
Abstract
Chromium substituted cobalt ferrites CoFe2−xCrxO4 (x = 0; 1; 1.5) were synthesized through a soft chemistry method—the gluconate precursor route. The gluconate precursors were characterized by infrared spectroscopy (IR), ultraviolet–visible spectroscopy and thermal analysis. The spinel oxide powders were investigated by X-ray diffraction (XRD), scanning electron microscopy (SEM), Mössbauer spectroscopy and Brunauer–Emmett–Teller N2 adsorption–desorption analyses. XRD indicated the formation of the spinel-type phase with good crystallinity. The mean crystalline domains size decreased from 23.8 to 17.8 nm with the increase in the chromium content. SEM revealed faceted particles for which the particle sizes varied significantly with the chromium content. The chromium substituted cobalt ferrites were found to have high catalytic performance.
Similar content being viewed by others

Introduction
Nanostructured spinel oxides such as ferrites, chromites or chromium substituted ferrites have been extensively studied due to their interesting magnetic, electrical and catalytic properties. These spinel oxides have various potential applications as components of electronic devices, magnetic storage devices, microwave and high frequency absorbing materials, active components of ferrofluids and catalysts (Goldman 2006; Jauhar and Singhal 2014; Hankare et al. 2011).
The nanosized cobalt-based spinel ferrites and chromites were found to be the best candidates for the catalytic combustion of methane; among them cobalt chromite (CoCr2O4) and cobalt ferrite (CoFe2O4). They have high activity, high stability and, also, low cost (Fino et al. 2007; Chen et al. 2013).
CoCr2O4 is a normal spinel due to the large octahedral ligand field stabilization energy of Cr3+. CoFe2O4 has a partial inverse spinel structure. The inversion degree of the spinel structure depends on the synthesis conditions.
It is well known that the addition of metal cations of different valence states in the tetrahedral and/or the octahedral sites may improve the properties of the ferrites. The substitution of Fe3+ with Cr3+ in the cobalt ferrite leads to changes in the A[B2]O4 structure, given that the Cr3+ ions have strong preference for the B sites (Vadivel et al. 2014).
Taking into account these considerations, it was considered interesting to study the catalytic activity of chromium-substituted ferrites in the methane combustion.
The method of preparation plays an important role in the composition, the structure, the morphology and implicitly, on the properties of the spinel oxides.
Several methods, such as the coprecipitation (Vadivel et al. 2014; Pervaiz and Gul 2012), the hydrothermal and micro-emulsion routes (Zhang et al. 2016; Koseoglu et al. 2012; Iqbal and Siddiquah 2008), the sol–gel/sol gel combustion method (Bhasker and Ramana Reddy 2015; Raghasudha et al. 2016; Toksha et al. 2011), the combustion process (Gingasu et al. 2015a; Sijo 2017; Sharma et al. 2017) and the citrate precursor route (Hankare et al. 2009, 2011), have been developed.
The precursor method can be easily used to obtain nanosized spinel oxides due to the simplicity of the process and the low-temperature treatment (Mindru et al. 2014).
The main goal of this work is the preparation of nanocrystalline CoFe2−xCrxO4 (x = 0; 1; 1.5) spinel-type oxide powders through thermal decomposition of multimetallic gluconate compounds—the gluconate precursor route. The catalytic performance of the chromium substituted cobalt ferrites in the total oxidation reaction of methane was evaluated.
Experimental
Reagents
All chemicals chromium nitrate (Cr(NO3)3·9H2O), iron nitrate (Fe(NO3)3·9H2O), cobalt nitrate (Co(NO3)2·6H2O) and δ-gluconolactone (C6H10O6) were of reagent quality (Merck).
CoFe2−xCrxO4 ferrites preparation
Chromium substituted cobalt ferrites have been obtained by the same procedure as for chromium substituted copper ferrites described in our previous work (Mindru et al. 2015).
The following systems were investigated for the preparation of the gluconate precursors:
2(Fe 3+2− x , Cr 3+ x ):1Co2+:8C6O7H11−, where x = 0; 1; 1.5; C6O7H11− = gluconate anion.
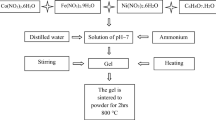
The iron(III), chromium(III), and cobalt(II) nitrates were dissolved in minimum amount of water and mixed with an aqueous solution of gluconic acid (C6O7H12), obtained by the hydrolysis of δ-gluconolactone (C6H10O6) at 80 °C. The molar ratio of metal nitrates to gluconic acid was 2:1:8. The pH was raised to 6 by adding NH4OH:CH3OH (1:1). Methanol was added until light-grey precipitates were formed. After 24 h/4 °C, the precipitates were filtered and dried over P4O10.
The elemental analysis was consistent with the formula:
(NH4)2[CoFe2(C6O7H11)4(OH)2]·2H2O I: Anal.: Calcd.: Fe% 10.60; Co%: 5.58; C%: 27.25; N%: 2.65; H%: 5.49; found: Fe% 10.35; Co%: 5.23; C%: 26.76; N%: 2.65; H%: 5.13.
(NH4)2[CoFeCr(C6O7H11)4(OH)2]·6H2O II: Anal.: Calcd.: Cr%: 4.62; Fe% 4.98; Co%: 5.24; C%: 25.60; N%: 2.49; H%: 5.69; found: Cr%: 4.79; Fe% 5.16; Co%: 5.43; C%: 25.95; N%: 2.80; H%: 5.21.
