
Abstract
The metabolic profiles of rice and barley plants constitutively expressing a sodium-pumping ATPase (PpENA1) isolated from the bryophyte Physcomitrella patens were examined using GC-MS. Quantitative real-time PCR (qRT-PCR) was used to determine the mRNA levels of PpENA1 in root and leaf tissues of the transgenic rice and barley lines. PpENA1 mRNA levels were significantly higher in rice lines than in barley lines with the same dual CaMV35S promoter controlling PpENA1 transcription in both species. In rice, PpENA1 mRNA levels were greatest in the shoot whilst levels were greatest in the roots of barley. Metabolite profiles were determined in the flag leaf of both rice and barley plants grown under controlled conditions. A large proportion of the measured metabolites were significantly altered in the transgenic lines compared to null-segregating lines, revealing a considerable impact of expression of the sodium-pumping ATPase (PpENA1) transgene on metabolism. Interestingly, the metabolite changes were different between rice and barley, indicating different responses of rice and barley to the introduction of this gene.
Similar content being viewed by others

Introduction
In the last decade there have been considerable efforts to develop new analytical technologies for the broader analysis of metabolites. Chromatographic techniques coupled to mass spectrometry (MS) have proven to be powerful and suitable for the separation, detection, identification and quantification of a large number of different metabolic compounds simultaneously extracted from biological material (for review see Bino et al., 2004; Kopka et al., 2004; Hall, 2006). In addition, NMR now plays a major role in metabolite analyses (Krishnan et al., 2005). The study of metabolite pools using these new technologies, in combination with sophisticated data analysis tools, has been referred to as metabolomics (Fiehn, 2002). The study of the metabolome using these technologies is being applied in many different fields and aspects of biological sciences. In plant sciences, metabolomics technologies are being extensively utilised in general phenotyping approaches, to broaden our understanding of plant metabolism and physiology, in gene function studies, for QTL analysis and in support of targeted breeding programs (for summary see Villas-Boas et al., 2006). Importantly, metabolomics can be used for the detailed characterisation of genetically altered plants. To complement metabolomics studies, the ionome (or ‘nutriome’) of higher plants is starting to be studied (Salt, 2004; http://www.acpfg.com.au/nutriomics/). Consistent with classical studies (e.g., Wild, 1988), this work indicates a fairly tight regulation of ion homeostasis networks in plants. Numerous mutants with defects in the homeostasis of specific ions have now been identified, but the interactions between the ionome and metabolome still require further systematic evaluation. Baxter et al. (2007) have recently introduced a new tool with great potential for the study of these interactions (Purdue Ionomics Information Management System, http:// www.purdue.edu/dp/ionomics). Currently the system contains publicly available data of 15 ions collected from over 60,000 tissue samples from Arabidopsis. It will allow the integration of comprehensive ionomics datasets with other Arabidopsis resources, such as transcriptomic or metabolomic data sets, and eventually to transfer this concept across to other organisms (for example, rice). Previous studies have already indicated numerous interactions between the accumulation of specific metabolites and elements, notably of Na+ and so-called compatible solutes such as proline and glycinebetaine but the combination of ionomic and metabolomics data on large-scale plant studies will allow researchers to decipher such interactions on a much greater scale and in a more systematic way.
There have been major advances over the last twenty years in our ability to create new genetic variation by the introduction of foreign genes, the manipulation of the expression of endogenous genes or by mutation. Concurrently, there is now an important role for scientists to evaluate, as well as describe, the effects of transgenic alterations on plant performance and possible impacts on human health and the environment. The development of routine methodologies for genetic transformation of plant genomes and community concern over this technology has introduced a greater need to monitor the effects of these genetic alterations. This not only includes changes in the visible phenotype but also changes in the biochemical composition of the cells and the appearance of new and unintended cell products (Cellini et al., 2004; Kuiper et al., 2003). Metabolomics can be used to monitor and evaluate the effects of transgenesis on a plant’s metabolism (Rischer and Oksman-Caldentey, 2006) and to make an assessment of potential risks associated with these changes. There have been some instances where the introduction or deletion of a gene in a plant has resulted in additional, unexpected alterations in plant metabolism, even when the target gene activity was not involved directly in metabolic processes but rather in cell development or plant structure (e.g., Takahashi et al., 2005; Long et al., 2006; for summary see Kuiper et al., 2001).
