously diabetic mice. Diabetes 27:969–974
Mundinger TO, Mei Q, Figlewicz DP, Lernmark A, Taborsky GJ Jr (2003) Impaired glucagon response to sympathetic nerve stimulation in the BB diabetic rat: effect of early sympathetic islet neuropathy. Am J Physiol Endocrinol Metab 285:E1047–E1054
Christianson JA, Riekhof JT, Wright DE (2003) Restorative effects of neurotrophin treatment on diabetes-induced cutaneous axon loss in mice. Exp Neurol 179:188–199
Archer AG, Watkins PJ, Thomas PK, Sharma AK, Payan J (1983) The natural history of acute painful neuropathy in diabetes mellitus. J Neurol Neurosurg Psychiatry 46:491–499
Sato J, Kumazawa T (1996) Sympathetic modulation of cutaneous polymodal receptors in chronically inflamed and diabetic rats. Prog Brain Res 113:153–159
Lynch JJ III, Jarvis MF, Kowaluk EA (1999) An adenosine kinase inhibitor attenuates tactile allodynia in a rat model of diabetic neuropathic pain. Eur J Pharmacol 364:141–146
Kiesman WF, Elzein E, Zablocki J (2009) A1 adenosine receptor antagonists, agonists, and allosteric enhancers. Handb Exp Pharmacol 193:25–58
Valls MD, Cronstein BN, Montesinos MC (2009) Adenosine receptor agonists for promotion of dermal wound healing. Biochem Pharmacol 77:1117–1124
Tsuda M, Tozaki-Saitoh H, Inoue K (2010) Pain and purinergic signaling. Brain Res Rev 63:222–232
Migita K, Moriyama T, Koguchi M, Honda K, Katsuragi T, Takano Y, Ueno S (2009) Modulation of P2X receptors in dorsal root ganglion neurons of streptozotocin-induced diabetic neuropathy. Neurosci Lett 452:200–203
Xu GY, Li G, Liu N, Huang LY (2011) Mechanisms underlying purinergic P2X3 receptor-mediated mechanical allodynia induced in diabetic rats. Mol Pain 7:60
Balasubramanyan S, Sharma SS (2008) Protective effect of adenosine in diabetic neuropathic pain is mediated through adenosine A1-receptors. Indian J Physiol Pharmacol 52:233–242
Finco C, Abbracchio MP, Malosio ML, Cattabeni F, Di Giulio AM, Paternieri B, Mantegazza P, Gorio A (1992) Diabetes-induced alterations of central nervous system G proteins. ADP-ribosylation, immunoreactivity, and gene-expression studies in rat striatum. Mol Chem Neuropathol 17:259–272
Duarte JM, Oses JP, Rodrigues RJ, Cunha RA (2007) Modification of purinergic signaling in the hippocampus of streptozotocin-induced diabetic rats. Neuroscience 149:382–391
Morrison PD, Mackinnon MW, Bartrup JT, Skett PG, Stone TW (1992) Changes in adenosine sensitivity in the hippocampus of rats with streptozotocin-induced diabetes. Br J Pharmacol 105:1004–1008
Cassar M, Jones MG, Szatkowski M (1998) Reduced adenosine uptake accelerates ischaemic block of population spikes in hippocampal slices from streptozotocin-treated diabetic rats. Eur J Neurosci 10:239–245
Duarte JM, Oliveira CR, Ambrósio AF, Cunha RA (2006) Modification of adenosine A1 and A2A receptor density in the hippocampus of streptozotocin-induced diabetic rats. Neurochem Int 48:144–150
Migdalis IN, Xenos K, Chairopoulos K, Varvarigos N, Leontiades E, Karmaniolas K (2000) Ca2+–Mg2+–ATPase activity and ionized calcium in type 2 diabetic patients with neuropathy. Diabetes Res Clin Pract 49:113–118
Shah CA (2008) Diabetic retinopathy: a comprehensive review. Indian J Med Sci 62:500–519