(NH4)2[CoFe0.5Cr1.5(C6O7H11)4(OH)2]·H2O III: Anal.: Calcd.: Cr%: 7.55; Fe% 2.71; Co%: 5.71; C%: 27.88; N%: 2.71; H%: 5.42; found: Cr%: 7.35; Fe% 2.39; Co%: 5.53; C%: 27.92; N%: 2.81; H%: 5.60.
The calcination of these precursors at 700 °C for 1 h leads to the formation of well-crystallized CoFe2−xCrxO4 (x = 0; 1; 1.5) spinel-type oxide powders.
Characterization techniques
The metal content of the compounds was determined by atomic absorption spectroscopy with a SAA1 instrument; the C, N and H values were obtained using a Carlo Erba Model 1108 CHNSO elemental analyzer.
The IR spectra were recorded on KBr pellets with a JASCO FTIR 4100 spectrophotometer in the 4000–400 cm−1 range.
The absorption spectra were recorded with a JASCO V-670 spectrophotometer in the domain 200–1800 nm.
The thermal measurements (TG and DSC) were performed using a Netzsch TG 449C STA Jupiter. Samples were placed in an open alumina crucible and heated with 10 °C min−1 from room temperature up to 900 °C, under a dried air flow of 20 mL min−1.
X-ray diffraction data were collected using Rigaku’s Ultima IV X-ray powder diffractometer, operating at 40 kV and 30 mA, with CuKα radiation (λ = 1.5406 Å) and graphite monochromator, in parallel beam geometry with CBO optics, 0.02° step size and 0.5° min−1 scan speed. Phase identification was performed using Rigaku’s PDXL software, with Whole Powder Pattern Fitting (WPPF) module, connected to ICDD-PDF-2 database.
The morphology of the spinel oxides was analyzed by SEM using a FEI Quanta 3D FEG apparatus. Micrographs were obtained from a secondary electron detector working at accelerating voltages of 5 kV. The elemental chemical composition was determined with an energy dispersive X-ray (EDX) spectrometer using an accelerating voltage of 30 kV.
The 57Fe Mössbauer spectra were recorded at room temperature in transmission geometry, using a WissEL-ICE Oxford Mössbauer cryomagnetic system (Wissenschaftliche Elektronik GmbH, Starnberg, Germany, and ICE Innovative cryogenic system, Oxford, UK) and a 15 mCi 57Co source in Rhodium matrix. α-Fe foil was used in a range of ± 10 mm s−1 for the velocity calibration.
Nitrogen adsorption/desorption isotherms were measured at 77 K using a Micromeritics ASAP 2020 automated gas adsorption system. The samples were degassed at 250 °C for 4 h before analysis. Specific surface areas (SBET) were calculated by the Brunauer–Emmett–Teller (BET) equation using adsorption data in the relative pressure range 0.05–0.3, and pore size distribution from the desorption branch using the Barrett–Joyner–Halenda (BJH) model. The total pore volume (Vtotal) was estimated from the amount adsorbed at p/p0 of 0.99.
The hydrogen temperature-programmed reduction (H2-TPR) measurements were carried out in a flow system with a Chembet 3000-Quantachrome instrument apparatus equipped with thermal conductivity detectors (TCD). The continuous flow of 5% volume H2 in Ar (70 mL min−1) was passed over 50 mg powder of catalyst. The temperature was increased to 850 °C at a heating rate of 10 °C min−1.
The catalytic properties of the obtained cobalt ferrites were tested in CH4 combustion. The reactions were carried out in a fixed bed micro reactor under atmospheric pressure and volume rate air/CH4 gas mixture 2/1. The composition of CH4 gas mixture was 10% CH4, 5% N2, 85% Ar. The quartz micro reactor (I.D. = 6 mm) with 40 mg catalyst was placed inside a tubular electrical furnace. The measurements were performed on the samples heated stepwise in the temperature range 200–600 °C in steps of 50 °C with 30 min at each temperature level. CH4 and CO2 in the effluent gas were analyzed online using a gas chromatograph equipped with Porapaq QS 80/100, carbon molecular sieve column and thermal conductivity detector (Gingasu et al. 2015b).
Results and discussion
The selection of the ligands is the most important step in the precursor method because the multimetallic complex compounds used as precursors must decompose at low temperatures and generate only volatile products. The ligands that largely satisfy these requirements are the anions of carboxylic/polycarboxylic and polyhydroxycarboxylic acids: citrate, malate, tartrate, gluconate, etc. (Mindru et al. 2014). The gluconic acid (C6H12O7) is well known as a very good metal sequestrant agent.
The following gluconate complex compounds have been isolated and investigated by infrared spectroscopy, ultraviolet–visible spectroscopy and thermal analysis:
(NH4)2[CoFe2(C6O7H11)4(OH)2]·2H2O (I), (NH4)2[CoFeCr(C6O7H11)4(OH)2]·6H2O (II) and (NH4)2[CoFe0.5Cr1.5(C6O7H11)4(OH)2]·H2O (III).
Characterization of the gluconate precursors
The IR spectra of the gluconate compounds show the presence of two strong bands assigned to the coordinated COO− groups—νasymOCO (at 1606 cm−1 for compound I, ~ 1614 cm−1 for compound II and 1616 cm−1 for compound III) and νsymOCO (1398 cm−1 for I and 1384 cm−1 for II and III) (Fig. 1).