In this work, we aimed to evaluate the metabolic alterations resulting from the insertion of a gene from the moss, Physcomitrella patens into higher plants. We have chosen a Physcomitrella-specific Na+-pumping ATPase (PpENA1), which is absent in flowering plants and appears likely to be responsible, in part, for the high salinity tolerance of the moss (Benito and Rodriguez-Navarro, 2003). In plants, the maintenance of non-toxic levels of cytosolic Na+ involves, at least in part, sequestration of Na+ into intracellular vacuoles, a process that relies upon Na+/H+ antiporters (Tester and Davenport, 2003). The recent identification of two genes encoding Na+-ATPases (PpENA1 and PpENA2) in Physcomitrella(Benito and Rodriguez-Navarro, 2003) suggests the ENA-type Na+-ATPases were lost during the evolution of higher plants or alternatively that Physcomitrella has gained these genes. PpENA1 was shown to act as a Na+-pump when expressed heterologously in yeast and was found to complement a salt sensitive yeast strain deficient in Na+ and K+ efflux (Benito and Rodriguez-Navarro, 2003). Furthermore, gene targeting experiments in Physcomitrella suggest that PpENA1 plays an essential role in salinity tolerance in moss under moderate stress (C. Lunde, unpublished results). In contrast, PpENA2 was unable to complement the salt-sensitive yeast strain; mRNA levels were present at low levels and expression was not induced in salt stressed Physcomitrella.
Metabolite levels in leaves of rice and barley plants constitutively expressing PpENA1 transcript were compared to control plants. Resulting data showed that there was a species-specific alteration of metabolism following transgenesis demonstrating the importance of comprehensive monitoring of genetic alterations.
Material and methods
Plant material and growth conditions
Physcomitrella patens (Hedw.) derived from a wild type specimen collected in Gransden Wood in Huntingdonshire, UK (Ashton and Cove, 1977) was grown at 22 °C on cellophane disks placed on solid minimal media in Petri dishes (Ashton et al., 1979), supplemented with ammonium tartrate (0.5 g/l). Standard growth conditions were 16 h white light (fluorescent tubes, GRO-LUX, 100 μmol m−2 sec−1) and 8 h darkness.
Agrobacterium tumefaciens-mediated barley (Hordeum vulgare cv. Golden Promise) transformation was performed as described by Tingey et al. (1997) and modified by Matthews et al. (2001). Immature embryos were taken from donor plants. Scutella were cultured on callus induction medium, based on the recipe of Wan and Lemaux (1994). An Agrobacterium solution of strain AGL-O was placed onto scutella and scutella were transferred to callus induction medium containing 95 μM hygromycin B (Becton Dickinson Biosciences) for 3 days in the dark at 22–24 °C. Callus derived from treated scutellum was transferred to shoot regeneration medium based on the FHG recipe of Wan and Lemaux (1994). Regenerated shoots were excised from the callus and transferred to culture boxes (Magenta Corporation) that contained hormone-free callus induction medium, supplemented with 95 μM hygromycin B. Tissue culture-derived plants were established in soil and grown to maturity (Singh et al., 1997). All media contained 150 mg/l Timentin (Smith Kline Beecham). Seed was harvested from fertile lines, dried for 3 days at 37 °C and was placed into cold storage (4 °C, 30% humidity).
Embryogenic nodular units arising from scutellum-derived callus of rice (Oryza sativa) L. cv. Nipponbare were inoculated with supervirulent A. tumefaciens strains EHA105 and AGL-1. Hygromycin-resistant shoots were regenerated after 9 weeks according to the protocol described by Sallaud et al. (2003; 2004). Rooted T0 plantlets were transferred to the greenhouse in Jiffy peat pots, and moved to soil after 15 days. Seed was harvested from fertile lines, dried for 3 days at 37 °C, and was placed into cold storage (4 °C, 30% humidity).
Segregating T1 rice and barley seed were germinated on wet filter paper in Petri dishes at 28 °C for 4 days. Seedlings were transplanted into PVC tubes, containing small plastic beads, supported in tubs containing a hydroponic growth medium as described by Roessner et al. (2006). Hydroponic growth solution was used to flood the tubs over 20 min every hour and tubs were left to drain. After 6 weeks, the plants were then placed into pots containing soil and barley plants were placed in the glasshouse (12–25 °C temp range) whilst rice plants were moved in to a growth chamber (14 h day, 28 °C, 85% humidity). A green flag leaf was removed from the plants before the heads became mature, frozen in liquid nitrogen and stored at −80 °C until metabolite extraction. We selected two transgenic barley and three transgenic rice lines, each representing an independent transgenic event, and the respective null segregating lines as control plants. Five replicates of each line were grown, with the flag leaf from each individual being assayed independently. This strategy has been chosen in order to firstly identify changes due purely to transgenesis when observed in the all the transgenic lines and not due to the site of insertion of the transgene. Secondly, biological replication is necessary to evaluate natural variation relative to changes due to the transgene.