Steinle JJ, Kern TS, Thomas SA, McFadyen-Ketchum LS, Smith CP (2009) Increased basement membrane thickness, pericyte ghosts, and loss of retinal thickness and cells in dopamine beta hydroxylase knockout mice. Exp Eye Res 88:1014–1019
Costa G, Pereira T, Neto AM, Cristóvão AJ, Ambrosio AF, Santos PF (2009) High glucose changes extracellular adenosine triphosphate levels in rat retinal cultures. J Neurosci Res 87:1375–1380
Pereira TO, da Costa GN, Santiago AR, Ambrosio AF, dos Santos PF (2010) High glucose enhances intracellular Ca2+ responses triggered by purinergic stimulation in retinal neurons and microglia. Brain Res 1316:129–138
Robertson PL, Ar D, Goldstein GW (1990) Phosphoinositide metabolism and prostacyclin formation in retinal microvascular endothelium: stimulation by adenine nucleotides. Exp Eye Res 50:37–44
Sugiyama T, Kobayashi M, Kawamura H, Li Q, Puro DG (2004) Enhancement of P2X7-induced pore formation and apoptosis: an early effect of diabetes on the retinal microvasculature. Invest Ophthalmol Vis Sci 45:1026–1032
Sugiyama T, Kawamura H, Yamanishi S, Kobayashi M, Katsumura K, Puro DG (2008) Regulation of P2X7-induced pore formation and cell death in pericyte-containing retinal microvessels. Am J Physiol Cell Physiol 288:C568–C576
Wurm A, Iandiev I, Hollborn M, Wiedemann P, Reichenbach A, Zimmermann H, Bringmann A, Pannicke T (2008) Purinergic receptor activation inhibits osmotic glial cell swelling in the diabetic rat retina. Exp Eye Res 87:385–393
Pflueger AC, Schenk F, Osswald H (1995) Increased sensitivity of the renal vasculature to adenosine in streptozotocin-induced diabetes mellitus rats. Am J Physiol 269:F529–F535
Pflueger AC, Osswald H, Knox FG (1999) Adenosine-induced renal vasoconstriction in diabetes mellitus rats: role of nitric oxide. Am J Physiol 276:F340–F346
Sällström J, Carlsson PO, Fredholm BB, Larsson E, Persson AE, Palm F (2007) Diabetes-induced hyperfiltration in adenosine A1-receptor deficient mice lacking the tubuloglomerular feedback mechanism. Acta Physiol (Oxf) 190:253–259
Vallon V, Schroth J, Satriano J, Blantz RC, Thomson SC, Rieg T (2009) Adenosine A1 receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus. Nephron Physiol 111:30–38
Oppermann M, Qin Y, Lai EY, Eisner C, Li L, Huang Y, Mizel D, Fryc J, Wilcox CS, Briggs J, Schnermann J, Castrop H (2009) Enhanced tubuloglomerular feedback in mice with vascular overexpression of A1 adenosine receptors. Am J Physiol Renal Physiol 297:F1256–F1264
Castrop H (2007) Mediators of tubuloglomerular feedback regulation of glomerular filtration: ATP and adenosine. Acta Physiol (Oxf) 189:3–14
Guan Z, Inscho EW (2011) Role of adenosine 5′-triphosphate in regulating renal microvascular function and in hypertension. Hypertension 58:333–340
San Martin R, Valladares D, Roa H, Troncoso E, Sobrevia L (2009) Do adenosine receptors offer new therapeutic options for diabetic nephropathy? Curr Vasc Pharmacol 7:450–459
Vallon V, Osswald H (1994) Dipyridamole prevents diabetes-induced alterations of kidney function in rats. Naunyn Schmiedebergs Arch Pharmacol 349:217–222
Morato M, Patinha D, Afonso J, Correia C, Albino-Teixeira A (2010) Endogenous and exogenous adenosine and structural alterations in diabetic nephropathy: a possible role of the renin-angiotensin system? Purinergic Signal 6:100