IR spectra of the gluconic acid (a) and gluconate precursor powders: b (NH4)2[CoFe2(C6O7H11)4(OH)2]·2H2O (I), c (NH4)2[CoFeCr(C6O7H11)4(OH)2]·6H2O (II) and d (NH4)2[CoFe0.5Cr1.5(C6O7H11)4(OH)2]·H2O (III)
The comparison of these spectra with the spectrum of the free acid emphasizes a change in the region 1030–1130 cm−1, a shift towards smaller values (1045–1090 cm−1), which is consistent with the coordination through one or several OH groups. This change demonstrates the formation of donor–acceptor bonds with metal ions and sustains the formation of a chelate ring upon complexation.
The spectra of the gluconate compounds also exhibit a broad and very intense band in the 3500–3000 cm−1 range which can be assigned to the vibration of water molecules/the formation of hydrogen bonds between water and/or hydroxyl groups. The presence on this band of a shoulder at ~ 2940 cm−1 supports the presence of NH4+ ions (ν3 stretching) (Nakamoto 1986; Petit et al. 1999).
The absorption spectra of gluconate compounds in the range 200–1800 nm were recorded to obtain information about the coordination modes of Co(II) (d7), Fe(III) (d5) and Cr(III) (d3) ions (Fig. 2).

Absorption spectra of gluconate precursor powders: (a) (NH4)2[CoFe2(C6O7H11)4(OH)2]·2H2O (I), (b) (NH4)2[CoFeCr(C6O7H11)4(OH)2]·6H2O (II) and (c) (NH4)2[CoFe0.5Cr1.5(C6O7H11)4(OH)2]·H2O (III)
These spectra evidence the characteristic features of Cr(III) (d3) and Co(II) (d7) ions in an octahedral symmetry. The intense band observed in the range 400–500 nm can be assigned to the spin allowed 4A2g → 4T1g (F) and 4T1g → 4T1g(P) (ν3) transitions characteristics of the Cr(III) and Co(II), in octahedral environment, respectively. The broadband in the range 600–800 nm is most probably a superposition of three bands assigned to: the 4A2g → 4T2g (F) transition of the Cr(III) (d3) ion, the 4T1g → 4A2g (ν2) transition of the Co(II) (d7) octahedral high spin ion and the forbidden transition of an octahedral Fe(III) (d5) high spin ion. A broad absorption band at ~ 1200 nm is present in all these spectra and can be assigned to a 4T1g → 4T2g (ν1) transition of the Co(II) (d7) ion. The band/shoulder below 400 nm can most likely be assigned to CT transitions (Lever 1984).
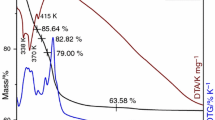
The thermal behavior of the gluconate precursors is very similar. Figure 3 presents, as an example, the thermal analysis TG-DSC recorded for the precursor (II).

TG, DTG and DSC curves of (NH4)2[CoFeCr(C6O7H11)4(OH)2]·6H2O (II)
The thermal decomposition of gluconate precursors takes place in the temperature range 30–400 °C. The elimination of 2NH3 molecules and 1H2O molecule (weight loss ~ 4.5 to 6% in TG curve) occurs below 140 °C. Above this temperature, the elimination of the remaining water molecules and the decomposition/oxidation of the gluconate ligands take place up to 400 °C with the formation of the oxidic phase (weight loss between 69 and 75%). This process is accompanied by a very strong exothermic peak on the DSC curve. For this reason, the thermal decomposition of the gluconate compounds can be considered a self-combustion process.
Characterization of CoFe2−xCrxO4 spinel powders
X-ray diffraction
To obtain well-crystallized chromium substituted cobalt ferrites, CoFe2−xCrxO4 (x = 0; 1; 1.5), the gluconate compounds were annealed at 700 °C/1 h. The X-ray diffractograms of the spinel oxides are presented in Fig. 4.

XRD patterns of spinel oxide powders: a CoFe2O4, b CoFeCrO4 and c CoFe0.5Cr1.5O4 obtained by thermal decomposition of gluconate precursors, calcined at 700 °C/1 h
The X-ray diffraction patterns of CoFe2−xCrxO4 (x = 0; 1; 1.5) powders confirm the formation of the spinel structure phase (ICDD 00-022-1086, S.G. Fd3m). No other phase was detected.
The lattice parameters decrease with the increase of the chromium content from 8.386 Å for sample x = 0 to 8.344 Å for x = 1.5. This decrease in the lattice parameter can be attributed to the substitution of the larger Fe3+ ion (0.78 Å) by the smaller Cr3+ ion (0.615 Å) (Koseoglu et al. 2012). The values of the lattice parameters were similar to the values reported in literature (Table 1). The values of the mean crystalline domains size were 23.8 nm for CoFe2O4, 20 nm for CoFeCrO4, and 17.8 nm for CoFe0.5Cr1.5O4.
Scanning electron microscopy
The SEM measurements of CoFe2−xCrxO4 (x = 0; 1; 1.5) powder samples (Fig. 5) revealed faceted particles of spinel phase crystallites. The particle size varies significantly with the Cr content. CoFe2O4 (Fig. 5a, d) exhibits typical sizes of a few hundred nanometers, CoFe0.5Cr1.5O4 (x = 1.5) shows similarly shaped, faceted particles of smaller size (Fig. 5b, e), while CoFeCrO4 (x = 1) has the finest particle sizes, with many primary particles in the nanometer range (Fig. 5c, f).

SEM micrographs and EDX spectra of spinel oxide powders: a, d, g, h CoFe2O4; b, e, i CoFeCrO4; c, f, j CoFe0.5Cr1.5O4
Dense agglomerates with well aligned crystalline domains can be observed in the SEM measurements of CoFe2O4 (Fig. 5g). This behavior is explained by the occurrence of a magnetic self-alignment of the domains that facilitates early sintering of polycrystalline aggregates and rapid grain growth.