All chemicals were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia). Derivatisation reagent N-methyl-N-(tert-butyldimethylsilyl)trifluoroaceteamide + 1% tert-butyldimethylchlorosilane was purchased by Pierce, distributed by Progen Bioscience (Australia). N-methyl-N-[trimethylsilyl]trifluoroacetamide was purchased by Biolab Ltd. (Clayton, Victoria, Australia).
RNA extraction, cDNA synthesis and quantitative real-time RT-PCR (qRT-PCR)
Total RNA was extracted from protonemal tissue of Physcomitrella and from leaf and root tissues of rice and barley using Trizol (Invitrogen) according to the manufacturer’s instructions. Purified RNA was treated with DNaseI using a DNA-free kit (Ambion, USA). RNA integrity was checked on a 1% (w/v) agarose gel containing ethidium bromide. cDNA was synthesised using SuperScript III First-Strand Synthesis System (Invitrogen) and 2.5 μM oligo-dT (18–20) primer according to the manufacturer’s protocol. PpENA1F 5′ CACCATGGAGGGCTCTGGGGAC and PpENA1R 5′ GCGGATTCTGCAACATGAGGT primers (200 nm) were used in PCR with 200 μM of dNTPs, 1 μl of Physcomitrella cDNA and Elongase enzyme and PCR reagents (Invitrogen) to amplify the PpENA1 open reading frame. qRT-PCR analysis and quantitation was performed as described in Burton et al. (2003). Additional primers specific to PpENA1 were synthesised for qRT-PCR experiments, PpENA1Fq 5′ AAGGCATTACCTGGGAGTGGAT and PpENA1Rq 5′ TCACATGTTGTAGGAGTT.
Plasmid construction
The PpENA1 PCR amplified 2938 bp cDNA was cloned into pENTR-D-TOPO (Invitrogen) according to the manufacturer’s directions and the PpENA1 fragment was recombined into pMDC32 (Curtis and Grossniklaus, 2003) via a Gateway LR recombination reaction (Invitrogen) according to the manufacturer’s directions to create the pAJ54 vector. The pAJ54 vector was used to transform Agrobacterium strains for plant transformation.
Extraction, derivatisation and GC-MS analysis of metabolites
Frozen barley and rice leaf tissue was homogenised using a mortar and pestle pre-cooled with liquid nitrogen and approximately 60 mg of powder were extracted in 350 μl of methanol; 20 μl of polar internal standard (0.2 mg ml−1 in water) was added as a quantification standard. The mixture was extracted for 15 min at 70 °C and subsequently mixed vigorously with 1 volume of water. After centrifugation at 2200g, the supernatant was transferred into a new tube and a 100 μl aliquot was taken and dried in vacuo for further derivatisation with TBS (see below). The rest of the supernatant was purged of non-polar metabolites by adding 300 μl chloroform. Following centrifugation, the upper methanol/water phase was taken and washed again with 300 μl chloroform. After centrifugation, an aliquot of 100 μl was taken and dried in vacuofor further derivatisation with TBS (see below). Both the TBS and TMS dry residues were re-dissolved and derivatised for 120 min at 37 °C (in 20 μl of 30 mg ml−1 methoxyamine hydrochloride in pyridine). The TBS aliquot was further treated with 40 μl MTBSTFA (N-methyl-N-(tert-butyldimethylsilyl)trifluoroaceteamide + 1% tert-butyldimethylchlorosilane) for 45 min at 65 °C and the TMS aliquot was treated with 40 μl MSTFA (N-methyl-N-[trimethylsilyl]trifluoroacetamide). To the derivatives, 5 μl of a retention time standard mixture (0.029% (v/v) n-dodecane, n-pentadecane, n-nonadecane, n-docosane, n-octacosane, n-dotracontane, n-hexatriacontane dissolved in pyridine) was added prior to derivatisation. Sample volumes of 1 μl were injected onto the GC column using a hot needle technique.
The GC-MS system comprised of an AS 3000 autosampler, a Trace gas chromatograph Ultra and a DSQ quadrupole mass spectrometer (ThermoElectron Cooperation, Austin, USA). The mass spectrometer was tuned according to the manufacturer’s recommendations using tris-(perfluorobutyl)-amine (CF43). Gas chromatography was performed on a 30 m VF-5MS column with 0.25 μm film thickness with a 10 m Integra guard column (Varian, Inc, Victoria, Australia). The injection temperature was set at 230 °C, the MS transfer line at 280 °C and the ion source adjusted to 250 °C. Helium was used as the carrier gas at a flow rate of 1ml min−1. The analysis of TBS samples was performed under the following temperature program; start at injection 100 °C, a hold for 1 min, followed by a 1 °C min−1 oven temperature ramp to 106 °C followed by 7 °C min−1 oven temperature ramp to 325 °C and a final 10 min heating at 325 °C. The system was temperature equilibrated for 1min at 100 °C prior to injection of the next sample. Mass spectra were recorded at 2scan s−1 with an m/z 70–600 scanning range. The analysis of TMS samples was performed under same conditions as described in Roessner et al. (2006). Both chromatograms and mass spectra were evaluated using the Xcalibur program (ThermoFinnigan, Manchester, UK). Mass spectra of eluting TMS compounds were identified using the commercial mass spectra library NIST (http://www.nist.gov) and the public domain mass spectra library of Max-Planck-Institute for Plant Physiology, Golm, Germany (http://csbdb.mpimp-golm.mpg.de/csbdb/dbma/msri.html). Mass spectra of eluting TBS compounds were identified using our in-house TBS mass spectral library. All matching mass spectra were additionally verified by determination of the retention time by analysis of authentic standard substances. Resulting relative response ratios normalised per gram extracted fresh weight for each analysed metabolite were prepared as described in Roessner et al. (2006).