Morato M, Patinha D, Afonso J, Sousa T, Albino-Teixeira A (2010) Renal effects of adenosine on diabetic Wistar and SHR rats. Purinergic Signal 6:140
Xia JF, Liang QL, Hu P, Wang YM, Li P, Luo GA (2009) Correlations of six related purine metabolites and diabetic nephropathy in Chinese type 2 diabetic patients. Clin Biochem 42:215–220
Roa H, Gajardo C, Troncoso E, Fuentealba V, Escudero C, Yáñez A, Sobrevia L, Pastor-Anglada M, Quezada C, San MR (2009) Adenosine mediates transforming growth factor-beta 1 release in kidney glomeruli of diabetic rats. FEBS Lett 583:3192–3198
Awad AS, Huang L, Ye H, Duong ET, Bolton WK, Linden J, Okusa MD (2006) Adenosine A2A receptor activation attenuates inflammation and injury in diabetic nephropathy. Am J Physiol Renal Physiol 290:F828–F837
Pawelczyk T, Grden M, Rzepko R, Sakowicz M, Szutowicz A (2005) Region-specific alterations of adenosine receptors expression level in kidney of diabetic rat. Am J Pathol 167:315–325
Pflueger AC, Berndt TJ, Knox FG (1998) Effect of renal interstitial adenosine infusion on phosphate excretion in diabetes mellitus rats. Am J Physiol 274:R1228–R1235
Leipziger J (2011) Luminal nucleotides are tonic inhibitors of renal tubular transport. Curr Opin Nephrol Hypertens 20:518–522
Friedman DJ, Rennke HG, Csizmadia E, Enjyoji K, Robson SC (2007) The vascular ectonucleotidase ENTPD1 is a novel renoprotective factor in diabetic nephropathy. Diabetes 56:2371–2379
Ellenberg M (1980) Development of urinary bladder dysfunction in diabetes mellitus. Ann Intern Med 92:321–323
Longhurst PA, Belis JA (1986) Abnormalities of rat bladder contractility in streptozotocin-induced diabetes mellitus. J Pharmacol Exp Ther 238:773–777
Luheshi GN, Zar MA (1990) Inhibitory effect of streptozotocin-induced diabetes on non-cholinergic motor transmission in rat detrusor and its prevention by sorbinil. Br J Pharmacol 101:411–417
Pak KJ, Ostrom RS, Matsui M, Ehlert FJ (2010) The M2-muscarinic receptor inhibits the development of streptozotocin-induced neuropathy in mouse urinary bladder. J Pharmacol Exp Ther 335:239–248
Liu G, Daneshgari F (2005) Alterations in neurogenically mediated contractile responses of urinary bladder in rats with diabetes. Am J Physiol Renal Physiol 288:F1220–F1226
Liu G, Daneshgari F (2008) Temporal expression of muscarinic and purinergic receptors in diabetic rat bladder. Neurourol Urodyn 27:594–595
Suadicani SO, Urban-Maldonado M, Tar MT, Melman A, Spray DC (2009) Effects of ageing and streptozotocin-induced diabetes on connexin43 and P2 purinoceptor expression in the rat corpora cavernosa and urinary bladder. BJU Int 103:1686–1693
Mumtaz FH, Lau DH, Siddiqui EJ, Morgan RJ, Thompson CS, Mikhailidis DP (2006) Changes in cholinergic and purinergic neurotransmission in the diabetic rabbit bladder. In Vivo 20:1–4
Benkó R, Lázár Z, Pórszasz R, Somogyi GT, Barthó L (2003) Effect of experimental diabetes on cholinergic, purinergic and peptidergic motor responses of the isolated rat bladder to electrical field stimulation or capsaicin. Eur J Pharmacol 478:73–80
Lee WC, Wu HP, Tai TY, Yu HJ, Chiang PH (2009) Investigation of urodynamic characteristics and bladder sensory function in the early stages of diabetic bladder dysfunction in women with type 2 diabetes. J Urol 181:198–203