The EDX elemental chemical analysis of all powders reveals the homogeneous composition of the spinel phase in each sample (Fig. 5h, j).
Mössbauer spectroscopy
Room temperature Mössbauer spectra of CoFe2−xCrxO4 (x = 0; 1; 1.5) powders, together with the computer fit (continuous lines) are presented in Fig. 6. Dramatic changes of the Mössbauer patterns can be observed with the increase of the Cr content in the samples. At x = 0, the spectrum exhibits a magnetic hyperfine like pattern with large and non Lorentzian line width (Fig. 6a).

Mössbauer spectra of spinel oxide powders: a CoFe2O4, b CoFeCrO4 and c CoFe0.5Cr1.5O4 obtained through thermal decomposition of gluconate precursors, calcined at 700 °C/1 h. Continuous lines represent the fit of the experimental points
To understand the deconvolution procedure of this spectrum, we have to remember some structural peculiarities of the iron-cobalt spinel. It is well known that the formula reflecting the cation distribution in the cobalt ferrite structure can be written as (Co1−yFey)[CoyFe2−y]O4, where () stands for the tetrahedral sites and [] stands for the octahedral ones (Petit and Forester 1971). The octahedral sites have six cations placed in tetrahedral sites as second nearest neighbors and each A sites has 12 B cations in the first sphere of coordination (Petit and Forester 1971). In an inverse spinel the divalent cations are placed only in the octahedral sites. In the cobalt ferrite, Co2+ ions can be also found in tetrahedral sites, being able to modify the magnetic hyperfine fields at octahedral Fe3+ sites via supertransfered interactions (Rao et al. 2014). It is natural to suppose a random distribution of Co2+ ions in the tetrahedral sites. The Mössbauer spectrum will reflect this distribution through a number of octahedral magnetic sublattices with decreasing magnetic fields, corresponding to the number of Co2+ ions in tetrahedral sites. The relative abundance of these magnetic sublattices depends on the probability of Co2+ cations to occupy the tetrahedral sites (Sawatzky et al. 1969).
The best fit through the experimental points was obtained by taking into account one tetrahedral magnetic sublattice and four octahedral sublattices B0, B1, B2, B3 ascribed to the presence of 0, 1, 2 and, respectively, 3 divalent cobalt ions in the tetrahedral positions of the cobalt spinel structure. From the fit of the tetrahedral and octahedral areas one can calculate the value of the inversion parameter y (Sawatzky et al. 1969; Liu et al. 2016; Carta et al. 2009). The calculated y is 0.27 and, consequently, the formula for our sample is (Co0.73Fe0.27)[Co0.27Fe1.73]O4.
The Mössbauer fit parameters: the isomer shift (IS), the quadrupole splitting (QS) and the magnetic hyperfine field (Hhf) are presented in Table 2. The tetrahedral magnetic sublattice was associated with lowest IS and smaller Hhf based on structural considerations regarding the electric charge density at the Fe3+ nucleus in the A sites and magnetic interactions in the system (Rao et al. 2014).
At x = 1, the Mössbauer spectrum is represented by a collapsed magnetic hyperfine component together with a central quadrupole doublet. The best fit was obtained with a rather large magnetic hyperfine field distribution (continuous green line in Fig. 6b) from ~ 4 to ~ 47 T and a central doublet (blue line in Fig. 6b) with IS = 0.245 mm s−1 and QS = 0.454 mm s−1. The magnetic field distribution could reflect a distribution in nanoparticle sizes, while the central doublet indicates the presence of a paramagnetic phase.
At x = 1.5, the Mössbauer spectrum consists only of a central quadrupole doublet with IS = 0.258 mm s−1 and QS = 0.451 mm s−1. Within the errors limits, these parameters are close to the parameters of the doublet in the spectrum at x = 1. Consequently, one can claim the appearance of the chromium rich phase CoFe0.5Cr1.5O4, even at lower Cr concentrations (x < 1.5).
Brunauer–Emmett–Teller (BET) N2 adsorption–desorption analyses
To determine the textural properties of the samples, the nitrogen adsorption–desorption isotherms were recorded (Fig. 7). All the samples exhibit similar type IV isotherms which are usually associated with slit-shaped pores (Sing et al. 1985). The hysteresis loops appear at very high relative pressure (> 0.9) which suggests that the porosity is generated by the spaces between laterally interconnected crystallites. CoFe2O4 has the smallest specific surface area (4 m2 g−1), while the Cr substituted samples have almost similar SBET values: 19.5 m2 g−1 for CoFeCrO4 and 19.8 m2 g−1 for CoFe0.5Cr1.5O4. These results indicate that the substitution with Cr induces a significant increase of surface area which can be mainly attributed to the decrease of both crystallite domains size (from XRD measurements) and particle size (from SEM measurements). The total volume of pores also increases with gradual increasing of the chromium substitution degree (0.04 cm3 g−1 for CoFe2O4, 0.1 cm3 g−1 for CoFeCrO4 and 0.18 cm3 g−1 for CoFe0.5Cr1.5O4). CoFeCrO4 contains a small fraction of micropores (~ 7% according to t-plot analysis). As the content of Cr increases the microporosity disappears and the mesopores become larger, as can be seen in the pore size distribution graphs (Fig. 7).

N2 adsorption desorption isotherms and pore size distribution (inset) of the spinel oxide samples
Catalytic activity of CoFe2−xCrxO4 for methane combustion reaction
The reducibility (oxygen mobility) of the cobalt ferrite (CoFe2O4) and the chromium substituted cobalt ferrites (CoFeCrO4, CoFe0.5Cr1.5O4) was evaluated by H2-TPR.