Statistical analysis
Data were prepared as described in Roessner et al. (2001) and presented as x-fold compared to the reference (segregating null) which is set to 1 following calculation of the mean of 5 individual replicates per genotype. If two observations are described in the text as significantly different, this means that their difference was determined to be statistically significant (P < 0.05) according to the t-test algorithm incorporated into Microsoft Excel (Microsoft Corp., Seattle, USA). Principal component analyses (PCA) were carried out on the response/gFW raw data for each individual metabolite and measurement following a log10 transformation. PCA are presented in a two-dimensional graphical display of the data in which a single sample is represented by a point in three-dimensional space. PCA was carried using the Pirouette 3.11 software (Infometrix Inc, Woodinville, US).
Results and discussion
Plant phenotypes
Two independent transgenic lines of barley, HvG40-2 and HvG40-4, and three independent transgenic lines of rice, OsG40-2, OsG40-15, and OsG40-23, all containing single transgene copies were developed. These did not show any abnormal growth phenotypes and plants of the same species developed at similar rates (data not shown).
Transgenic plants developed flowers at the same time as wild type controls and produced fertile seed. Flag leaves taken for metabolic analysis were of a similar developmental stage based upon location and size. Leaves showing any visible signs of senescence were not taken for metabolic analyses. Leaves were taken for metabolite analysis because they show the greatest differences in sodium accumulation and it is generally agreed that they are the primary site of sodium toxicity (Tester and Davenport, 2003).
qRT-PCR analysis of transgene expression
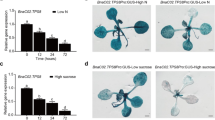
The levels of PpENA1 mRNA were quantified in the shoots and roots of the segregating barley and rice lines by qRT-PCR. The same transformation construct was used for both barley and rice, but there was a significantly higher level of transgene expression driven by the dual 35S cauliflower mosaic viral promoter in rice (Table 1). Variable expression levels have previously been noted with the 35S promoter in monocot species (Schledzewski et al., 1994). Levels of PpENA1 mRNA were comparable in the same tissue types of the same species but the rice line OsG40-23 had 2- to 3-fold more PpENA1 mRNA in its shoots when compared to the other two rice lines. Expression was also higher in barley roots than in shoots in contrast to the rice lines which showed maximal expression in the shoot.
Metabolite analysis in rice and barley leaves
Metabolite levels were determined using a recently established GC-MS method for barley tissues (Roessner et al., 2006) which was also optimised for rice leaf tissue. The GC-MS profiles differed markedly between barley and rice leaves and this observation was supported following principal component analysis showing that the first component separated the two species (figure 3). This suggests that each species is characterised by a specific distribution of metabolite levels although being genetically quite related. The levels of 129 metabolites were quantified and 110 were unambiguously identified with respect to their chemical nature which was based upon mass spectra and retention time indices matching our in-house mass spectral library and the library obtained from the Golm Metabolome Database (MSRI, Kopka et al., 2005; Schauer et al., 2005; Roessner et al., 2006) (Table 2). The following discussion will only describe statistically significant (based on Student’s t-test with P-value < 0.05) changes in levels of identified metabolites. For comparisons to the respective null segregants, data from the two independent barley lines were assessed individually as well as after data averaging. This approach was also applied to the data from the three independent rice lines.
Metabolite changes in transgenic barley lines
The constitutive over-expression of the Physcomitrella Na+-pumping ATPase (PpENA1) in barley resulted in a statistically significant (P < 0.05) alteration in about half of the metabolites analysed (Table 2, figure 1). The largest impact was observed in the levels of free amino acids with marked increases in alanine, GABA, glutamine, glycine, homoserine and phenylalanine in both transgenic lines. β-alanine, glutamate, isoleucine, proline, 5-oxoproline, serine, threonine and valine were additionally increased only in line HvG40-4 and allantoin only in line HvG40-2. Most pronounced increases were in alanine (8-fold), GABA (9-fold), glutamine (22-fold), proline (85-fold) and threonine (11-fold).