Munoz A, Romain Z, Munch E, Gangitano D, Boone T, Smith C, Somogyi G (2009) Changes in purinergic and nitrergic sensory signals in female rats during early diabetes. Neurol Urodyn 28:110–111
Pinna C, Zanardo R, Puglisi L (2000) Prostaglandin-release impairment in the bladder epithelium of streptozotocin-induced diabetic rats. Eur J Pharmacol 388:267–273
Vlaskovska M, Kasakov L, Rong W, Bodin P, Bardini M, Cockayne DA, Ford AP, Burnstock G (2001) P2X3 knock-out mice reveal a major sensory role for urothelially released ATP. J Neurosci 21:5670–5677
Munoz A, Smith CP, Boone TB, Somogyi GT (2011) Overactive and underactive bladder dysfunction is reflected by alterations in urothelial ATP and NO release. Neurochem Int 58:295–300
Ayan S, Yildirim S, Ucar C, Sarioglu Y, Gültekin Y, Bütüner C (1999) Corporal reactivity to adenosine and prostaglandin E1 in alloxan-induced diabetic rabbit corpus cavernosum, and the effect of insulin therapy. BJU Int 83:108–112
Carneiro FS, Giachini FR, Lima VV, Carneiro ZN, Leite R, Inscho EW, Tostes RC, Webb RC (2008) Adenosine actions are preserved in corpus cavernosum from obese and type II diabetic db/db mouse. J Sex Med 5:1156–1166
Gür S, Öztürk B (2000) Altered relaxant responses to adenosine and adenosine 5′-triphosphate in the corpus cavernosum from men and rats with diabetes. Pharmacology 60:105–112
Calvert RC, Khan MA, Thompson CS, Mikhailidis DP, Burnstock G (2008) A functional study of purinergic signalling in the normal and pathological rabbit corpus cavernosum. BJU Int 101:1043–1047
Gür S, Kadowitz PJ, Abdel-Mageed AS, Kendirci M, Sikka SC, Burnstock G, Hellstrom WJG (2009) Management of erectile function by purinergic P2 receptors in diabetic rat. J Urol 181:2382
D’Amato M, Currò D (1990) Non-adrenergic non-cholinergic inhibitory innervation of the gastric fundus in streptozotocin-diabetic rats. Acta Physiol Hung 75(Suppl):77–78
Jenkinson KM, Reid JJ (2000) Altered non-adrenergic non-cholinergic neurotransmission in gastric fundus from streptozotocin-diabetic rats. Eur J Pharmacol 401:251–258
Belai A, Lefebvre RA, Burnstock G (1991) Motor activity and neurotransmitter release in the gastric fundus of streptozotocin-diabetic rats. Eur J Pharmacol 194:225–234
Jenkinson KM, Reid JJ (1995) Effect of diabetes on relaxations to non-adrenergic, non-cholinergic nerve stimulation in longitudinal muscle of the rat gastric fundus. Br J Pharmacol 116:1551–1556
Martinez-Cuesta MA, Massuda H, Whittle BJ, Moncada S (1995) Impairment of nitrergic-mediated relaxation of rat isolated duodenum by experimental diabetes. Br J Pharmacol 114:919–924
Hoyle CHV, Reilly WM, Lincoln J, Burnstock G (1988) Adrenergic, but not cholinergic or purinergic, responses are potentiated in the cecum of diabetic rats. Gastroenterology 94:1357–1367
Tahani H, Samia M, Rizk S, Habib YA, Tallaat M (1977) Effect of repeated doses of ATP on serum protein pattern and fat content of the liver in experimental diabetes. Z Ernährungswiss 16:120–127
Grden M, Podgorska M, Szutowicz A, Pawelczyk T (2007) Diabetes-induced alterations of adenosine receptors expression level in rat liver. Exp Mol Pathol 83:392–398