Figure 8 suggests that these ferrites are stable up to 200 °C (CoFeCrO4, CoFe0.5Cr1.5O4) and 300 °C (CoFe2O4).

H2-TPR profiles of cobalt ferrite and chromium substituted cobalt ferrites
Above 300 °C, a set of four reduction peaks related to the reduction of the spinel phase can be observed for CoFe2O4 sample.
As illustrate in the literature, H2-TPR shows the effect of substituent on the reducibility and the influence of reduction conditions on the ferrite structure (Bellal et al. 2017; Meshkani and Rezaei 2015; Devaiah and Smirniotis 2017; Sastri et al. 1982; Qwabe et al. 2015; Chagas et al. 2016). H2-TPR of the pure iron oxide (α-Fe2O3) shows a characteristic curve with three peaks attributed to the partial reduction of Fe3+ to Fe2+, total reduction of Fe3+ to Fe2+ and the reduction of Fe2+ to Fe0 (Sastri et al. 1982; Dumitru et al. 2013; Nogueira et al. 2011). The formation of the CoFe2O4 spinel phase changes the H2-TPR profile; a broad peak and two or three peaks were identified. These peaks are associated with the reduction of CoO to metallic cobalt in conjunction with the reduction of Fe2O3 to Fe3O4, both present in ferrite phase.
The reduction peaks detected for the CoFe2O4 sample occur at high temperatures of 514, 602, 715, 775 °C. These peaks are attributed to the iron reduction in the cobalt ferrite. The H2 consumption for the cobalt ferrite was 1.4 μmol of H2/μmol which is lower than the theoretical value (3 μmol H2/μmol cobalt ferrite) required for complete reduction of iron (Dumitru et al. 2013). This indicates that not all iron was reduced.
The chromium substituted cobalt ferrites display different profiles of bulk reduction from 200 to 800 °C. The first two peaks with very low intensity can be associated with the removal of oxygen species adsorbed in oxygen vacancies and the partial reduction of cobalt to Co0 (Sastri et al. 1982; Tasca et al. 2011). The H2 consumption is the same for these processes. The next three peaks are associated with the partial reduction of Fe3+ to Fe2+ and partial reduction of Fe2+ to Fe0.
The theoretical value of H2 consumption required for the complete reduction of cobalt and iron in CoFe2−xCrxO4 (x = 1; 1.5) varies between 1.25 to 2.50 μmol of H2/μmol ferrite. TPR results show lower H2 consumption (0.9 and 0.5 μmol of H2/μmol) for CoFeCrO4, respectively, CoFe0.5Cr1.5O4 ferrites.
The CoFeCrO4 recovered after H2-TPR sample was characterized by XRD and SEM. Besides spinel phase, Co0 (ICDD 00-015-0806) and Fe0 (ICDD 00-006-0696) were identified (Fig. S1). The SEM measurements (Fig. S2) showed a noticeable refinement of particle size, and primary particle sizes corresponds very well with crystalline domains size calculated from XRD. Therefore, TPR causes the disaggregation of polycrystalline aggregates into individual nanoparticles with similar size (~ 20 nm) than XRD-determined size of coherent crystallite domains.
TPR confirm the stability of the chromium cations in +3 oxidation state (Meshkani and Rezaei 2015), emphasize the different Fe-Co interactions after chromium substitution and evidence the oxygen mobility—very important for applications in oxidation reactions. The reduction is reversible in the oxidation reactions. In air flow and high temperature, the reoxidation occurs more rapidly than the reduction (Qwabe et al. 2015; Nogueira et al. 2011).
The H2-TPR for the CoFe2O4 sample shows the reduction peaks at high temperatures. In contrast, CoFeCrO4 starts reducing at lower temperatures (Fig. 8). These results, together with the activity data, indicate a significant increase in conversion above 400 °C (CoFe2−xCrxO4; x = 1; 1.5) and 450 °C (CoFe2O4), when partial reduction of cobalt and iron cations occurs (Fig. 9).

Methane conversion as function of temperature over CoFe2−xCrxO4 (x = 0; 1; 1.5)
Figure 9 shows the variation of methane conversion with the composition of CoFe2−xCrxO4 (x = 0; 1; 1.5) ferrites. Better activity was obtained for the chromium substituted cobalt ferrite samples. Total conversion was obtained for CoFeCrO4 between 400 and 600 °C. The CoFeCrO4 sample after catalytic test was characterized by XRD (Fig. S1) and SEM (Fig. S2). In the X-ray diffraction, single-phase spinel was detected. The SEM measurements showed the same morphology and texture of the polycrystalline powder than the sample before catalytic test.
Three temperature/conversion stages of the methane oxidation reaction can be observed in Fig. 9. This could be explained by the coexistence of Langmuir–Hinshelwood and the Mars–van Krevelen mechanisms during the reaction progress (Zasada et al. 2017). At low temperatures (200 °C < T < 400–450 °C) dominant is Langmuir–Hinshelwood mechanism. The oxidation is the result of CH4 and O2 reaction adsorbed on the ferrite surface. At higher temperature the mechanism of methane oxidation is according to the Mars–van Krevelen scheme and the conversion increase till 100%. The Mars–van Krevelen redox type mechanism considers the redox nature of the catalyst surface (Bellal et al. 2017; Dumitru et al. 2013; Royer et al. 2005). The catalyst activity depends strongly on the mobility of the lattice oxygen and on the reducibility (valence change) of the lattice metal ions. The oxygen species [O2−]s interact with reduced metal ions, forming oxygen vacancies on surface [O2−]s + M → [*]S + [MO], and are being reformed by the reaction of the last with O2 molecules [[*]S + 1/2O2 → [O2−]s].