Mapping of metabolite changes of barley leaves expressing PpENA1 compared to leaves of null segregating plants onto known metabolic pathways. Data from 2 transgenic barley lines were averaged and normalised to data from null segregating lines (Table 2, Supplementary Data). X-fold values are presented as the mean ± %SE of five independent determinations for each line. Those values that are significantly different to control are colored in red for increases and blue for decreases. Those values which are significantly changed only in one of the transgenic lines but not after averaging of the two lines are colored in orange for increase and light blue for decrease. Metabolites not determined are marked in italics.
There were also statistically significant changes (P < 0.05) in the levels of 23 organic acids. Most pronounced changes were found in 4-hydroxy-cinnamic acid (4-fold) in line HvG40-4, citric acid (0.05-fold) in line HvG40-2 and isocitric acid (between 0.25 and 0.3-fold) in both lines (Table 2). In addition, salicylic acid was increased in both lines, but only found to be significant in line HvG40-4 (3-fold).
Out of the 25 sugars analysed, only 10 were increased significantly (P < 0.05) but a strong and significant decrease was observed in glucose-6-P (0.14 and 0.24-fold in both lines) and fructose-6-P (0.23-fold in line HvG40-4). Trehalose was strongly increased in line HvG40-2 (5-fold) but due to high standard deviation was not statistically significant (Table 2). When levels were averaged over the replicates of the two lines, the increase became significant (P < 0.05) as indicated in figure 1.
Furthermore, a number of fatty acids increased between 1.5 and 3-fold with strongest increases in 9,12-(Z,Z)-octadecanoic acid (4-fold) and tetradecanoic acid (5-fold). In addition, a 3-fold increase was observed in ethanolamine.
The data indicated that glycolytic and TCA cycle intermediates were decreased whereas a range of free amino acids were elevated. One possible explanation would be that the carbon structures needed for the synthesis of free amino acids (e.g., proline, GABA) maybe acting as osmoprotectants are withdrawn from the TCA cycle. In addition, salicylic acid was increased concurrently with its biosynthetic intermediates phenylalanine and benzoic acid. Salicylic acid is a phytohormone and has been shown to be involved in mediating or orchestrating stress responses in plants, especially in pathogen defence responses (Halim et al., 2006). There was also a slight, but significant increase in the levels of allantoin in one of the transgenic lines (HvG40-2). Allantoin is considered to be a major player in nitrogen metabolism in plants and may act as a nitrogen transport compound throughout the plant (Desimone et al., 2002). A disruption in nitrogen metabolism could also be explained by the elevated levels of glutamine and glutamate. Another striking increase in both transgenic lines was seen for ethanolamine which is produced by the decarboxylation of serine (http://www.arabidopsis.org). Serine levels were found to be dramatically elevated in the transgenic barley lines. Ethanolamine is further used for the biosynthesis of choline, an important intermediate both for the synthesis of the membrane phospholipid phosphatidylcholine and of the osmoprotectant glycine betaine (Rontein et al., 2001; Rontein et al., 2003). Further studies are required to determine if the elevated levels of ethanolamine are indeed responsible for increased choline and/or glycine betaine production.
Metabolite changes in transgenic rice lines
In general there were fewer metabolites affected by the constitutive over-expression of the Na+-pumping ATPase (PpENA1) in rice leaves compared with barley leaves (Table 2, figure 2). Interestingly, amino acid levels decreased in contrast to barley with leucine (between 0.3 and 0.2-fold in line OsG40-2, OsG40-15 and OsG40-23), β-alanine, isoleucine, threonine, tyrosine and valine (0.2-fold in line OsG40-15 and OsG40-23), proline (between 0.4 and 0.3-fold in line OsG40-15 and OsG40-23) and asparagine (0.5-fold in line OsG40-2 only).

Mapping of metabolite changes of rice leaves expressing PpENA1 compared to leaves of null segregating plants onto known metabolic pathways. Data from three transgenic rice lines were averaged and normalised to data from null segregating lines (Table 2, Supplementary Data). X-fold values are presented as the mean ± %SE of five independent determinations for each line. Those values that are significantly different to control are colored in red for increases and blue for decreases. Those values which are significantly changed only in one or two of the transgenic lines but not after averaging of the two lines are colored in orange for increase and light blue for decrease. Metabolites not determined are marked in italics.