Harada H, Asano O, Hoshino Y, Yoshikawa S, Matsukura M, Kabasawa Y, Niijima J, Kotake Y, Watanabe N, Kawata T, Inoue T, Horizoe T, Yasuda N, Minami H, Nagata K, Murakami M, Nagaoka J, Kobayashi S, Tanaka I, Abe S (2001) 2-Alkynyl-8-aryl-9-methyladenines as novel adenosine receptor antagonists: their synthesis and structure-activity relationships toward hepatic glucose production induced via agonism of the A2B receptor. J Med Chem 44:170–179
Liu IM, Tzeng TF, Tsai CC, Lai TY, Chang CT, Cheng JT (2003) Increase in adenosine A1 receptor gene expression in the liver of streptozotocin-induced diabetic rats. Diabetes Metab Res Rev 19:209–215
Hashimoto N, Robinson FW, Shibata Y, Flanagan JE, Kono T (1987) Diversity in the effects of extracellular ATP and adenosine on the cellular processing and physiologic actions of insulin in rat adipocytes. J Biol Chem 262:15026–15032
Camberos MC, Perez AA, Udrisar DP, Wanderley MI, Cresto JC (2001) ATP inhibits insulin-degrading enzyme activity. Exp Biol Med (Maywood) 226:334–341
Song ES, Juliano MA, Juliano L, Fried MG, Wagner SL, Hersh LB (2004) ATP effects on insulin-degrading enzyme are mediated primarily through its triphosphate moiety. J Biol Chem 279:54216–54220
Foley JE (1992) Rationale and application of fatty acid oxidation inhibitors in treatment of diabetes mellitus. Diabetes Care 15:773–784
Dong Q, Ginsberg HN, Erlanger BF (2001) Overexpression of the A1 adenosine receptor in adipose tissue protects mice from obesity-related insulin resistance. Diabetes Obes Metab 3:360–366
Cox BF, Clark KL, Perrone MH, Welzel GE, Greenland BD, Colussi DJ, Merkel LA (1997) Cardiovascular and metabolic effects of adenosine A1-receptor agonists in streptozotocin-treated rats. J Cardiovasc Pharmacol 29:417–426
Dhalla AK, Chisholm JW, Reaven GM, Belardinelli L (2009) A1 adenosine receptor: role in diabetes and obesity. Handb Exp Pharmacol 193:271–295
Wojcik M, Zieleniak A, Wozniak LA (2010) New insight into A1 adenosine receptors in diabetes treatment. Curr Pharm Des 16:4237–4242
Gharibi B, Abraham AA, Ham J, Evans BA (2012) Contrasting effects of A1 and A2b adenosine receptors on adipogenesis. Int J Obes (Lond) 36:397–406
Lee H, Jun DJ, Suh BC, Choi BH, Lee JH, Do MS, Suh BS, Ha H, Kim KT (2005) Dual roles of P2 purinergic receptors in insulin-stimulated leptin production and lipolysis in differentiated rat white adipocytes. J Biol Chem 280:28556–28563
Laplante MA, Monassier L, Freund M, Bousquet P, Gachet C (2010) The purinergic P2Y1 receptor supports leptin secretion in adipose tissue. Endocrinology 151:2060–2070
Yu Z, Jin T (2010) Extracellular high dosages of adenosine triphosphate induce inflammatory response and insulin resistance in rat adipocytes. Biochem Biophys Res Commun 402:455–460
Madec S, Rossi C, Chiarugi M, Santini E, Salvati A, Ferrannini E, Solini A (2011) Adipocyte P2X7 receptors expression: a role in modulating inflammatory response in subjects with metabolic syndrome? Atherosclerosis 219:552–558
Lee SC, Vielhauer NS, Leaver EV, Pappone PA (2005) Differential regulation of Ca2+ signaling and membrane trafficking by multiple p2 receptors in brown adipocytes. J Membr Biol 207:131–142
Schmid AI, Szendroedi J, Chmelik M, Krssak M, Moser E, Roden M (2011) Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 34:448–453
Minet AD, Gaster M (2010) ATP synthesis is impaired in isolated mitochondria from myotubes established from type 2 diabetic subjects. Biochem Biophys Res Commun 402:70–74