In the case of the chromium substituted cobalt ferrites, H2-TPR results evidence easily reducible cobalt ions. At lower temperatures (T < 400 °C) TPR profiles are similar for these ferrites. Significant variations can be observed at higher temperatures (Fig. 8). However, the conversion values are different for these samples at lower temperature (Fig. 9). This is due to the different mechanisms of the reactions that dominate before and after 400 °C temperature. These metal ions (Co, Cr, Fe) which exhibit more than one oxidation state, may participate in the cyclic electron process of the reaction. Simultaneously, Cr3+ and Fe3+ cations influence the properties regardless of Co2+ properties in the ferrite structure.
Significantly lower conversion was obtained for CoFe2O4 catalysts (65%). It can also be observed that the maximum conversion is shifted to higher temperatures (550–600 °C).
According to literature (Chagas et al. 2016; Tasca et al. 2011; Royer et al. 2005; Carta et al. 2009; Kantserova et al. 2003), the ions located in the octahedral B-sites behave as active sites, whereas the ions in the tetrahedral A-sites are catalytically inactive. The catalytic activity is due to the octahedrally coordinated cations which are almost exclusively on the surface of spinel crystallites. The chromium substituted cobalt ferrites have an inverse spinel structure, indicating that the difference in their catalytic activity is most likely determined by the properties of the metal ions M2+ in the composition of the spinels (Olar et al. 2004). The higher activity of the ferrite may be explained by the cations environment in the complex oxide, which facilitates the transfer of the transition metal ion into a higher oxidation state.
The inversion parameter, y, for the CoFe2O4 sample was calculated from the Mössbauer spectrum. The formula proposed for this sample is (Co0.73Fe0.27)[Co0.27Fe1.73]O4, indicating a small number of cobalt ions and a higher number of iron ions in the octahedral sites. H2-TPR shows that the ferrite is stable up to 500 °C, indicating the presence of stable Co2+ ions. In this case, the catalytic effect is due to the Co2+ ions from octahedral sites, their influence on the iron ions from the ferrite and the oxygen adsorption.
The catalytic tests indicate that all the catalysts show 100% selectivity towards CO2 in the investigated temperature range. No CO was observed during the reaction.
Conclusions
To summarize, we have successfully synthesized nanocrystalline CoFe2−xCrxO4 (x = 0; 1; 1.5) spinel-type oxide powders through the gluconate precursor route. Three new compounds have been isolated and characterized: (NH4)2[CoFe2(C6O7H11)4(OH)2]·2H2O, (NH4)2[CoFeCr(C6O7H11)4(OH)2]·6H2O and (NH4)2[CoFe0.5Cr1.5(C6O7H11)4(OH)2]·H2O.
The structural, morphological and textural properties of the CoFe2−xCrxO4 spinel oxide powders have been investigated. The X-ray diffractograms indicate the crystallization of a cubic spinel structure; no other phases were identified in the samples. The lattice constant and average crystallite size are found to decrease with the increase in chromium content. The SEM measurements revealed faceted particles of spinel phase crystallites.
With the increase of the chromium content in the analyzed samples, changes of the Mössbauer patterns can be observed. At x = 0, from Mössbauer data, the inversion parameter y was found to be 0.27; the corresponding stoichiometric formula was calculated as (Co0.73Fe0.27)[Co0.27Fe1.73]O4. At x = 1, Mössbauer spectra evidenced a magnetic hyperfine field distribution accompanied by a quadrupole doublet. Finally, at x = 1.5, the Mössbauer spectrum consists in a quadrupole doublet with the hyperfine parameters IS and QS close to the doublet appearing at x = 1; this behavior suggests the initiation of CoFe0.5Cr1.5O4 structure crystallization even at x = 1 (CoFeCrO4).
All chromium substituted cobalt ferrites CoFe2−xCrxO4 (x = 0; 1; 1.5) have been tested in methane oxidation reaction. The maximum values of the methane conversion were reached for chromium substituted cobalt ferrites after 450 °C temperature of reaction.