In rice as seen in barley, isocitric acid (0.3-fold in line OsG40-2) and citric acid (0.3-fold in all three lines) were dramatically decreased but only quinic and salicylic acid (3 and 4-fold in line OsG40-2 and OsG40-23, respectively) and erythronic and glyceric acid (2-fold in line OsG40-23) were increased. In addition, pentonic acid-1,4-lactone was only detectable in the transgenic lines.
There were no significant differences in the levels of sugars or fatty acids in the rice leaves. Levels of ethanolamine were elevated, as they were in barley, by 2- and 4-fold in lines OsG40-15 and OsG40-23, respectively. Other commonalities between the rice and barley transgenics included reductions in TCA cycle intermediates.
Surprisingly, the osmoprotectants proline and GABA decreased in transgenic rice lines when compared to the control. This is in contrast to the results observed in the barley transgenic lines. This may be related to the higher intrinsic tolerance to high tissue Na+ in barley, and thus a greater ability to respond to perturbations in plant Na+ levels. In rice, proline has been shown to have little or no effect on levels of salinity tolerance and may even have an exacerbating effect (Garcia et al., 1997), which is in stark contrast with results obtained for many other plant species in which elevated levels of proline reduce salt stress. If a rice plant was stressed as a result of expressing the PpENA1 transgene, then it might make sense to down regulate proline synthesis.
In the rice lines there were elevated levels of salicylic acid and this was also the case in the barley lines but in the rice the biosynthetic intermediates phenylalanine and benzoic acid were not significantly changed. In contrast to the observations in barley, compounds involved in nitrogen metabolism, such as allantoin and glutamine remained unchanged in transgenic rice lines when compared to the controls.
Comparison of metabolite changes using principal component analysis
When principal component analysis (PCA) was applied to the metabolite profiles, a clear separation of profiles from both species could be observed in PC1, indicating that most metabolite differences were between the species rather than due to transgene expression (figure 3). The second component (PC2) separated the transgenic rice lines from the null segregating control plants, whereas there was no clear separation between the profiles from either transgenic or control barley leaves. These were separated by the third component (PC3) but the distance of clusters was not as large as observed for the rice profiles. It can be concluded that the overall differences between the rice transgenic lines and their controls is larger than those observed in barley.

Principal component analysis (PCA) of metabolite profiles of leaves of both rice and barley expressing PpENA1. PCA of metabolite profiles of rice and barley leaves expressing PpENA1 and null segregating control plants. The data originated from Table 2 (supplementary data) and are log transformed. PCA vectors span a 9-dimensional space to give the best sample separation with each point representing a linear combination of all the metabolites from an individual sample. (A) Representation of plotting PC 1 and 2 for visualisation which include 53.8% of the information derived from metabolic variances. (B) Representation of plotting PC 1 and 3 for visualisation which include 42.2% of the information derived from metabolic variances.
Concluding remarks
The work described here clearly highlighted that metabolic profiling enabled the discovery that heterologous expression of the same transgene in rice and barley had different effects on the metabolome. The most pronounced differences between the two species was that free amino acids were increased in transgenic barley lines upon expression of PpENA1, compared to the controls whereas in rice transgenic lines, free amino acids were reduced. In addition, from Table 2 it is clear that there are a smaller number of metabolites affected by transgene expression in rice but the scale of the changes is greater. There was also some commonality observed between the two species – e.g., citric and isocitric acid were decreased in both barley and rice transgenic lines and salicylic acid increased in all transgenic lines irrespective of species. The observed differences between the two species would be difficult to predict based solely on the known function of a Na+ pumping ATPase and as such, our results highlight the value of metabolic profiling in assessing the effects of genetic modification. The changes appear to correlate with the higher abundance of PpENA1 mRNA in the rice lines compared to barley. Further work is required to determine protein abundance and location of activity, both sub-cellularly as well as in specific cell types. This will also clarify if the metabolite changes observed are caused by the transgene or the different transformation events.
In conclusion, the data demonstrates that it is essential to investigate the metabolic consequences of the introduction of a gene into a plant’s genome even when the gene is involved in a general biochemical process such as ion homeostasis rather than encoding for a biosynthetic enzyme with known catalytic function within a known pathway. Our data demonstrated that potential interactions between the ionome and metabolome appear likely to be important. Metabolomics as a tool to analyse alterations in a large number of metabolites simultaneously in genetically modified plants has the potential to give a fast and comprehensive impression about the impact and unintended effects of a transgenic event. Together with other ‘omics’ approaches, such as transcriptomics and proteomics, metabolomics provides an important tool in the investigation of the substantial equivalence of genetically modified crops with their progenitor genotypes.