Thong FS, Lally JS, Dyck DJ, Greer F, Bonen A, Graham TE (2007) Activation of the A1 adenosine receptor increases insulin-stimulated glucose transport in isolated rat soleus muscle. Appl Physiol Nutr Metab 32:701–710
Challis RA, Budohoski L, McManus B, Newsholme EA (1984) Effects of an adenosine-receptor antagonist on insulin-resistance in soleus muscle from obese Zucker rats. Biochem J 221:915–917
Kim MS, Lee J, Ha J, Kim SS, Kong Y, Cho YH, Baik HH, Kang I (2002) ATP stimulates glucose transport through activation of P2 purinergic receptors in C2C12 skeletal muscle cells. Arch Biochem Biophys 401:205–214
Mortensen SP, Gonzalez-Alonso J, Nielsen JJ, Saltin B, Hellsten Y (2009) Muscle interstitial ATP and norepinephrine concentrations in the human leg during exercise and ATP infusion. J Appl Physiol 107:1757–1762
Borno A, Ploug T, Bune LT, Rosenmeier JB, Thaning P (2012) Purinergic receptors expressed in human skeletal muscle fibres. Purinergic Signal 8:255–264
Ellis CG, Goldman D, Hanson M, Stephenson AH, Milkovich S, Benlamri A, Ellsworth ML, Sprague RS (2010) Defects in oxygen supply to skeletal muscle of prediabetic ZDF rats. Am J Physiol Heart Circ Physiol 298:H1661–H1670
Sprague RS, Bowles EA, Achilleus D, Ellsworth ML (2011) Erythrocytes as controllers of perfusion distribution in the microvasculature of skeletal muscle. Acta Physiol (Oxf) 202:285–292
Bienso RS et al (2012) GLUT4 and glycogen synthase are key players in bed rest-induced insulin resistance. Diabetes 61:1090–1099
Altavilla D, Squadrito F, Polito F, Irrera N, Calo M, Lo Cascio P, Galeano M, La Cava L, Minutoli L, Marini H, Bitto A (2011) Activation of adenosine A2A receptors restores the altered cell-cycle machinery during impaired wound healing in genetically diabetic mice. Surgery 149:253–261
Wang J, Wan R, Mo Y, Li M, Zhang Q, Chien S (2010) Intracellular delivery of adenosine triphosphate enhanced healing process in full-thickness skin wounds in diabetic rabbits. Am J Surg 199:823–832
Yamauchi R, Kobayashi M, Matsuda Y, Ojika M, Shigeoka S, Yamamoto Y, Tou Y, Inoue T, Katagiri T, Murai A, Horio F (2010) Coffee and caffeine ameliorate hyperglycemia, fatty liver, and inflammatory adipocytokine expression in spontaneously diabetic KK-Ay mice. J Agric Food Chem 58:5597–5603
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63:1–34
Muller CE, Jacobson KA (2011) Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta 1808:1290–1308
Sone H, Sasaki Y, Komai M, Toyomizu M, Kagawa Y, Furukawa Y (2004) Biotin enhances ATP synthesis in pancreatic islets of the rat, resulting in reinforcement of glucose-induced insulin secretion. Biochem Biophys Res Commun 314:824–829
Conflict of Interest
The authors report no conflict of interest.
Funding
This work was supported by The Danish Council for Independent Research Natural Sciences (IN).
Author information
Authors and Affiliations
Corresponding author
Additional information
G. Burnstock and I. Novak contributed equally in writing the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Burnstock, G., Novak, I. Purinergic signalling and diabetes. Purinergic Signalling 9, 307–324 (2013). https://doi.org/10.1007/s11302-013-9359-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-013-9359-2