References
Bellal YH, Zouaoui-Mahzoul N, Lounas I, Benadda A, Benrabaa R, Auroux A, Meddour-Boukhobza L, Djadoun A (2017) Cobalt and cobalt-iron spinel oxides as bulk and silica supported catalysts in the ethanol combustion reaction. J Mol Catal A Chem 426:97–106. https://doi.org/10.1016/j.molcata.2016.11.005
Bhasker SU, Ramana Reddy MV (2015) Effect of chromium substitution on structural, magnetic and electrical properties of magneto-ceramic cobalt ferrite nano-particles. J Sol Gel Sci Technol 73:396–402. https://doi.org/10.1007/s10971-014-3546-7
Carta D, Casula MF, Falqui A, Loche D, Mountjoy G, Sangregorio C, Corrias A (2009) A structural and magnetic investigation of the inversion degree in ferrite nanocrystals MFe2O4 (M = Mn Co, Ni). J Phys Chem C 113:8606–8615. https://doi.org/10.1021/jp901077c
Chagas CA, De Souza EF, De Carvalho MCNA, Martins RL, Schmal M (2016) Cobalt ferrite nanoparticles for the preferential oxidation of CO. Appl Catal A 519:139–145. https://doi.org/10.1016/j.apcata.2016.03.024
Chen J, Zhang X, Arandiyan H, Peng Y, Chang H, Li J (2013) Low temperature complete combustion of methane over cobalt chromium oxides catalysts. Catal Today 201:12–18. https://doi.org/10.1016/j.cattod.2012.03.026
Devaiah D, Smirniotis PG (2017) Effects of the Ce and Cr contents in Fe–Ce–Cr ferrite spinels on the high-temperature water–gas shift reaction. Ind Eng Chem Res 56:1772–1781. https://doi.org/10.1021/acs.iecr.6b04707
Dumitru R, Papa F, Balint I, Culita DC, Munteanu C, Stanica N, Ianculescu A, Diamandescu L, Carp O (2013) Mesoporous cobalt ferrite: a rival of platinum catalyst in methane combustion reaction. Appl Catal A 467:178–186. https://doi.org/10.1016/j.apcata.2013.07.013
Fino D, Solaro S, Russo N, Saracco G, Specchia V (2007) Catalytic removal of methane over thermal-proof nanostructured catalysts for CNG engines. Top Catal 42–43:449–454. https://doi.org/10.1007/s11244-007-0223-x
Gingasu D, Diamandescu L, Mindru I, Marinescu G, Culita DC, Calderon-Moreno JM, Preda S, Bartha C, Patron L (2015a) Chromium substituted cobalt ferrites by glycine–nitrates process. Croat Chem Acta 88:445–451. https://doi.org/10.5562/cca2743
Gingasu D, Mindru I, Culita DC, Patron L, Calderon-Moreno JM, Osiceanu P, Preda S, Oprea O, Parvulescu V, Teodorescu V, Walsh JPS (2015b) Structural, magnetic and catalytic properties of cobalt chromite through precursor method. Mater Res Bull 62:52–64. https://doi.org/10.1016/j.materresbull.2014.11.009
Goldman A (2006) Modern ferrite technology, 2nd edn. Springer, New York
Hankare PP, Sankpal UB, Patil RP, Mulla IS, Lokhande PD, Gajbhiye NS (2009) Synthesis and characterization of CoCxFe2−xO4 nanoparticles. J Alloys Compd 485:98–801. https://doi.org/10.1016/j.jallcom.2009.06.087
Hankare PP, Sankpal UB, Patil RP, Lokhande PD, Sasikala R (2011) Synthesis, characterization and catalytic activity of chromium substituted cobalt ferrospinels. Mater Sci Eng B 176:103–109. https://doi.org/10.1016/j.mseb.2010.10.005
Iqbal MJ, Siddiquah MR (2008) Electrical and magnetic properties of chromium-substituted cobalt ferrite nanomaterials. J Alloys Compd 453:513–518. https://doi.org/10.1016/j.jallcom.2007.06.105
Jauhar S, Singhal S (2014) Substituted cobalt nano-ferrites, CoMxFe2−xO4 (M = Cr3+, Ni2+, Cu2+, Zn2+; 0.2 ≤ x ≤ 1.0) as heterogeneous catalysts for modified Fenton’s reaction. Ceram Int 40:11845–11855. https://doi.org/10.1016/j.ceramint.2014.04.019
Kantserova MR, Gavrilenko KS, Kosmambetova GR, Il’in VG, Orlik SN (2003) Deep oxidation of methane over nano-sized ferrites with spinel structures. Theor Exp Chem 39:322–329. https://doi.org/10.1023/B:THEC.0000003494.21579.14
Koseoglu Y, Oleiwi MIO, Yilgin R, Kocbay AN (2012) Effect of chromium addition on the structural, morphological and magnetic properties of nano-crystalline cobalt ferrite system. Ceram Int 38:6671–6676. https://doi.org/10.1016/j.ceramint.2012.05.055
Lever ABP (1984) Inorganic electronic spectroscopy, 2nd edn. Elsevier Publishing Company, Amsterdam
Liu M, Lu M, Wang L, Xu S, Zhao J, Li H (2016) Mössbauer study on the magnetic properties and cation distribution of CoFe2O4 nanoparticles synthesized by hydrothermal method. J Mater Sci 51:5487–5492. https://doi.org/10.1007/s10853-016-9853-3
Meshkani F, Rezaei M (2015) Preparation of nanocrystalline metal (Cr, Al, Mn, Ce, Ni, Co and Cu) modified ferrite catalysts for the high temperature water gas shift reaction. Renew Energy 74:588–598. https://doi.org/10.1016/j.renene.2014.08.037
Mindru I, Gingasu D, Culita DC, Marinescu G, Patron L (2014) Magnetic ferrites: design and synthesis. In: Lyshevski SE (ed) Dekker encyclopedia of nanoscience and nanotechnology, 3rd edn. CRC Press, New York, pp 2176–2189
Mindru I, Gingasu D, Patron L, Marinescu G, Calderon-Moreno JM, Diamandescu L, Preda S, Oprea O (2015) Chromium substituted copper ferrites via gluconate precursor route. Ceram Int 41:5318–5330. https://doi.org/10.1016/j.ceramint.2014.12.081
Nakamoto K (1986) Infrared and Raman spectra of inorganic and coordination compounds, 4th edn. Wiley, New York