References
Ashton N.W., Cove D.J. (1977) Isolation and preliminary characterization of auxotrophic and analog resistant mutants of moss, Physcomitrella patens. Mol. Gen. Genet. 154:87–95
Ashton N.W., Grimsley N.H, Cove D.J. (1979) Analysis of gametophytic development in the moss Physcomitrella patens using auxin and cytokinin resistant mutants. Planta 144:427–435
Baxter I., Ouzzani M., Orcun S., Kennedy B., Jandhyala S.S., Salt D.E. (2007) Purdue Ionomics Information Management System. An Integrated Functional Genomics Platform. Plant Physiol. 143:600–6100
Benito B, Rodriguez-Navarro A. (2003) Molecular cloning and characterization of a sodium-pump ATPase of the moss Physcomitrella patens. Plant J. 36:382–389
Bino R.J., Hall R.H., Fiehn O., Kopka J., Saito K., Draper J., Nikolau B., Mendes P., Roessner-Tunali U., Beale M., Trethewey R.N., Lange B.M., Syrkin Wurtele E., Sumner L. (2004) Opinion: Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9:418–425
Brand L., Hoerler M., Nueesch E., Vassalli S., Barrell P., Yang W., Jefferson R.A., Grossniklaus U, Curtis M.D. (2006) A versatile and reliable two-component system for tissue-specific gene induction in Arabidopsis. Plant Physiol. 141:1194–1204
Burton R.A., Shirley N.J., King B.J., Harvey A.J, Fincher G.B. (2004) The CesA gene family of barley. Quantitative analysis of transcripts reveals two groups of co-expressed genes. Plant Physiol. 134:224–236
Cellini F., Chesson A., Colquhoun I., Constable A., Davies H.V., Engel K.H., Gatehouse A.M., Karenlampi S., Kok E.J., Leguay J.J., Lehesranta S., Noteborn H.P., Pedersen J, Smith M. (2004) Unintended effects and their detection in genetically modified crops. Food Chem. Toxicol. 42:1089–1125
Curtis M.D., Grossniklaus U. (2003) A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol. 133:462–469
Desimone M., Catoni E., Ludewig U., Hilpert M., Schneider A., Kunze R., Tegeder M., Frommer W.B., Schumacher K. (2002) A novel superfamily of transporters for allantoin and other oxo derivatives of nitrogen heterocyclic compounds in Arabidopsis. Plant Cell 14:847–856
Fiehn O. (2002) Metabolomics – the link between genotypes and phenotypes. Plant Mol. Biol. 48:155–171
Garcia A.B., Engler J.A., lyer S., Tom Cerats T., Montagu M.V., Caplan A.B. (1997) Effects of osmoprotectants upon NaCl stress in rice. Plant Physiol. 115:159–169
Halim V.A., Vess A., Scheel D., Rosahl S. (2006) The role of salicylic acid and jasmonic acid in pathogen defence. Plant Biol. 8:307–313
Hall R.D. (2006) Plant metabolomics: from holistic hope, to hype, to hot topic. New Phytol. 169:453–68
Kopka J., Fernie A.R., Weckwerth W., Gibon Y, Stitt M. (2004) Metabolite profiling in plant biology: Platforms and destinations. Genome Biol. 5: 109–117
Kopka J., Schauer N., Krueger S., Birkemeyer C., Usadel B., Bergmüller E., Dörmann P., Gibon Y., Stitt M., Willmitzer L., Fernie A.R., Steinhauser D. (2005) GMD@CSB.DB: the golm metabolome database. Bioinformatics 21:1635–16358
Krishnan P., Kruger N.J., Ratcliffe R.G. (2005) Metabolite fingerprinting and profiling in plants using NMR. J. Exp. Bot. 56:255–265
Kuiper H.A., Kleter G.A., Noteborn H.P., Kok E.J. (2001) Assessment of the food safety issues related to genetically modified foods. Plant J. 27:503–528
Kuiper H.A., Kok E.J, Engel K.H. (2003) Exploitation of molecular profiling techniques for GM food safety assessment. Curr. Opin. Biotechnol. 14:238–243
Long M., Millar D.J., Kimura Y., Donovan G., Rees J., Fraser P.D., Bramley P.M., Bolwell G.P. (2006) Metabolite profiling of carotenoid and phenolic pathways in mutant and transgenic lines of tomato: identification of a high antioxidant fruit line. Phytochem. 67:1740–1757
Matthews P.R., Wang M.-B., Waterhouse P.M., Thornton S., Fieg S.J., Gubler F., Jacobsen J.V. (2001) Marker gene elimination from transgenic barley, using co-transformation with adjacent ’twin T-DNAs’ on a standard Agrobacterium transformation vector. Mol. Breeding 7:195–202
Pardo J.M., Cubero B., Leidi E.O, Quintero F.J. (2006) Alkali cation exchangers: roles in cellular homeostasis and stress tolerance. J. Exp. Bot. 57:1181–1199
Rischer H., Oksman-Caldentey K.M. (2006) Unintended effects in genetically modified crops: revealed by metabolomics? Trends Biotechnol. 24:102–104
Roessner U., Luedemann A., Brust D., Fiehn O., Linke T., Willmitzer L., Fernie A.R. (2001). Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 13:11–29
Roessner U., Patterson J.H., Forbes M.G., Fincher G.B., Langridge P, Bacic A. (2006) An investigation of boron toxicity in barley using metabolomics. Plant Physiol. 142: 1087–1101
Rontein D., Nishida I., Tashiro G., Yoshioka K., Wu W.I., Voelker D.R., Basset G., Hanson A.D. (2001) Plants synthesize ethanolamine by direct decarboxylation of serine using a pyridoxal phosphate enzyme. J. Biol. Chem. 276:35523–35529
Rontein D., Rhodes D., Hanson A.D. (2003) Evidence from engineering that decarboxylation of free serine is the major source of ethanolamine moieties in plants. Plant Cell Physiol. 44:1185–1191
Sallaud C., Gay C., Larmande P., Bes M., Piffanelli P., Piegu B., Droc G., Regad F., Bourgeois E., Meynard D., Perin C., Sabau X., Ghesquiere A., Glaszmann J.-C., Delseny M., Guiderdoni E. (2004) High throughput T-DNA insertion mutagenesis in rice: a first step towards in silico reverse genetics. Plant J. 39:450–464
Sallaud C., Meynard D., van-Boxtel J., Gay C., Bes M., Brizard J.P., Larmande P., Ortega D., Raynal M., Portefaix M., Ouwerkerk P.B.F., Rueb S., Delseny M., Guiderdoni E. (2003) Highly efficient production and characterization of T-DNA plants for rice (Oryza sativa L.) functional genomics. Theoretic App. Gen. 106: 1396–1408
Salt D.E. (2004) Update on plant ionomics. Plant Physiol. 136:2451–2456
Schauer N., Steinhauser D., Strelkov S., Schomburg D., Allison G., Moritz T., Lundgren K., Roessner-Tunali U., Forbes M.G., Willmitzer L., Fernie A.R., Kopka J. (2005) GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 579:1332–1337
Schledzewski K, Mendel R. (1994) Quantitative transient gene expression: comparison of the promoters for maize polyubiquitin 1, rice actin 1, maize derived Emu and CaMV 35S in cells of barley, maize and tobacco. Transgenic Res. 4:249–255
Singh R.-R., Kemp J.A., Kollmorgen J.F., Qureshi J.A., Fincher G.B. (1997) Fertile plant regeneration from cell suspension and protoplast cultures of barley (Hordeum vulgare cv. Schooner). Plant Cell Tissue and Organ Culture 49:121–127
Takahashi N., Nakazawa M., Shibata K., Yokota T., Ishikawa A., Suzuki K., Kawashima M., Ichikawa T., Shimada H., Matsui M. (2005) shk1-D, a dwarf Arabidopsis mutant caused by activation of the CYP72C1 gene, has altered brassinosteroid levels. Plant J 42:13–22
Tester M., Davenport R. (2003) Na+ tolerance and Na+ transport in higher plants. Ann. Bot. 91:503–527
Tingey S., McElroy D., Kalla R., Fieg S., Wang M., Thornton S., Brettell R. (1997) Agrobacterium tumefaciens-mediated barley transformation. Plant J. 11:1369–1376
Villas-Boas S.G., Roessner U., Hansen M., Smedsgaard J, Nielsen J. (2006) Metabolome Analysis. Publ. Wiley & Sons, New Jersey, USA
Wan Y., Lemaux P.G. (1994) Generation of large numbers of independently transformed fertile barley plants. Plant Physiol. 104:37–48
Wild, A. (Ed) (1988). Russell’s soil conditions and plant growth, 11th edn. Longman, pp. 991.
Acknowledgements
This work is supported by funding to the Australian Centre for Plant Functional Genomics from the Australian Research Council, Grain Research Development Cooperation, South Australian Government, Victorian Government, Department for Primary Industry, University of Melbourne, University of Adelaide and University of Queensland. CL thanks the Danish Research Council for funding. Special thanks to Suganthi Suren for GC-MS analysis assistance, to Alex Johnson for rice transformation, to Rohan Singh and Konny Beck-Oldach for barley transformation, to Jodie Kretschmer for technical assistance and Ursula Langridge for plant maintenance in the glasshouse.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jacobs, A., Lunde, C., Bacic, A. et al. The impact of constitutive heterologous expression of a moss Na+ transporter on the metabolomes of rice and barley. Metabolomics 3, 307–317 (2007). https://doi.org/10.1007/s11306-007-0056-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-007-0056-4