Nogueira IM, Sabadia GQ, Moreira AA, Filho JM, Oliveira AC (2011) Investigation of the deactivation of iron nanocomposites by coking in the dehydrogenation of ethylbenzene. J Mol Catal A Chem 351:81–92. https://doi.org/10.1016/j.molcata.2011.09.020
Olar R, Badea M, Diamandescu L, Cristurean E, Brezeanu M (2004) Soft chemical synthesis and characterisation of some substituted ferrites. J Alloys Compd 363:262–267. https://doi.org/10.1016/S0925-8388(03)00452-3
Pervaiz E, Gul IH (2012) Enhancement of electrical properties due to Cr3+ substitution in Co-ferrite nanoparticles synthesized by two chemical techniques. J Magn Magn Mater 324:3695–3703. https://doi.org/10.1016/j.jmmm.2012.05.050
Petit GA, Forester DW (1971) Mössbauer study of cobalt zinc ferrites. Phys Rev B 4:3912–3926. https://doi.org/10.1103/PhysRevB.4.3912
Petit S, Righi D, Madejova J, Decarreau A (1999) Interpretation of the infrared NH4 + spectrum of the NH4 +-clays: application to the evaluation of the layer charge. Clay Miner 34:543–549. https://doi.org/10.1180/000985599546433
Qwabe LQ, Friedrich HB, Singh S (2015) Preferential oxidation of CO in a hydrogen rich feed stream using Co–Fe mixed metal oxide catalysts prepared from hydrotalcite precursors. J Mol Catal A Chem 404-405:167–177. https://doi.org/10.1016/j.molcata.2015.04.020
Raghasudha M, Ravinder D, Veerasomaiah P (2016) Investigation of superparamagnetism in pure and chromium substituted cobalt nanoferrite. J Magn Magn Mater 420:45–50. https://doi.org/10.1016/j.jmmm.2016.06.090
Rao GSN, Rao BP, Hamdeh HH (2014) Mössbauer spectroscopic study of high magnetostrictive cobalt chromium ferrites for automobile torque sensors. Procedia Mater Sci 6:1511–1515. https://doi.org/10.1016/j.mspro.2014.07.131
Royer S, Alamdari H, Duprez D, Kaliaguine S (2005) Oxygen storage capacity of La1−xA′xBO3 perovskites (with A′ = Sr, Ce; B = Co, Mn)—relation with catalytic activity in the CH4 oxidation reaction. Appl Catal B 58:273–288. https://doi.org/10.1016/j.apcatb.2004.12.010
Sastri MVC, Viswanath RP, Visvanathan B (1982) Studies on the reduction of iron oxide with hydrogen. Int J Hydrog Energy 7:951–955. https://doi.org/10.1016/0360-3199(82)90163-X
Sawatzky GA, Van Der Woude F, Morrish AH (1969) Mössbauer study of several ferrimagnetic spinels. Phys Rev 187:747–757. https://doi.org/10.1103/PhysRev.187.747
Sharma S, Choudhary N, Verma MK, Sharma ND, Singh D (2017) Cation distribution and magnetic properties of nano and bulk CoCrFeO4 ferrite synthesized by glycine-nitrate combustion method. Ceram Int 43:11083–11089. https://doi.org/10.1016/j.ceramint.2017.05.154
Sijo AK (2017) Magnetic and structural properties of CoCrxFe2−xO4 spinels prepared by solution self combustion method. Ceram Int 43:2288–2290. https://doi.org/10.1016/j.ceramint.2016.11.010
Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquerol J, Siemieniewska T (1985) Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (IUPAC recommendations 1984). Pure Appl Chem 57:603–619. https://doi.org/10.1351/pac198557040603
Tasca JE, Quincoces CE, Lavat A, Alvarez AM, Gonzalez MG (2011) Preparation and characterization of CuFe2O4 bulk catalysts. Ceram Int 37:803–812. https://doi.org/10.1016/j.ceramint.2010.10.023
Toksha BG, Shirsath SE, Mane ML, Patange SM, Jadhav SS, Jadhav KM (2011) Autocombustion high-temperature synthesis, structural, and magnetic properties of CoCrxFe2−xO4 (0 ≤ x ≤ 1.0). J Phys Chem C 115:20905–20912. https://doi.org/10.1021/jp205572m
Vadivel M, Babu RR, Sethuraman K, Ramamurthi K, Arivanandhan M (2014) Synthesis, structural, dielectric, magnetic and optical properties of Cr substituted CoFe2O4 nanoparticles by co-precipitation method. J Magn Magn Mater 362:122–129. https://doi.org/10.1016/j.jmmm.2014.03.016
Zasada F, Janas J, Piskorz W, Gorczyńska M, Sojka Z (2017) Total oxidation of lean methane over cobalt spinel nanocubes controlled by the self-adjusted redox state of the catalyst: experimental and theoretical account for interplay between the Langmuir–Hinshelwood and Mars–Van Krevelen mechanisms. ACS Catal 7:853–2867. https://doi.org/10.1021/acscatal.6b03139
Zhang W, Zuo X, Zhang D, Wu C, Silva SRP (2016) Cr3+ substituted spinel ferrite nanoparticles with high coercivity. Nanotechnology 27:245707. https://doi.org/10.1088/0957-4484/27/24/245707
Acknowledgments
Support of the EU (ERDF) and Romanian Government, allowing for the acquisition of the research infrastructure under POS-CCE O 2.2.1 project INFRANANOCHEM-Nr. 19/01.03.2009, is gratefully acknowledged. The work also benefits from the support of the “Materials Science and Advanced Characterization Methods” Programme of the “Ilie Murgulescu” Institute of Physical Chemistry, financed by the Romanian Academy. One of the authors (LD) acknowledges the financial support under the Core Program PN 10N/2017.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mindru, I., Gingasu, D., Diamandescu, L. et al. CoFe2−xCrxO4 ferrites: synthesis, characterization and their catalytic activity. Chem. Pap. 72, 3203–3213 (2018). https://doi.org/10.1007/s11696-018-0553-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-018-0553-0